

Cells during mitosis and meiosis undergo chromosome condensation through a process that depends on evolutionary conserved large protein complexes known as condensins. There are two condensing complexes, condensin I and II, each of which is composed of five subunits. Condensin I and II play essential, yet distinct, roles in chromosome condensation and segregation during mitosis. Recent findings also revealed that condensin complexes contribute to a variety of interphase chromosome functions, such as gene regulation, recombination, and repair.1
Murakami-Tonami et al.2 in this issue provide evidence for a crosstalk between SMC2-mediated chromosome condensation and MYCN amplified neuroblastoma cells. They demonstrate that SMC2, a condensin core subunit, is regulated by MYCN, a MYC family member. The MYC family members (MYC, MYCN, and MYCL) are transcription factors, which regulate the expression of a wide range of genes involved in different cellular functions. Amplification of MYC family members has oncogenic properties and has been found in carcinomas of the cervix, colon, breast, lung, and stomach. The MYC family members have been explored in recent years as promising targets for anti-cancer drugs.
While studying the molecular signatures underlying neuroblastoma development, Murakami-Tonami et al. observed higher expression levels of 79 genes in MYCN-amplified tumor mice. The authors have chosen the SMC2 gene as a candidate of interest and by further characterizing it they were able to unmask a novel MYCN-SMC2 axis influencing the DNA damage response (DDR).
MYC overexpression is known to cause DNA damage through induction of reactive oxygen species (ROS) production and replication stress.3,4 Murakami-Tonami et al. showed that MYCN amplified tumor cells depend on SMC2 for survival, as these cells undergo massive DNA damage and apoptosis following SMC2 depletion. To further investigate the connection between SMC2, MYCN expression, and DDR, the authors analyzed the data of three different cohort sources of neuroblastoma patients and found that DDR genes expression increased in parallel with SMC2 levels. The importance of the current study, from a clinical perspective, relies on the finding that lower SMC2 expression is beneficial to patients bearing MYCN-amplified tumors. Altogether these data emphasize the synthetic lethal therapeutic potential of targeting SMC2 in MYCN amplified tumors.
The oncogenic role of SMC2 in MYCN overexpressing cells remains to be elucidated.
Remarkably, treatments with cisplatin, a DNA cross-linking agent or camptotechin, a DNA topoisomerase I inhibitor, have synergistic effects with SMC2 depletion in cells overexpressing MYCN. Since cisplatin and camptotechin cause replication fork damage by generating topological stress and hindering fork progression, these observations strongly suggest that, in MYCN overexpressing cells, SMC2 plays a protective role in response to intra-S DNA damage.
Given that oncogene overexpression causes transcription deregulation associated with replication stress, one possibility is that in MYCN amplified cells, the unscheduled hyperactivation of transcriptional units, in the absence of a correct epigenetic and cotranscriptional program, generates toxic topological stress5 and, possibly, substrates for SMC2 (Fig. 1). Interestingly, recent observations have linked chromatin condensation events to R-loop structures,6 which result from aberrant topological events at transcribed units.5 In summary, the work presented by Murakami-Tonami et al. provides evidence for an oncogenic function of SMC2 in MYCN amplified tumors. These findings also provide first hand evidence that patients bearing MYCN-amplified tumors might benefit from low SMC2 levels, thereby making SMC2 and/or the condensin complex a novel pharmacological target for the treatment of MYCN-amplified neuroblostoma. MYCN is well studied for its oncogenic properties; it represents an important biomarker and a potential target for therapeutic anti-cancer agents. However, considering the diversity of the MYCN transcriptional targets, its complex regulation and, more importantly, its functional overlap with other members of the Myc family, the identification of those MYCN-targets that actively promote tumorigenesis remains a real challenge. The study presented by Murakami-Tonami et al. highlights a promising therapeutic strategy for treating MYCN-amplified neuroblastomas.
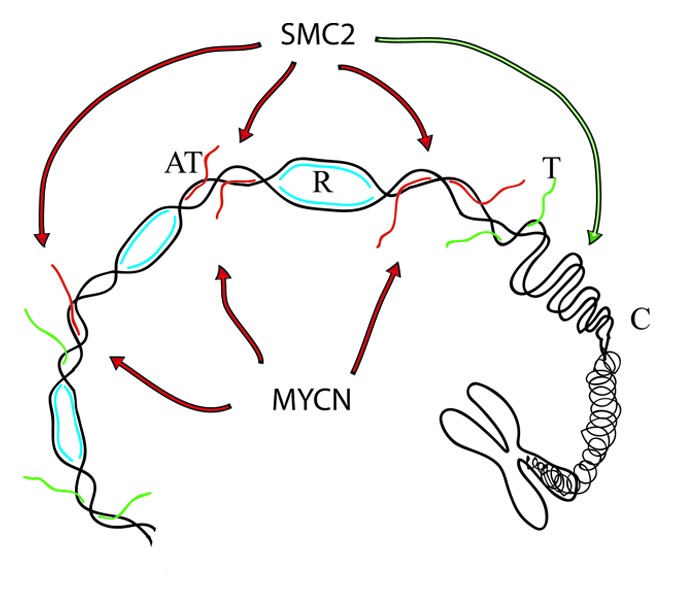
Figure 1. A model to explain the MYCN-SMC2 oncogenic axis. The model shows chromatin undergoing replication (R, in blue), transcription (T, in green), and condensation (C). MYCN overexpression causes replication stress by inducing unscheduled hyperactivation of transcription units (UT, in red). SMC2, is involved in chromatin condensation (green arrow) but may also participate in aberrant condensation events at MYCN-induced transcription units (red arrows).
Murakami-Tonami Y, et al. Cell Cycle. 2014;13:1115–31. doi: 10.4161/cc.27983.
References
- 1.Hirano T. Genes Dev. 2012;26:1659–78. doi: 10.1101/gad.194746.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murakami-Tonami Y., et al. Cell cycle. 2014;13:24–40. doi: 10.4161/cc.27983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vafa O, et al. Mol Cell. 2002;9:1031–44. doi: 10.1016/S1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 4.Herold S, et al. Nat Rev Cancer. 2009;9:441–4. doi: 10.1038/nrc2640. [DOI] [PubMed] [Google Scholar]
- 5.Bermejo R, et al. Mol Cell. 2012;45:710–8. doi: 10.1016/j.molcel.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Castellano-Pozo M, et al. Mol Cell. 2013;52:583–90. doi: 10.1016/j.molcel.2013.10.006. [DOI] [PubMed] [Google Scholar]