

Bone morphogenetic proteins (BMPs), a class of secreted TGF-β-related proteins, play key roles in many cell differentiation decisions in metazoan development. BMP signaling is dysregulated in developmental syndromes and contributes to progressive diseases, including cancers.
BMP ligands activate signaling responses through tetrameric complexes consisting of 2 type II (RII) and 2 type I (RI) transmembrane receptor kinases. Ligand binding induces RII receptors to phosphorylate and activate RI receptors, and these then activate the intracellular effectors Smad1 and Smad5 through C-terminal serine phosphorylation, which stands in contrast to activation of Smad2 and Smad3 by TGF-βs and activins. Activated Smads form trimeric complexes with one Smad4 that activate or repress target gene transcription. Smad activation is inhibited by “inhibitory Smads”, i.e., Smad6 and Smad7, which bind to RI receptors, thus preventing activation of Smad1 and Smad5 in response to BMPs, or Smad2 and Smad3 in response to TGF-βs or activins. Smad6 inhibits primarily BMP signaling, and Smad7 inhibits BMP- and TGF-β/activin-induced Smad signaling.
We reported that arginine methylation of Smad6 by the methyltransferase PRMT1 initiates BMP-induced Smad signaling, prior to Smad1/5 activation, through C-terminal phosphorylation.1 In the absence of BMP signaling, Smad6 binds to the BMP-RI, thus preventing leaky BMP signaling, and PRMT1 binds to the BMP-RII. In response to BMP binding to and stabilization of the RII-RI complex, PRMT1 methylates the BMP-RI-associated Smad6 at Arg74. This critical step reduces the affinity of Smad6 for the RI receptors, resulting in Smad6 dissociation and enabling BMP-induced Smad1/5 activation. Dissociation of Smad6 from RI receptors moves Smad6 toward association with the RII receptors. Our study introduces the notion that Smad6 safeguards against BMP signaling activation, complementing its role in a negative feedback loop following Smad activation, and defines a novel role for protein methylation in initiating a signal transduction pathway.
In addition to Arg methylation, Smad6 is targeted for mono-ubiquitylation at Lys174. Also, this modification decreases the affinity of Smad6 for the BMP-RI and enhances BMP-induced Smad signaling.2 Whether Smad6 methylation and mono-ubiquitylation functionally crosstalk remains to be seen.
The functional linkage of PRMT1 and Smad6 most likely impacts cancer initiation and progression, since Smad6 and PRMT1 expression are deregulated in cancers. In lung3 and pancreatic ductal4 adenocarcinomas, increased Smad6 expression links with poor prognosis.3,4 Furthermore, increased Smad6 expression contributes to the malignant phenotype, apparent by dramatically increased anchorage-independent growth of pancreatic ductal cancer cells.4 Considering the epistatic relationship between Smad6 and PRMT1,1 these findings suggest that decreased Smad6 methylation, e.g., due to decreased PRMT1 expression, attenuates BMP signaling with an outcome that is similar to increased Smad6 expression. It will be interesting to correlate Arg methylation of Smad6 with BMP responsiveness in cancer cell proliferation.
Evaluating the role of PRMT1 in cancer is complex because of its many roles and targets. PRMT1 accounts for about 80% of all arginine methyltransferase activity in cells. It methylates histone 4 (H4) on Arg3 (R3) and thus allows the activation of many genes. In fact, PRMT1 was first linked to malignant transformation of hematopoietic cells when it was identified as a component of a transcription complex that mediates H4R3 methylation and H4 acetylation, linking an oncogenic role to its methyltransferase activity. PRMT1 also methylates non-histone proteins, including key mediators of cell proliferation, RNA processing, and DNA repair. Therefore, any change in PRMT1 expression or activity is expected to regulate many targets. PRMT1 expression was increased in bladder and colon cancer samples, which may correlate with stimulation of cell proliferation,5 but is downregulated in some carcinomas, such as breast carcinoma samples.6 Decreased PRMT expression in these tumors is expected to attenuate BMP signaling and benefit cancer initiation and behavior, yet this needs to be verified.
PRMT1 is expressed as different splicing isoforms, with PRMT1v1, the isoform that we showed to methylate Smad6,1 as the predominant isoform.5 Changes in the relative levels of PRMT1 isoforms were seen in cancers, with increased expression of PRMT1v2, a predominantly cytosolic isoform, seen in breast cancers. PRMT1v2 is unable to methylate histones but targets cytosolic proteins and promotes the survival and invasiveness of breast cancer cells.7 The role of PRMT1v2 in Smad6 methylation and BMP responsiveness will be interesting areas of research.
Finally, the role of PRMT1-mediated Smad6 methylation in cancer cells may depend on the stage of cancer. As the role of BMP signaling in cancer progression is not well defined, yet increasingly appreciated, we may draw parallels with the more extensive knowledge of TGF-β signaling in cancer progression.8 Early in carcinoma progression, cells deactivate the anti-proliferative component of TGF-β signaling, and increased TGF-β signaling promotes epithelial–mesenchymal transdifferentiation and cancer cell invasion, setting the stage for cancer dissemination.8 By analogy, the roles of BMP signaling and dysregulation of Smad6 methylation by PRMT1 may be stage-dependent, and attenuated Smad6 methylation may benefit cancers at an early stage. Whether PRMT1 also regulates Smad7 and defines the role of TGF-β signaling in cancer progression is an exciting concept that is worth exploring. (Fig. 1)
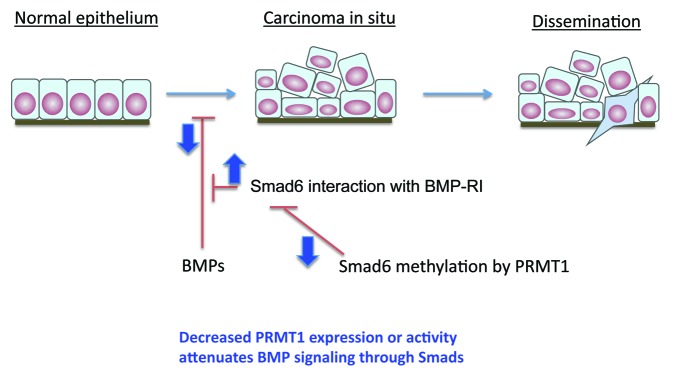
Figure 1. Roles of BMP signaling and deregulation of Smad6 methylation by PRMT1 may depend on the stage of cancer progression. BMP signaling may inhibit cancer development at an early stage. Impaired Smad6 methylation through dowregulation of PRMT1 expression or through changes in differential PRMT1 isoform generation may benefit cancer progression at an early stage.
Xu J, et al. Mol Cell. 2013;51:5–19. doi: 10.1016/j.molcel.2013.05.004.
References
- 1.Xu J, et al. Mol Cell. 2013;51:5–19. doi: 10.1016/j.molcel.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang X, et al. EMBO J. 2013;32:996–1007. doi: 10.1038/emboj.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeon HS, et al. Cancer Res. 2008;68:9686–92. doi: 10.1158/0008-5472.CAN-08-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kleeff J, et al. Biochem Biophys Res Commun. 1999;255:268–73. doi: 10.1006/bbrc.1999.0171. [DOI] [PubMed] [Google Scholar]
- 5.Yoshimatsu M, et al. Int J Cancer. 2011;128:562–73. doi: 10.1002/ijc.25366. [DOI] [PubMed] [Google Scholar]
- 6.Mathioudaki K, et al. Br J Cancer. 2008;99:2094–9. doi: 10.1038/sj.bjc.6604807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goulet I, et al. J Biol Chem. 2007;282:33009–21. doi: 10.1074/jbc.M704349200. [DOI] [PubMed] [Google Scholar]
- 8.Katsuno Y, et al. Curr Opin Oncol. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]