

Abstract
Heterochromatin protein 1 (HP1) interacts with various proteins, including lamins, to play versatile functions within nuclei, such as chromatin remodeling and DNA repair. Accumulation of prelamin A leads to misshapen nuclei, heterochromatin disorganization, genomic instability, and premature aging in Zmpste24-null mice. Here, we investigated the effects of prelamin A on HP1α homeostasis, subcellular distribution, phosphorylation, and their contribution to accelerated senescence in mouse embryonic fibroblasts (MEFs) derived from Zmpste24−/− mice. The results showed that the level of HP1α was significantly increased in Zmpste24−/− cells. Although prelamin A interacted with HP1α in a manner similar to lamin A, HP1α associated with the nuclease-resistant nuclear matrix fraction was remarkably increased in Zmpste24−/− MEFs compared with that in wild-type littermate controls. In wild-type cells, HP1α was phosphorylated at Thr50, and the phosphorylation was maximized around 30 min, gradually dispersed 2 h after DNA damage induced by camptothecin. However, the peak of HP1α phosphorylation was significantly compromised and appeared until 2 h, which is correlated with the delayed maximal formation of γ-H2AX foci in Zmpste24−/− MEFs. Furthermore, knocking down HP1α by siRNA alleviated the delayed DNA damage response and accelerated senescence in Zmpste24−/− MEFs, evidenced by the rescue of the delayed γ-H2AX foci formation, downregulation of p16, and reduction of senescence-associated β-galactosidase activity. Taken together, these findings establish a functional link between prelamin A, HP1α, chromatin remodeling, DNA repair, and early senescence in Zmpste24-deficient mice, suggesting a potential therapeutic strategy for laminopathy-based premature aging via the intervention of HP1α.
Keywords: HP1, lamin A, progeria, DNA damage, chromatin remodeling, cellular senescence
Introduction
Hutchinson–Gilford progeria syndrome (HGPS), an autosomal dominant human disorder with disastrous premature aging features, is predominantly caused by a de novo G608G point mutation in LMNA gene. LMNA gene encodes for A-type lamin proteins, which are components of the nuclear lamina, a scaffolding network lying beneath the inner nuclear membrane and further extending into the nucleoplasm. The G608G mutation activates a cryptic splice donor in exon 11 of LMNA gene, resulting in a 50-aa internal deletion (Δ50) in prelamin A that contains a proteolytic cleavage site required for lamin A maturation. The Δ50 prelamin A left behind is permanently modified by a farnesyl group, also called progerin.1,2 A more severe progeria disorder, restrictive dermopathy (RD), has homozygous deficiency in Zmpste24, a zinc metalloproteinase responsible for the final cleavage step of prelamin A, resulting in a partially processed farnesyl full-length prelamin A instead of mature lamin A.3 Recapitulating HGPS, Zmpste24-deficient mice exhibit severe growth retardation, hair loss, osteoporosis, dilated cardiomyopathy, muscular dystrophy, lipodystrophy, and their lifespan is shortened to 4–6 mo.4 Thus one common characteristic of HGPS and RD is incomplete processing of prelamin A into mature lamin A. Lamin A as a type-V intermediate filament protein has head, tail, and connected rod domain structure and usually forms a head–tail dimer, which plays essential roles in nuclear membrane strength and shape, positioning of the nuclear pore complexes, lamina assembly, anchoring chromatin, and dynamic chromatin organization.5 Progerin interferes with the integrity of the nuclear envelope, thus leading to misshapen nuclei with lobulation, thickening, and blebbing. Such irregular nuclei are also observed in cells derived from old healthy individuals.6-8 Reducing farnesyl prelamin A accumulation on the nuclear lamina by farnesyltransferase inhibitor (FTI) or accelerating autophagic degradation of progerin through rapamycin ameliorates cellular senescence in HGPS cells and premature aging in Zmpste24-deficient mice.9-13 Further studies revealed interphase aneuploidy chromatin, loss or aggregation of peripheral heterochromatin, abnormally clustered centromeres, and mislocalized telomeres in fibroblasts isolated from HGPS patients and Zmpste24-deficient mice.14 These abnormalities in chromatin organization are related to epigenetic changes, including the trimethylation of histone H3 at Lys9 (H3K9me3) and Lys27 (H3K27me3), histone H4 at Lys20 (H4K20me3) and the acetylation of histone H4 at Lys16 (H4K16ac).15,16 These histone modifications are key factors for heterochromatin organization or chromatin remodeling. Changes in H3K9me3 patterns have also been observed in fibroblasts isolated from another progeriod syndrome mandibuloacral dysplasia (MAD) and in cells derived from old healthy individuals.7,17 These findings clearly demonstrate that prelamin A accumulation leads to abnormal organization and function of heterochromatin, which may contribute to the phenotypes in progeria.
Mammalian heterochromatin protein 1 (HP1), including HP1α, HP1β, and HP1γ, are non-histone chromatin-associated proteins, which bind to histone H3 that is tri-methylated at lysine 9 (H3K9me3) via their N-terminal chromo domain (CD) to control heterochromatin high-order organization.18,19 HP1 requires dimerization via their C-terminal chromo shadow domain (CSD) and interacts with many nuclear proteins that possess a conserved PxVxL penta-peptide motif to trigger a variety of functions, ranging from replication, transcription, chromatin organization, and nuclear architecture, to chromosomal maintenance.20,21 Via the CD region, HP1 directly or indirectly associates with lamins, such as lamin B receptor (LBR), LAP2β and lamina-associated polypeptides (LAP2α).22-24 It not only anchors the peripheral heterochromatin to the inner nuclear membrane, but also mediates nuclear envelope reassembly in a dynamic manner.25
Progeriod cells are characterized by accumulation of irreparable DNA damages and defective DNA repair.26 Recently, heterochromatin-mediated DNA damage response (DDR) has been found important for genome integrity; the superstructure of heterochromatin impedes DNA repair as a barrier, and most of heterochromatin components, such as KRAB-associated protein 1 (KAP-1), HP1, play important roles in the heterochromatin DDR.27-29 ATM phosphorylates KAP-1 at Ser824 (pS824-KAP-1), alters KAP-1 affinity for chromatin, and facilitates heterochromatin remodeling in DDR.27,30 Recent findings also link HP1 proteins to the DNA damage response. All 3 HP1 isoforms are mobilized and recruited to various types of DNA damage sites.31,32 In response to DNA damage, HP1β is rapidly mobilized by casein kinase 2 (CK2)-mediated phosphorylation of its CD region at Thr 51 (pT51-HP1β). The phosphorylation disrupts hydrogen bonds that fold the CD around H3K9me3, releasing HP1β from heterochromatin and possibly leading to heterochromatin relaxation.29 Inhibition of CK2 or a constitutively Thr51 mutant HP1β diminishes γ-H2AX signals.29 Moreover, KAP-1 directly binds to HP1α CSD region.27 Cells lacking HP1 fail to form discreet pS824-KAP-1 foci after DNA damage.33 Previously we have shown that KAP-1 phosphorylation is compromised and leads to delayed DNA damage-induced chromatin remodeling and accelerated senescence in Zmpste24-deficient mice.34 However, it is still unclear whether the defective KAP-1 phosphorylation is the unique regulatory mechanism underlying the genomic instability and accelerated senescence in progeria mice. Therefore it would be interesting to explore the roles of HP1 in mechanisms linking abnormal lamin A, delayed chromatin remodeling, defective DNA repair, and premature aging. In this study, we investigated the effects of prelamin A accumulation on HP1 expression, subcellular distribution, phosphorylation, and their impacts on premature senescence. We found that the level of HP1α associated with the nuclear matrix was significantly increased in Zmpste24−/− cells. Upon DNA damage, HP1α phosphorylation was significantly compromised, leading to delayed formation of γ-H2AX foci in Zmpste24−/− MEFs. Knocking down HP1α alleviated the delayed DNA damage response and accelerated senescence in Zmpste24−/− MEFs. Our findings establish a functional link between prelamin A, HP1α, heterochromatin repair, and early senescence in Zmpste24-deficient mice and suggest a potential therapeutic strategy for laminopathy-based premature aging via the intervention of HP1α.
Results
The level of HP1α is increased in Zmpste24−/− MEFs
HP1 proteins serve as linkers, connecting peripheral heterochromatin to inner nuclear membranes.24,25 To investigate whether HP1s are involved in defective DNA repair and premature aging, we first examined levels of HP1 isoforms and H3K9me3 by western blotting in Zmpste24−/− MEFs and wild-type littermate controls. To eliminate epigenetic alterations induced by cellular senescence, early-passage Zmpste24−/− MEFs (passage 3), which have similar proliferative potential as wild-type littermate controls (data not shown and ref. 15), were used throughout this study. Compared with wild-type littermate controls, a more than 40% increase in HP1α level was observed in Zmpste24−/− MEFs, whereas HP1β and HP1γ were much less affected (Fig. 1A and B). Consistent with our previous report,35 the level of H3K9me3, a HP1-binding heterochromatin marker, was significantly elevated, whereas levels of H3K9me1 and H3K9me2 were hardly affected (Fig. S1 A and B). To evaluate whether the elevated HP1α protein is attributable to the transcriptional regulation of HP1α by prelamin A, the mRNA level of HP1α was examined by real-time reverse-transcription PCR. As shown, the level of HP1α mRNA was not affected in Zmpste24−/− MEFs (Fig. 1C), indicating that prelamin A modulates HP1α in a post-translational manner. Collectively, these data suggest that the presence of prelamin A stabilizes HP1α protein in Zmpste24−/− MEFs.
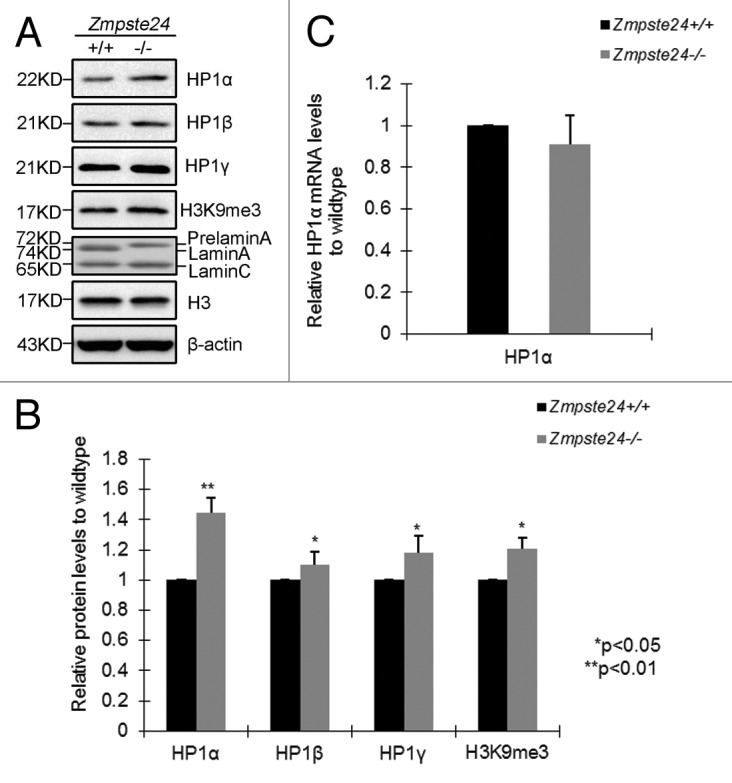
Figure 1. Increased level of HP1α in Zmpste24−/− MEFs. (A) Representative immunoblots showing protein levels of HP1α, HP1β, HP1γ, H3K9me3 in Zmpste24−/− MEFs and wild-type littermate controls at passage 3. (B) Relative levels of HP1α, HP1β, HP1γ, H3K9me3 in Zmpste24−/− MEFs comparing with wild-type littermate controls. Data (mean ± s.e.m.) represent 3 independently derived lines of MEFs in separate experiments. *P < 0.05, **P < 0.01, 2-tailed Student t test. (C) Real-time RT-PCR analysis of HP1α mRNA expression in Zmpste24−/− MEFs and wild-type littermate controls. HP1α mRNA abundance was calculated by 2-△△Ct normalized to β-actin. Data represent 3 independently derived lines of MEFs in separate experiments.
HP1α interacts with prelamin A in Zmpste24−/− MEFs
Given that lamin A and prelamin A bind to HP1α and LAP2α in HEK293 cells,36 we reasoned that prelamin A might increase the binding capacity to HP1α, thus stabling it. To this end, co-immunoprecipitation (Co-IP) was performed in identical early passage of Zmpste24−/− MEFs and wild-type littermate controls to investigate the interaction of endogenous prelamin A and HP1. Since lamin-containing nuclear matrix (NM) is insoluble in RIPA buffer containing 150 mM NaCl, and raising the salt concentration to 500 mM completely abolishes the interaction between A-type lamins and HP1, RIPA buffer containing 350 mM NaCl was used for the Co-IP. As shown in Figure 2A, HP1α, HP1β, and HP1γ were detected in the anti-lamin A/C immunoprecipitates by western blotting. However, the pull-down levels are comparable between Zmpste24−/− MEFs and wild-type littermate controls, suggesting that prelamin A might bind to HP1 isoforms in a similar capacity as lamin A. Reciprocally, endogenous lamin A/prelamin A was detected in the anti-HP1α immunoprecipitates. Interestingly, the anti-HP1α immunoprecipitates also pull down lamin C (Fig. 2B), which is encoded by an alternate splicing variant of LMNA gene and shares the N-terminal 566 aa with lamin A,37 suggesting that HP1 interacts with lamin A/prelamin A through domains beyond C-terminal lamin A-specific region. Nonetheless, these data have rolled out the possibility that prelamin A directly regulates HP1 protein stability.
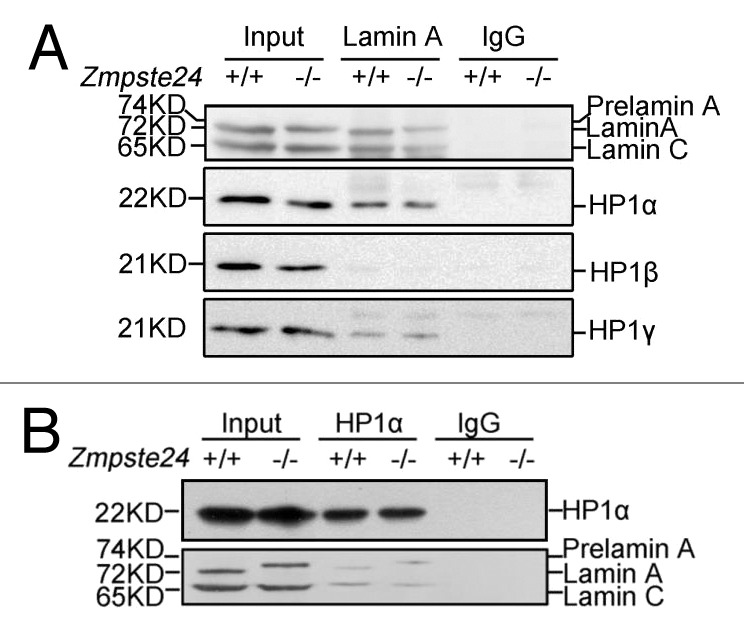
Figure 2. HP1 interacts with endogenous prelamin A in a similar manner as lamin A. (A) Immunoblots showing endogenous HP1α, HP1β, HP1γ in the anti-lamin A/C immunoprecipitates from Zmpste24−/− MEFs and wild-type littermate controls. Data represent 1 of 3 independent experiments. (B) Immunoblots showing endogenous lamin A/prelamin A/lamin C in the anti-HP1α immunoprecipitates in Zmpste24−/− MEFs and wild-type littermate controls. Data represent 1 of 3 independent experiments.
HP1α is mainly distributed in nuclease-resistant nuclear matrix and fraction in Zmpste24−/− MEFs
Accumulation of prelamin A jeopardizes the nuclear lamina and matrix structure, resulting in disturbed heterochromatin organization.14,15,38 Although prelamin A has similar binding capacity to HP1 as lamin A, it may jeopardize the overall nuclear matrix structure and consequently mis-localize HP1 proteins and, thus, modulate their stability. Therefore, we further investigate whether HP1α is mislocalized in the nuclear matrix fraction of Zmpste24−/− MEFs. A well-established subcellular fractionation strategy was applied to separate cell extracts into fractions including cytoplasm (S1), nucleoplasm (S2), chromatin and nuclear matrix (P2), nucleoplasm and nuclease susceptible chromatin (S2'), and micrococcal nuclease (MNase)-resistant nuclear matrix (P2').16 α-tubulin, β-actin, high-mobility group protein B1 (HMGB1), lamin A/C served as controls for the cytoplasmic, nucleoplasmic, euchromatin-bound, and nuclear matrix fractions, respectively. As expected, in wild-type MEFs, α-tubulin was detected only in cytoplasm fraction, whereas β-actin was present in all cytoplasmic and nuclear fractions (Fig. 3A). For nuclear proteins, lamin A/C was resistant to MNase digestion and remained exclusively in nuclear matrix fraction (P2'). In contrast, the euchromatin-bound protein HMGB1 was absent from the nuclear matrix fraction (P2') and was completely released into nucleoplasmic fraction (S2') after MNase treatment. Although sensitive to MNase, after digestion, HP1 and H3K9me3 were still partially retained in the nuclear matrix fraction (P2') (Fig. 3A). Similarly, HP1 distribution was examined in Zmpste24−/− cells; as shown in Figure 3B, the level of HP1α protein retained in the nuclear matrix fraction (P2') of Zmpste24−/− MEFs was much higher than that of wild-type controls. Concomitantly, the level of HP1α released into the nucleoplasm and suspensible chromatin fraction (S2') was much lower in progeriod cells than that in wild-type cells. Likewise, H3K9me3 distribution was shown in a similar manner as HP1α (Fig. 3B). Notably, subcellular distributions of HP1β and HP1γ were not much affected in Zmpste24−/− MEFs. Altogether, these findings suggest that the increased HP1α in progeriod cells is mainly distributed in prelamin A-enriched nuclear matrix fraction (P2'), which might mislead the heterochromatin to the dis-organized nuclear matrix, thus disturbing the formation and function of heterochromatin.
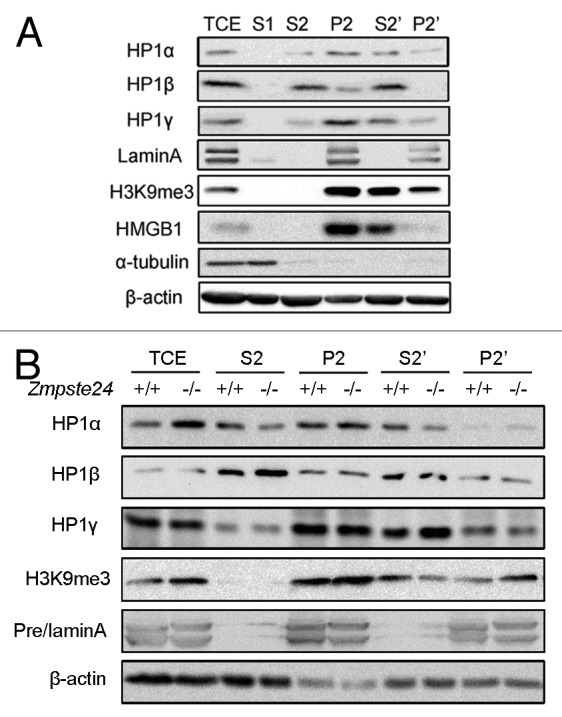
Figure 3. Increased HP1α was mainly localized in nuclease-resistant chromatin and nuclear matrix fraction in Zmpste24−/− MEFs. (A) Subcellular fractions of MEFs: S1 (cytoplasmic), S2 (nucleoplasmic), P2 (chromatin + nuclear matrix), S2' (nucleoplasmic + nuclease susceptible chromatin), P2' (nuclear matrix + nuclease resistant chromatin), and TCE (total cell extraction) as described in the “Materials and Methods”. Representative immunoblots showing the subcellular distribution of HP1α, HP1β, HP1γ, HMGB1 (euchromatin marker), H3K9me3 (heterochromatin marker), and LaminA/C (nuclear matrix marker). (B) Immunoblots showing increased levels of HP1α in nuclease-resistant chromatin and nuclear matrix fraction, whereas no significant changes of HP1β and HP1γ in various nuclear fractions. Data represent 1 of 3 independently derived lines of MEFs in separate experiments.
HP1α phosphorylation is compromised following DNA damage in Zmpste24−/− MEFs
In addition to heterochromatin mis-organization, sustained DNA damage checkpoint response is another hallmark of progeriod cells, which is a driving factor of cellular senescence.26 We and others recently found that the irreparable DNA damages accumulated in Zmpste24-dificient mice are indeed associated with heterochromatin, indicating a defective heterochromatin repair.34,35 Recent reports showed that HP1 is mobilized via phosphorylation, which alters heterochromatin organization, and then is recruited to DNA damage sites.29,32 Therefore we asked whether HP1α phosphorylation is affected in progeriod cells. It has been reported that residues Thr 51 in human HP1β and Thr50 in HP1α are both crucial in regulating the binding of HP1 to H3K9me3 and the remodeling of heterochromatin in response DNA damage.29 Given the high similarity of chromodomain regions surrounding Thr 51 in HP1β and Thr50 in HP1α in both humans and mice (Fig. S2A), a polyclonal antibody that recognizes mouse HP1α phosphorylated at residue Thr50 (pT50-HP1α) was raised as described in “Materials and Methods”. The specificity of the anti-pT50-HP1α antibody was tested by SAP (shrimp alkaline phosphatase) treatment (Fig. S2B). By western blotting, pT50-HP1α dynamics were examined in Zmpste24−/− MEFs with Camptothecin (CPT) treatment, a radiomimetic reagent, which induces DSBs into progeriod cells as described before.39 γ-H2AX was examined as a sensitive marker for the presence of DSBs. CPT treatment induced rapid increase of γ-H2AX level in both Zmpste24−/− MEFs and wild-type controls (Fig. 4A). Thereafter, the level of γ-H2AX decreased at 4 h and returned to almost background level at 8 h in wild-type controls, but it remained significantly higher in Zmpste24−/− MEFs at 4 and 8 h (Fig. 4A). Although total level of HP1α did not show obvious changes within 8 h, CPT treatment induced a dynamic change of pT50-HP1α in wild-type MEFs, which peaked at 30 min and gradually decreased thereafter (Fig. 4A and B). However, the level of pT50-HP1α was significantly reduced and the kinetics delayed in Zmpste24−/− MEFs, whereby it maximized around 2–4 h after CPT treatment (Fig. 4A and B). Of note, the delayed pT50-HP1α kinetics in Zmpste24−/− MEFs is consistent with the delayed γ-H2AX level upon CPT treatment. To confirm the delayed pT50-HP1α kinetics after DNA damage in progeriod cells, we further employed immunofluorescence confocal microscopy. Within 30 min post-CPT treatment, pT50-HP1α and γH2AX foci were stochastically distributed throughout the nucleus in both Zmpste24−/− and wild-type control cells, and most of them co-localized with each other (Fig. 4C). The number of pT50-HP1α foci maximized around 30 min after DNA damage in wild-type cells; however, this was significantly delayed, until 2 h, in progeriod cells (Fig. 4D). Collectively, these findings confirm defective DNA repair evidenced by defective H2AX phosphorylation in progeriod cells and further imply that delayed pT50-HP1α might lead to defective mobilization of HP1α and cause delayed chromatin remodeling and, therefore, delayed recruitment of DNA damage response factors.

Figure 4. DNA damage-induced pT50-HP1α phosphorylation is compromised in Zmpste24−/− MEFs. (A) Representative immunoblots showing levels of γH2AX, HP1α and pT50-HP1α at indicated time points in Zmpste24−/− MEFs and wild-type littermate controls treated with 4 µM camptothecin (CPT). (B) Relative levels of HP1α and pT50-HP1α after CPT treatment in Zmpste24−/− MEFs and wild-type littermate controls. Data represent 3 independently derived lines of MEFs in separate experiments. (C) Representative photos of immunofluorescence confocal microscopy showing pT50-HP1α and γH2AX foci after CPT treatment at indicated time points. Scale bar, 5 μm. (D) The number of pT50-HP1α foci per cell after CPT treatment in Zmpste24−/− MEFs and wild-type controls. At least 100 cells were counted. Data represent mean ± s.e.m., *P < 0.05, **P < 0.01, 2-tailed Student t test.
HP1α knockdown attenuates defective DNA damage response and cellular senescence
DNA breaks in heterochromatin can be abrogated by knocking down HP1 or other heterochromatin components.28 To gain further insight into whether increased HP1α is the critical link between prelamin A and delayed DNA damage response and accelerated aging phenotypes in Zmpste24−/− MEFs, we explored if endogenous HP1α knockdown might alleviate the delayed DNA damage response and early onset of cellular senescence in Zmpste24−/− MEFs. In cells treated with short interfering RNAs (siRNAs) specifically targeting HP1α, although level of β-actin was quite stable for as long as 48 h, diminished HP1α expression was confirmed (Fig. S3). We next evaluated the effects of HP1α knockdown on γ-H2AX foci after CPT treatment. As shown in Figure 5A and B, a large number of γ-H2AX foci still persisted at 8 h in Zmpste24−/− MEFs, and this was significantly reduced in HP1α siRNA-treated Zmpste24−/− MEFs. Additionally, HP1α knockdown considerably reduced the expression of cellular senescence marker p16 expression (Fig. 5C and D). When cells were monitored for senescence-associated β-galactosidase (SA-β-gal) activity, the number of positively blue-stained cells dropped from more than 50% in scramble-treated to less than 25% in HP1α siRNA-treated Zmpste24−/− MEFs (Fig. 5E and F). Collectively, HP1α knockdown rescues defective DNA repair and attenuates the accelerated cellular senescence in Zmpste24−/− MEFs.

Figure 5. Knocking down HP1α promotes DNA damage response and attenuates cellular senescence in Zmpste24−/− MEFs. (A) Representative photos showing γH2AX foci at indicated time points in Zmpste24−/− MEFs and wild-type littermate controls treated with HP1α siRNA or mock siRNA. Scale bar, 5 μm. (B) The number of γH2AX foci per cell at indicated time points in Zmpste24−/− MEFs treated with HP1α siRNA. At least 100 cells were counted. Data represent mean ± s.e.m., *P < 0.05, **P < 0.01, 2-tailed Student t test. (C) Representative immunoblots showing levels of HP1α and P16 in Zmpste24−/− MEFs and wild-type littermate controls treated with HP1α siRNA or mock siRNA. (D) Quantification of P16 level in (C). Data represent mean ± s.e.m., n = 3, *P < 0.05, 2-tailed Student t test. (E) Representative photos of SA-β-gal activity showing the blue-stained senescent cells in Zmpste24−/− MEFs and wild-type littermate controls at passage 5. (F) Quantification of (E) showing the percentage of senescent cells. Data represent mean ± s.e.m., n = 3, ** P < 0.01, ***P < 0.001, 2-tailed Student t test.
Discussion
In this report, we have shown that nuclear matrix-associated HP1α level was remarkably elevated in Zmpste24−/− MEFs. Upon DNA damage, a significantly compromised HP1α phosphorylation on Thr50 was observed in Zmpste24−/− MEFs, which correlates with the delayed γ-H2AX foci formation. Knocking down HP1α improved γ-H2AX foci formation and rescued cellular senescence in Zmpste24−/− MEFs. Our data suggest a critical role of HP1α in prelamin A-regulated genomic instability and premature aging.
Heterochromatin perturbation and epigenetic mis-regulation are hallmarks of progeria.6,15,17 However, how various components functionally affect heterochromatin dynamics in the presence of prelamin A or progerin remains largely unknown. Here we found that prelamin A accumulation resulted in an increase of HP1α protein level, while the mRNA was hardly affected, suggesting a potential role for prelamin A in stabilizing HP1α protein. It has been shown that lamin A and prelamin A binds to HP1α in vitro, and farnesylated prelamin A showed reduced binding capacity to HP1α compared with the non-farnesylated form and wild-type lamin A.36 However, a comparable binding affinity of HP1α to prelaminA and lamin A was found in our study, although prelamin A is farnesylated in Zmpste24−/− MEFs. One possible interpretation of this seeming discordance is that ectopic farnesylated prelamin A might be functionally different from the endogenous one caused by Zmpste24 deficiency. Consistent with this notion is the finding that different strategies in generating unfarnesylatable progerin may result in opposite phenotypes in knock-in mouse models.40-42 It has also been widely accepted that there is reduced of heterochromatin in HGPS cells, supported by decreased H3K9me3, H3K27me3, and HP1α.15 However, we have previously shown that prelamin A might elicit functional difference with progerin in addition to the structural differences, i.e., loss of 50 amino acids, which may cause loss of function of lamin A.35 Interestingly, several reports found that lamin A regulates HP1 stability through inhibition of specific ubiquitin ligase activities, and loss of lamin A accelerates HP1α degradation in Lmna−/− MEFs.43,44 In this regard, the seemingly contradictory observation might be explained by the structural and functional difference between prelamin A in Zmpste24-null cells and progerin in human HGPS cells. The level of HP1α in the presence of progerin could be attributable to a combined gain of function of the partially processed C tail, which elevates the HP1α level, and loss of function of lamin A, which is due to 50-aa-truncation and accelerates the degradation of HP1α. Nevertheless, the lack of difference between prelamin A and lamin A in their binding capacity to HP1α suggests that it is unlikely that prelamin A regulates HP1α stability in a similar manner as it does on Suv39h1. On the other front, HP1, H3K9me3, and its principal methyltransferase Suv39h1 are all enriched in the pericentric heterochromatin region, and it is considered that N-terminal upstream of Suv39h1 CD region possess interaction site with HP1 CSD region, recruiting HP1α to bind H3K9me3 within its CD region.45 Consistent with our previous report, we observed a remarkably increased level of H3K9me3 in Zmpste24-null cells and a significant increase of association of HP1α with the nuclear matrix fraction, wherein H3K9me3 is highly enriched. Therefore, it is also plausible to speculate that prelamin A might interfere with the correct positioning and stability of HP1α/Suv39h1/H3K9me3 heterochromatic complex, thus impairing the localization of pericentric heterochromatin domains at the nuclear periphery and finally resulting in disorganization of heterochromatin.
Although initial studies revealed that HP1 plays a key role in heterochromatin formation and gene silencing, recent studies have highlighted the importance of HP1-mediated chromatin remodeling and DNA damage response. In this study, we found that the level of pT50-HP1α, which co-localizes with γ-H2AX, is compromised in Zmpste24−/− MEFs after DNA damage, suggesting defective mobilization of HP1α and, thereafter, compromised chromatin remodeling. Another heterochromatin protein KAP-1, which promotes heterochromatic DSB repair by ATM-mediated phosphorylation and therefore facilitates the release of KAP-1 and CHD3 from heterochromatin and global chromatin relaxation, directly binds to HP1α CSD.27,28 We have previously shown that pS824-KAP-1 level is significantly reduced, preventing the release of CHD3 from heterochromatin and global chromatin relaxation in Zmpste24−/− MEFs.34 We therefore speculate that nuclear matrix fraction enrichment of HP1α in Zmpste24−/− MEFs might be also involved in the retention of KAP-1 on heterochromatin. Notably, the decreased level of pS824-KAP-1 in our previous study is not totally in accordance with the dynamic changes of chromatin relaxation in Zmpste24−/− MEFs. Although the level of pS824-KAP-1 was decreased, it peaked 30 min after DNA damage, which is similar to wild-type cells. Interestingly, although delayed, the chromatin still underwent global relaxation at 2 h and, concomitantly, the recruitment of 53BP1 after irradiation in Zmpste24–/– MEFs, indicating a backup mechanism regulating the late-stage global chromatin relaxation. In this study, we found that the peak level of pT50-HP1α was postponed to 2 h, which correlates well with the delayed relaxation of chromatin in progeriod cells after DNA damage, implicating that pT50-HP1α could be the backup mechanism mediating chromatin remodeling in progeroid cells.
The rescue of cellular senescence by HP1α knockdown suggests potential links between chromatin remodeling, DNA repair, and premature aging. Interestingly, a recent report by Lattanzi and colleagues showed de-condensation of heterochromatin and enhanced DNA repair efficacy in centenarian fibroblasts and in normal cells treated with rapamycin, highlighting the pivotal roles of chromatin remodeling and DNA repair in aging/longevity.46 Rapamycin targets mTOR pathways, inhibits DNA damage response (or pseudo-DNA damage response), and therefore slows down cellular senescence.47-49 In this scenario, it would be interesting to examine whether rapamycin could rescue the defective chromatin remodeling and defective DNA repair in Zmpste24-null cells in future study. Nevertheless, our data shed new light into molecular basis of HP1α in heterochromatin mis-organization, delayed DNA damage response, and early senescence in Zmpste24-deficient progeria mouse model, providing an additional intervention target for progeria and normal aging.
Materials and Methods
Cell culture and drug treatments
Zmpste24−/− mice were generated by Zhou et al. previously.4 Genotyping of mice for the mutant Zmpste24 allele was performed by PCR as described previously.15 Mouse embryonic fibroblasts (MEFs) were obtained from 13.5-d Zmpste24+/+ and Zmpste24−/− embryos. Littermate-matched Zmpste24+/+ and Zmpste24−/− MEFs were cultured in complete Dulbecco modified Eagle medium (DMEM) (pH 7.4) supplemented with 10% fetal bovine serum. Early passage (passage 3) of Zmpste24−/− MEFs without showing senescence phenotypes were employed in all experiments.15 To induce DNA damage, MEFs were treated with either 4 μM Camptothecin (CPT, Sigma C9911) or DMSO for 1 h. Afterward, CPT was removed by washing 3 times with PBS and replacing with fresh medium.
Antibodies
Commercial available antibodies used in this experiments are as follow: lamin A/C (Santa Cruz Biotechnology, sc-20681, http://www.scbt.com/datasheet-20681-lamin-a-c-h-110-antibody.html), HP1α (Millipore, 05–689, http://www.millipore.com/catalogue/item/05-689; Abcam, ab77256, http://www.abcam.com/hp1-alpha-antibody-chip-grade-ab77256.html), HP1β (Abcam, ab10811, http://www.abcam.com/CBX1-HP1-beta-MAC353-antibody-ChIP-Grade-ab10811.html), HP1γ (Millipore, 05–690, http://www.millipore.com/catalogue/item/05-690), γ-H2AX (Upstate, 05–636, http://www.millipore.com/catalogue/item/05-636), H3K9me3 (Abcam, ab8898, http://www.abcam.com/histone-h3-tri-methyl-k9-antibody-chip-grade-ab8898.html), H3 (Abcam, ab1791, http://www.abcam.com/histone-h3-antibody-chip-grade-ab1791.html), HMGB1 (Cell Signaling, 3935, https://www.cellsignal.com/products/3935.html), P16 (Santa Cruz Biotechnology, sc-81156, http://www.scbt.com/datasheet-81156-p16-1e12e10-antibody.html), α-tubulin (Cell Signaling, 2144, http://www.cellsignal.com/products/2144.html), β-actin (cwbiotech, CW0096A, http://www.cwbiotech.com/en/scart/cproduct.aspx?id=4283). pT50-HP1α polyclonal antibody was made by Abmart (Shanghai, http://www.ab-mart.com/index.php) against the phospho-peptide, C-SEEHN(pT)WEPEK. It was diluted 1:300 for immunoblotting and 1:100 for immunofluorescence staining. Secondary antibodies used are as follows: Horseradish peroxidase (HRP) labeled Goat anti-rabbit (Pierce, 31460, http://www.pierce-antibodies.com/Goat-anti-Rabbit-IgG-H-L-Secondary-Antibody-Polyclonal-31460.html), HRP labeled Goat anti-Mouse IgG (Pierce, 32430 http://www.pierce-antibodies.com/Glutamic-Acid-Decarboxylase-65-antibody-Polyclonal-PA532430.html), HRP labeled Goat anti-Rat IgG (Pierce, 31470, http://www.pierce-antibodies.com/Goat-anti-Rat-IgG-H-L-Secondary-Antibody-Polyclonal-31470.html), HRP labeled Rabbit anti-Goat IgG (Pierce, 31402, http://www.pierce-antibodies.com/Rabbit-anti-Goat-IgG-H-L-Secondary-Antibody-Polyclonal-31402.html), Alexa Fluor 488 donkey anti-goat IgG (H+L) (Invitrogen, A11055s, http://www.lifetechnologies.com/order/catalog/product/A11055), Alexa Fluor 488 donkey anti-rat IgG (H+L) (Invitrogen, A21208, http://www.lifetechnologies.com/order/catalog/product/A21208), Alexa Fluor 488 donkey anti-mouse IgG (H+L) (Invitrogen, A21202, http://www.lifetechnologies.com/order/catalog/product/A21202), Alexa Fluor 555 donkey anti-mouse IgG (H+L) (Invitrogen, A31570, http://www.lifetechnologies.com/order/catalog/product/A31570), Alexa Fluor 594 goat anti-rabbit IgG (H+L) (Invitrogen, A11012, http://www.lifetechnologies.com/order/catalog/product/A11012). All reagents were used at the dilutions recommended by the manufacturers or mentioned above.
Co-immunoprecipitation
Cell extracts were prepared in modified RIPA buffer (20 mM Tris, pH 7.5, 300 mM NaCl, 0.5% Nonidet P-40, 1% Triton X-100, 1 mM EGTA, 1 mM DTT). Protein A conjugated Sepharose beads (Roche 11719394001) were washed twice with ice-cold PBS and pelleted by centrifugation at 2000 × g for 1 min at 4 °C. Two micrograms of the antibody of interest and lysates from 1 × 107 MEFs were incubated for 2 h at 4 °C with rotation. The antibody conjugate was then incubated with 50 μL of the pre-washed Sepharose beads overnight at 4 °C with rotation. The excess unbound protein was removed by washing 3 times with 500 μL modified RIPA buffer; Sepharose beads were pelleted by centrifugation at 2000 × g for 2 min at 4 °C. Co-immunoprecipitated proteins were eluted off the Sepharose beads by adding 50 μL of 1 × loading buffer (1% SDS, 100 mM DTT, 50 mM Tris, pH 6.8) and boiling for 3 min. The elute was centrifuged at 2000 × g for 4 min, and the supernatant containing co-immunoprecipitated proteins was collected and analyzed by western blotting.
Western blotting
Whole-cell lysate was prepared by suspending the MEFs in 1 volume of suspension buffer (10 mM Tris-HCl, pH 7.5, 0.1 M NaCl, 1mM EDTA, 1 mM DTT, pH 8.0, protease inhibitors), followed by adding 1 volume of Laemmli buffer (0.1M Tris-HCl, pH 7.0, 4% SDS, 20% glycerol,1 mM DTT, protease inhibitors) and boiling for 5 min. Twenty micrograms of total proteins were electrophoresed through 10% SDS polyacrylamide gels and transferred to PVDF membrane (Millipore). The membrane was incubated with primary antibodies overnight at 4 °C, and then probed with appropriate secondary antibodies linked to HRP for 1 h at room temperature. Antibody binding was visualized by enhanced chemiluminiscence kit (Pierce 34095). Relative blot band intensity was measured by ImageJ and normalized to corresponding controls as indicated. For statistical analysis, at least 3 independent immunoblots were quantified and 2-tailed t test was used for P values.
Real-time PCR
Total RNA was extracted with the Trizol® reagent (Invitrogen, 15596-026) according to the manufacturer’s protocol. RNA quantity was monitored by UV spectrophotometry and agarose gel electrophoresis. One microgram of total RNA was transcribed to cDNA using the iScript™ cDNA Synthesis Kit (Bio-rad, 70-8891). Quantitative RT-PCR (Q-PCR) was run on the 7500 Fast Real-Time PCR System (Applied Biosystems), with the use of the SsoFast™ EvaGreen Supermix kit (Bio-rad, 172-5200). Mouse HP1α and β-actin primers are as follows: HP1α (232 bp) forward, 5′-GAAAGAAGAC CAAGAGGACA G C-3′; reverse, 5′-TTGTTTCACC CTCCTTCATC T-3′. β-actin (517 bp) forward, 5′-ATATCGCTGC GCTGGTCGTC-3′; reverse, 5′-AGGATGGCGT GAGGGAGAGC-3′. All Q-PCR reactions were performed in triplicate in the final volume 10 μl for 40 cycles according to the following protocol: each cycle at 95 °C for the initial 30 s, then at 95 °C for 5 s, and 60 °C for 5 s. The obtained results were averaged, and gene expression levels were normalized to β-actin.
Subcellular fractionation
MEFs were suspended (4 × 107 cells/ml) in buffer A [10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, complete protease inhibitors (Roche), and 1 mM PMSF (Sigma)]. After the addition of 0.1% Triton X-100, cells were incubated on ice for 5 min. Nuclei were obtained by centrifugation at 1300 × g for 4 min at 4 °C. The supernatant containing the cytosolic extract (S1) was collected by centrifugation at 20 000 × g for 15 min at 4 °C. Nuclei were then washed twice with ice-cold buffer A and lysed in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, complete protease inhibitors [Roche], and 1 mM PMSF [Sigma]) for 5 min on ice. The pellet (P2) containing the chromatin and nuclear matrix protein was obtained by centrifugation at 1700 × g for 4 min at 4 °C. The supernatant consisting of nucleoplasmic protein (S2) was also collected. To obtain nuclear matrix-associated proteins, cell nuclei (P1) were resuspended in buffer A plus 1 mM CaCl2 and digested with 5 units of micrococcal nuclease (MNase, Sigma, N3755) at 37 °C for 5 min; the reaction was stopped by the addition of 1 mM EGTA. By low-speed centrifugation as described above, the pellet obtained after digestion consisted of the nuclease-resistant chromatin and nuclear matrix-associated proteins (P2'); the supernatant contained the nucleoplasmic and nuclease susceptible chromatin-associated proteins (S2'). The fractions were dissolved in 1× loading buffer (1% SDS, 100 mM DTT, 50 mM Tris, pH 6.8) and sonicated for 15 s. Equal amounts were resolved using SDS-PAGE, and the levels of protein were further detected by western blotting.
Immunofluorescence microscopy
MEFs were seeded in 8-chamber slides to 70% confluence, treated with CPT for 1 h and allowed to recover for indicated time. The slides were washed twice with PBS and fixed with 4% paraformaldehyde in PBS for 10 min at 4 °C, followed by 10 min permeation with 0.2% Triton X-100/PBS. The slides were blocked in 1% BSA/PBS with 5% normal serum overnight at 4 °C, and then incubated with primary antibody diluted in 1% BSA/PBS overnight at 4 °C in a humid box. After washing 3 times with PBS, the slides were incubated with FITC- or TRITC-coupled secondary antibodies diluted in 1% BSA/PBS for 40 min at R.T. and then washed 3 times with 1%Tween20/PBS to remove unbound antibodies. The slides were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen, P36935), sealed with nail polish, and analyzed by Olympus Fluoview FV1000 immunofluorescence microscopy. Photos were processed with Photoshop CS®. Focus counting was performed by 100 cells randomly chosen for each experiment. Two-tailed Student t test was used for P values where applied.
RNA interference
HP1α sense and antisense RNA are 5′-GGAUACAGUC UGAGAGUUAT T-3′ and 5′-UAACUCUCAG ACUGUAUCCT T-3′ respectively. Transfection was performed in triplicate in 6-well plates with lipofectamine RNAiMAX Reagent (Invitrogen, 13778-100) according to the manufacturer’s instructions. Forty-eight hours post-transfection, the cells were treated with 4 μM CPT for 1 h, and harvested at indicated times for western blotting and immunofluorescence microscopy analysis as described above.
Senescence-associated β-gal assay
Senescence-associated β-Galactosidase activity was accessed with cellular senescence assay kit (Mirus, MiR2600). Briefly, cells were seeded in 6-well plates, fixed at room temperature for 5 min with 2 mL of 1× fixing solution, washed twice with 1× PBS, and stained with 2 mL of freshly prepared 1× β-Galactosidase detection solution at 37 °C for 16 h in dark room. The cells were washed twice with 1× PBS, overlaid with 70% glycerol/PBS, and photographed. Blue-stained MEFs were counted in more than 200 randomly chosen cells. Two-tailed t test was applied for P values.
Supplementary Material
Acknowledgments
We thank Drs Ashok R Venkitaraman and Venkat Pisupati (Hutchison/MRC Research Centre, Cambridge Univerisity) for providing pT51-HP1β polyclonal antibody. This study was supported by grants from the National Natural Science Foundation of China (NSFC 81070270) and Science and Technology Bureau of Shenzhen City Grants (JC201005280552A). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author contributions
Z.W. and B.L. designed experiments. J.L., X.Y., X.L., Y.W., and J.H. conducted experiments. H.Z. provided technology supports on MEFs. J.L., X.Y., G.Z., L.G., M.L., and V.K. analyzed data. Z.W., B.L., and Z.Z. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Glossary
Abbreviations:
- ATM
ataxia-telangiectasia mutated
- CD
chromo domain
- CK2
casein kinase 2
- CSD
chromo shadow domain
- CHD3
chromodomain-helicase-DNA-binding protein 3
- Co-IP
co-immunoprecipitation
- CPT
camptothecin
- DDR
DNA damage response
- DSBs
double-strand DNA breaks
- FTI
farnesyltransferase inhibitor
- HGPS
Hutchinson–Gilford progeria syndrome
- HP1
heterochromatin protein 1
- H3K9me3
histone H3 tri-methylated at lysine 9
- KAP-1
KRAB-associated protein 1
- MAD
mandibuloacral dysplasia
- MEFs
mouse embryonic fibroblasts
- MNase
micrococcal nuclease
- NM
nuclear matrix
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- pS824-KAP-1
phosphorylation of KAP-1 at Ser824
- pT50-HP1α
phosphorylation of HP1α at Thr50
- pT51-HP1β
phosphorylation of HP1β at Thr51
- RD
restrictive dermopathy
- SA-β-gal
senescence-associated β-galactosidase
- siRNA
small interfering RNA
- Zmpste24
zinc metallopeptidase STE24 homolog
- γ-H2AX
phospho-histone H2A.X at Ser139
References
- 1.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature. 2003;423:293–8. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al. Lamin a truncation in Hutchinson–Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 3.Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Geneviève D, Hadj-Rabia S, Gaudy-Marqueste C, Smitt HS, Vabres P, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 4.Pendás AM, Zhou Z, Cadiñanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodríguez F, Tryggvason K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–9. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 5.Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 2002;16:533–47. doi: 10.1101/gad.960502. [DOI] [PubMed] [Google Scholar]
- 6.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–63. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 2011;121:2833–44. doi: 10.1172/JCI43578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson–Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879–84. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–3. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 11.Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson–Gilford progeria syndrome cells. Sci Transl Med. 2011;3:89ra58. doi: 10.1126/scitranslmed.3002346. [DOI] [PubMed] [Google Scholar]
- 12.Blagosklonny MV. Progeria, rapamycin and normal aging: recent breakthrough. Aging (Albany NY) 2011;3:685–91. doi: 10.18632/aging.100352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cenni V, Capanni C, Columbaro M, Ortolani M, D’Apice MR, Novelli G, Fini M, Marmiroli S, Scarano E, Maraldi NM, et al. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur J Histochem. 2011;55:e36. doi: 10.4081/ejh.2011.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taimen P, Pfleghaar K, Shimi T, Möller D, Ben-Harush K, Erdos MR, Adam SA, Herrmann H, Medalia O, Collins FS, et al. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc Natl Acad Sci U S A. 2009;106:20788–93. doi: 10.1073/pnas.0911895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103:8703–8. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, Zhou Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci U S A. 2011;108:12325–30. doi: 10.1073/pnas.1102789108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filesi I, Gullotta F, Lattanzi G, D’Apice MR, Capanni C, Nardone AM, Columbaro M, Scarano G, Mattioli E, Sabatelli P, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23:150–8. doi: 10.1152/physiolgenomics.00060.2005. [DOI] [PubMed] [Google Scholar]
- 18.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 19.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 20.Brasher SV, Smith BO, Fogh RH, Nietlispach D, Thiru A, Nielsen PR, Broadhurst RW, Ball LJ, Murzina NV, Laue ED. The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J. 2000;19:1587–97. doi: 10.1093/emboj/19.7.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thiru A, Nietlispach D, Mott HR, Okuwaki M, Lyon D, Nielsen PR, Hirshberg M, Verreault A, Murzina NV, Laue ED. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J. 2004;23:489–99. doi: 10.1038/sj.emboj.7600088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duband-Goulet I, Courvalin JC. Inner nuclear membrane protein LBR preferentially interacts with DNA secondary structures and nucleosomal linker. Biochemistry. 2000;39:6483–8. doi: 10.1021/bi992908b. [DOI] [PubMed] [Google Scholar]
- 23.Ye Q, Callebaut I, Pezhman A, Courvalin JC, Worman HJ. Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J Biol Chem. 1997;272:14983–9. doi: 10.1074/jbc.272.23.14983. [DOI] [PubMed] [Google Scholar]
- 24.Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. 1996;271:14653–6. doi: 10.1074/jbc.271.25.14653. [DOI] [PubMed] [Google Scholar]
- 25.Kourmouli N, Theodoropoulos PA, Dialynas G, Bakou A, Politou AS, Cowell IG, Singh PB, Georgatos SD. Dynamic associations of heterochromatin protein 1 with the nuclear envelope. EMBO J. 2000;19:6558–68. doi: 10.1093/emboj/19.23.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–5. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- 27.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–6. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 28.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Löbrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 29.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–6. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- 30.Goodarzi AA, Noon AT, Jeggo PA. The impact of heterochromatin on DSB repair. Biochem Soc Trans. 2009;37:569–76. doi: 10.1042/BST0370569. [DOI] [PubMed] [Google Scholar]
- 31.Luijsterburg MS, Dinant C, Lans H, Stap J, Wiernasz E, Lagerwerf S, Warmerdam DO, Lindh M, Brink MC, Dobrucki JW, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol. 2009;185:577–86. doi: 10.1083/jcb.200810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayoub N, Jeyasekharan AD, Venkitaraman AR. Mobilization and recruitment of HP1: a bimodal response to DNA breakage. Cell Cycle. 2009;8:2945–50. doi: 10.4161/cc.8.18.9486. [DOI] [PubMed] [Google Scholar]
- 33.White D, Rafalska-Metcalf IU, Ivanov AV, Corsinotti A, Peng H, Lee SC, Trono D, Janicki SM, Rauscher FJ., 3rd The ATM substrate KAP1 controls DNA repair in heterochromatin: regulation by HP1 proteins and serine 473/824 phosphorylation. Mol Cancer Res. 2012;10:401–14. doi: 10.1158/1541-7786.MCR-11-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu B, Wang Z, Ghosh S, Zhou Z. Defective ATM-Kap-1-mediated chromatin remodeling impairs DNA repair and accelerates senescence in progeria mouse model. Aging Cell. 2013;12:316–8. doi: 10.1111/acel.12035. [DOI] [PubMed] [Google Scholar]
- 35.Liu B, Wang Z, Zhang L, Ghosh S, Zheng H, Zhou Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat Commun. 2013;4:1868. doi: 10.1038/ncomms2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lattanzi G, Columbaro M, Mattioli E, Cenni V, Camozzi D, Wehnert M, Santi S, Riccio M, Del Coco R, Maraldi NM, et al. Pre-Lamin A processing is linked to heterochromatin organization. J Cell Biochem. 2007;102:1149–59. doi: 10.1002/jcb.21467. [DOI] [PubMed] [Google Scholar]
- 37.Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol. 1998;122:42–66. doi: 10.1006/jsbi.1998.3987. [DOI] [PubMed] [Google Scholar]
- 38.Maraldi NM, Lattanzi G, Capanni C, Columbaro M, Merlini L, Mattioli E, Sabatelli P, Squarzoni S, Manzoli FA. Nuclear envelope proteins and chromatin arrangement: a pathogenic mechanism for laminopathies. Eur J Histochem. 2006;50:1–8. [PubMed] [Google Scholar]
- 39.Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, Zou Y. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J. 2008;22:603–11. doi: 10.1096/fj.07-8598com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang SH, Andres DA, Spielmann HP, Young SG, Fong LG. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J Clin Invest. 2008;118:3291–300. doi: 10.1172/JCI35876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies BS, Barnes RH, 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet. 2010;19:2682–94. doi: 10.1093/hmg/ddq158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang SH, Chang SY, Ren S, Wang Y, Andres DA, Spielmann HP, Fong LG, Young SG. Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum Mol Genet. 2011;20:436–44. doi: 10.1093/hmg/ddq490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaturvedi P, Parnaik VK. Lamin A rod domain mutants target heterochromatin protein 1α and β for proteasomal degradation by activation of F-box protein, FBXW10. PLoS One. 2010;5:e10620. doi: 10.1371/journal.pone.0010620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaturvedi P, Khanna R, Parnaik VK. Ubiquitin ligase RNF123 mediates degradation of heterochromatin protein 1α and β in lamin A/C knock-down cells. PLoS One. 2012;7:e47558. doi: 10.1371/journal.pone.0047558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto K, Sonoda M. Self-interaction of heterochromatin protein 1 is required for direct binding to histone methyltransferase, SUV39H1. Biochem Biophys Res Commun. 2003;301:287–92. doi: 10.1016/S0006-291X(02)03021-8. [DOI] [PubMed] [Google Scholar]
- 46.Lattanzi G, Ortolani M, Columbaro M, Prencipe S, Mattioli E, Lanzarini C, Maraldi NM, Cenni V, Garagnani P, Salvioli S, et al. Lamins are rapamycin targets that impact human longevity: a study in centenarians. J Cell Sci. 2014;127:147–57. doi: 10.1242/jcs.133983. [DOI] [PubMed] [Google Scholar]
- 47.Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009;8:4112–8. doi: 10.4161/cc.8.24.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–95. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 49.Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012;11:4642–9. doi: 10.4161/cc.22937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.