

Abstract
Cyclin D1 is a mitogenic sensor that responds to growth signals from the extracellular environment and regulates the G1-to-S cell cycle transition. When cells are acutely irradiated with a single dose of 10 Gy, cyclin D1 is degraded, causing cell cycle arrest at the G1/S checkpoint. In contrast, cyclin D1 accumulates in human tumor cells that are exposed to long-term fractionated radiation (0.5 Gy/fraction of X-rays). In this study we investigated the effect of fractionated low-dose radiation exposure on cyclin D1 localization in 3 strains of normal human fibroblasts. To specifically examine the nuclear accumulation of cyclin D1, cells were treated with a hypotonic buffer containing detergent to remove cytoplasmic cyclin D1. Proliferating cell nuclear antigen (PCNA) immunofluorescence was used to identify cells in S phase. With this approach, we observed S-phase nuclear retention of cyclin D1 following low-dose fractionated exposures, and found that cyclin D1 nuclear retention increased with exposure time. Cells that retained nuclear cyclin D1 were more likely to have micronuclei than non-retaining cells, indicating that the accumulation of nuclear cyclin D1 was associated with genomic instability. Moreover, inhibition of the v-akt murine thymoma viral oncogene homolog (AKT) pathway facilitated cyclin D1 degradation and eliminated cyclin D1 nuclear retention in cells exposed to fractionated radiation. Thus, cyclin D1 may represent a useful marker for monitoring long-term effects associated with exposure to low levels of radiation.
Keywords: Cyclin D1, low-dose radiation, long-term exposure, double-strand breaks, AKT
Introduction
Cyclin D1 is a regulatory subunit of cyclin-dependent kinases (CDKs). Levels of CDKs remain constant throughout the cell cycle, and their activity is regulated by partner cyclins.1 In contrast to other cyclins, cyclin D1 expression is induced by growth factors through the Ras signaling pathway, which involves Ras, Raf, mitogen-activated protein kinase/ERK kinase (MEK), and extracellular signal-regulated kinase (ERK).2,3 Therefore, cyclin D1 is considered a mitogenic sensor that responds to growth signals from the extracellular environment.4 Cyclin D1/CDK4 and cyclin D1/CDK6 complexes phosphorylate retinoblastoma 1 (RB1), which releases E2F proteins, leading to the transactivation of genes required for the G1/S transition.5,6
Ionizing radiation causes a genotoxic stress that results in various types of DNA damage, such as DNA base damage, single-strand breaks, and double-strand breaks (DSBs). Of these, DSBs are the most critical, and trigger genomic instability. To analyze the biological effects of long-term radiation exposure, cell cultures were exposed to fractionated radiation (FR) (0.5 Gy X-rays per fraction) at 1 Gy/min, twice per day, 6 d per week for 31 d, and the effect on cyclin D1 localization was analyzed.7,8 Cyclin D1 undergoes rapid degradation by the ubiquitin proteasome pathway following a single 10-Gy dose of radiation (SR), and this degradation causes cell cycle arrest at the G1/S checkpoint.7,9 Conversely, cyclin D1 is stabilized within the nuclei of human tumor cells that are exposed to FR over 31 d.7,8 The v-akt murine thymoma viral oncogene homolog (AKT) pathway regulates the cyclin D1 radioresponse. AKT phosphorylates residue 9 of glycogen synthase kinase 3 β (GSK3β), which prevents GSK3β from phosphorylating Thr286 of cyclin D1.10-12 This blocks the nuclear export of cyclin D1, and cytoplasmic cyclin D1 is subsequently subjected to proteasomal degradation.12 AKT is transiently activated after SR exposure13,14 but constitutively activated in cells exposed to FR for >14 d.7 These results suggest that exposure to FR affects the AKT signaling pathway.7,15 Constitutive AKT activation following FR exposures downregulates the nuclear export and proteolysis of cyclin D1, which results in the nuclear retention of cyclin D1 during S phase.7 We recently reported that the abnormal expression of cyclin D1 during S phase damages DNA by blocking DNA replication.15,16 These cyclin D1-mediated DSBs activate DNA-activated protein kinase (DNA-PK). DNA-PK, in turn, activates the AKT pathway and establishes a positive feedback loop that results in increased levels of cyclin D1.7,8 Thus, prolonged FR exposure causes the long-term deregulation of cyclin D1 expression, even after the radiation exposures have ended.
Cancer risks associated with exposure to low-dose radiation are likely lower than risks associated with high doses, and the effects of long-term radiation exposure are generally milder than for an acute exposure involving the same total dose. However, because epidemiological studies require a large sample size to accurately quantify risks, the effects of low-dose radiation remain unclear. Moreover, to understand the effects of low-dose radiation it is important to characterize the molecular mechanisms underlying the DNA damage response that follows this type of long-term radiation exposure. Cyclin D1 is a stable marker of long-term FR exposure, because it persists in the nucleus after FR exposures have stopped.7,8 It is therefore important to determine if cyclin D1 accumulates within the nucleus of normal human cells after long-term exposure to low doses of radiation. In this study, 3 strains of normal human diploid fetal lung fibroblast (WI-38, MRC5, and TIG3) were exposed to various levels of FR for 31 d, and the effects on cyclin D1 localization were analyzed.
Results
X-ray fractionation regimes and evaluating the effects of low-dose radiation
For 31 d, we exposed WI-38 cells to 0.01-, 0.05-, 0.5-, 1-, or 2-Gy fractions of X-rays. MRC5 and TIG3 cells were exposed to 0.01-, 0.05-, or 2-Gy fractions. These cells were referred to as 31FR cells. Cells for which the 31 d of FR exposure were followed by a 31-d non-radiation (NR) period are denoted 31FR-31NR. We analyzed cell growth during FR exposure (Fig. 1). The growth rate plateaued if cells were exposed to ≥0.5 Gy per fraction, indicating that the cells could no longer divide under FR exposure. In contrast, cells continued to grow exponentially for 31 d if exposed to 0.01 or 0.05 Gy FR (Fig. 1).

Figure 1. Effect of long-term FR on cell growth. Cell growth of un-irradiated cells (open circles) and cells exposed to 0.01-Gy (open triangles), 0.05-Gy (open squares), 0.5-Gy (closed circles), 1-Gy (asterisks), and 2-Gy (open diamonds) fractions. Growth curves for WI-38, MRC5, and TIG3 cells are shown.
Levels of nuclear cyclin D1 following long-term FR
We tested whether cyclin D1 accumulates in the nucleus of S-phase cells exposed to low-dose FR. To identify cells in S phase, immunofluorescence was used to label cells for proliferating cell nuclear antigen (PCNA), which localizes to the replication fork during S phase.17 Microscopic examination of control WI-38 cells double-labeled for PCNA and cyclin D1 revealed diffuse PCNA and cyclin D1 throughout the nucleus and cytoplasm (Fig. 2A). To specifically examine nuclear PCNA and cyclin D1, proteins within the cell membrane and cytoplasm were washed away using a hypotonic buffer containing detergent, and isolated nuclei were examined. For control cells, PCNA localized to replication sites within nuclei17, but cyclin D1 was absent (Fig. 2A). For cells exposed to 0.05 Gy of FR for 31 d, however, detergent-insoluble nuclear cyclin D1 was detected in PCNA-positive nuclei (Fig. 2A, arrow). The percentage of cells positive for both PCNA and cyclin D1 increased with the duration of radiation exposure for the 3 strains of cells (Fig. 2B). In contrast, when WI-38 cells were exposed to SR, double-positive cells were not detected 24 h later (Fig. S1).

Figure 2. Nuclear accumulation of cyclin D1 in cells exposed to long-term FR. (A) Immunofluorescence localization of cyclin D1 and PCNA is shown for WI-38 cells (upper panels), WI-38 cells treated with a detergent-containing hypotonic buffer (middle panels), and 31FR-WI-38 cells treated with a detergent-containing hypotonic buffer (lower panels). DNA was stained with Hoechst. (B) Cells double positive for cyclin D1 and PCNA were scored 24 h following the indicated FR exposure. Data for WI-38, MRC5, and TIG3 cells exposed to 0.01-Gy or 0.05-Gy fractions are shown.
Irreversible nuclear accumulation of cyclin D1 after long-term FR
We then asked whether cyclin D1 levels would return to baseline 31 d after the last FR exposure (31FR-31NR cells). Immunofluorescence was used to assess cyclin D1 levels in 31FR-31NR-WI-38, 31FR-31NR-MRC5, and 31FR-31NR-TIG3 cells. Interestingly, nuclear cyclin D1 was retained in 31FR-31NR cells (Fig. 3). Percentages of double-positive 31FR-31NR cells varied between the 3 cell lines (Fig. 3). As shown in Figure 1, differences in growth rates between these cell lines may affect the cyclin D1 response after long-term FR.
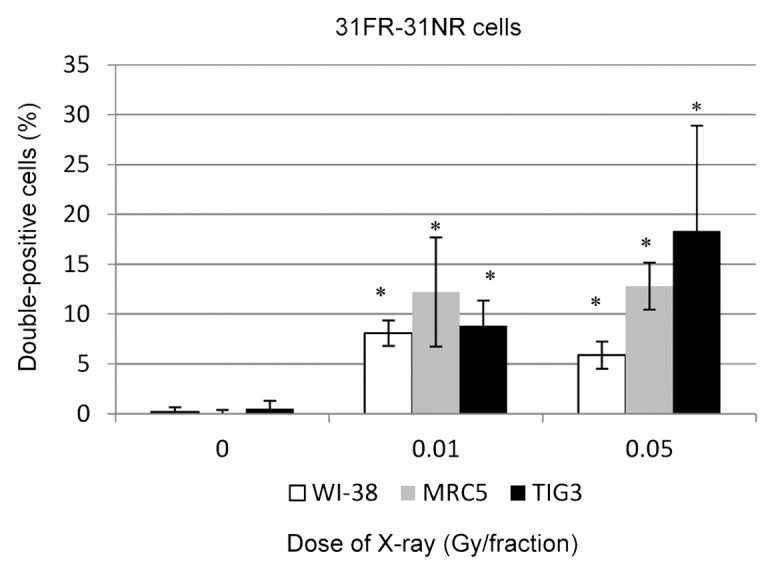
Figure 3. Retention of nuclear cyclin D1 in 31FR-31NR cells. Cells double positive for cyclin D1 and PCNA were scored in 31FR-31NR cells. Data for WI-38, MRC5, and TIG3 cells exposed to 0.01-Gy or 0.05-Gy fractions are shown.
Dose response of nuclear cyclin D1 accumulation
A hypotonic buffer was used to isolate nuclear fractions from WI-38 cells exposed to FR,18 and cyclin D1 levels were examined by western blotting. FR increased levels of nuclear cyclin D1 in a dose-dependent fashion (Fig. 4). Even the lowest doses of FR (0.01 and 0.05 Gy per fraction) affected the nuclear accumulation of cyclin D1.
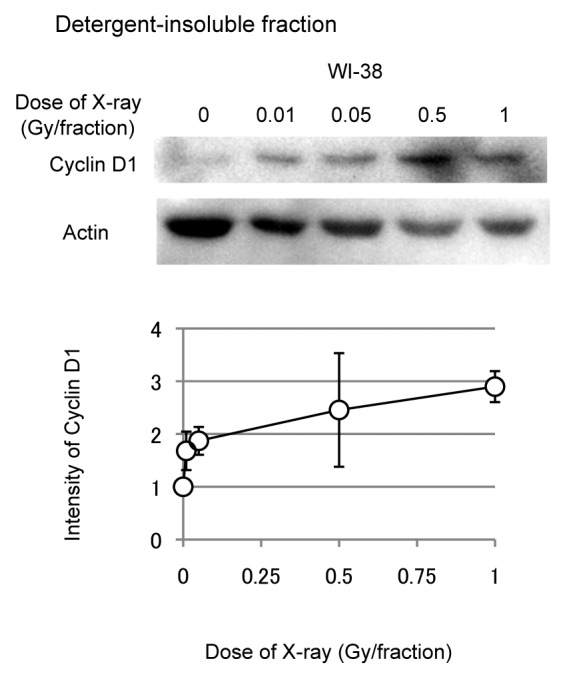
Figure 4. Dose response of nuclear cyclin D1 in WI-38 cells. Detergent-insoluble fractions of cell lysates were prepared 24 h after the last radiation exposure, and western blotting was performed. Levels of nuclear cyclin D1 were determined for 31FR-WI-38 cells exposed to indicated FR. Cyclin D1 levels were normalized to actin, β, and values are expressed relative to un-irradiated controls (0 FR). Data are means and standard deviations for 4 experiments using independent samples.
Nuclear cyclin D1 induced the formation of micronuclei
Nuclear accumulation of cyclin D1 induces genomic instability and is strongly linked to cancer development.19,20 Here, we analyzed micronucleus formation in WI-38 cells (Fig. 5A), as micronuclei represent potential cancer biomarkers.21 Micronuclei were observed in cyclin D1-positive 31FR-WI-38 cells (0.05 Gy per fraction; Fig. 5A and B) and even 31FR-31NR-WI-38 cells (Fig. 5B). Thus, long-term low-dose FR exposure induced genome instability that persisted after the exposures were stopped. We counted micronuclei in cyclin D1-negative and cyclin D1-positive cells, and found that the percentage of cells with micronuclei was higher for cyclin D1-positive cells (both 31FR-WI-38 and 31FR-31NR-WI-38) than for cyclin D1-negative counterparts (Fig. 5B). These results demonstrated that the nuclear accumulation of cyclin D1 was associated with the formation of micronuclei in 31FR-WI-38 and 31FR-31NR-WI-38 cells.

Figure 5. Micronucleus formation in 31FR-WI-38 cells. (A) Images of cyclin D1-positive (green) 31FR-WI-38 cells with micronuclei. DNA was stained with Hoechst. Magnified images of a particular cell (arrow) are shown in the insert. Arrow within the insert indicates the micronucleus. (B) The percentage of cells with a micronucleus is shown for total cells (left panel), cyclin D1-negative cells (middle panel), and cyclin D1-positive cells (right panel). Data for 31FR-WI-38 and 31FR-31NR-WI-38 cells are shown. Approximately 30% of 31FR and 15% of 31FR-31NR cells were positive for cyclin D1.
AKT inhibition prevents the nuclear accumulation of cyclin D1
The AKT inhibitor API-2 facilitates cyclin D1 degradation in 31FR and 31FR-31NR cells established from HepG2 and HeLa cells.7 As such, we expected that API-2 could prevent the nuclear accumulation of cyclin D1 in 31FR-WI-38 cells. To test this hypothesis, 31FR-WI-38 cells were treated with API-2 for 24 h, and then levels of PCNA and cyclin D1 were examined. API-2 treatment eliminated double-positive 31FR-WI-38 cells. We further confirmed that double-positive cells did not reappear 7 d after the API-2 was removed (Fig. 6).

Figure 6. API-2 prevents the nuclear accumulation of cyclin D1. Percentages of 31FR-WI-38 cells (doses are indicated) double positive for cyclin D1 and PCNA were determined for cells treated with API-2 (closed circles) and for cells not treated with API-2 (open circles). Cells were analyzed 24 h (left panel) or 7 d (right panel) after drug treatment.
Discussion
Nuclear accumulation of cyclin D1 following FR exposure
Here we demonstrated that exposing normal human fibroblasts to long-term, low-dose FR induced the nuclear accumulation of cyclin D1 during S phase (Fig. 2). Cyclin D1 levels normally fluctuate during the cell cycle. Cyclin D1 levels increase in the nucleus during the G1 phase and subsequently decline when cells enter S phase. This decline results from the nuclear exclusion of cyclin D1 and its degradation within the cytoplasm, both of which correlate with increased levels of GSK3β.12 Cell cycle-dependent proteolysis of cyclin D1 is essential for S-phase progression, and cyclin D1 accumulation during S phase perturbs DNA replication.15,20 These perturbations in DNA replication, e.g., DNA re-replication and the suppression of replication-fork progression, lead to DSBs.15,20 Here we observed that normal human fibroblasts exposed to long-term FR accumulated nuclear cyclin D1 and suffered chromosome damage in the form of micronuclei (Fig. 5). These micronuclei were likely generated as nuclear cyclin D1-retaining 31FR-31NR cells proliferated, considering the efficient cell cycle arrest of micronucleated cells. These results indicated that nuclear retention of cyclin D1 affects the genomic integrity of cells exposed to FR.
Irreversible nuclear cyclin D1 accumulation following long-term radiation exposure
Phosphorylation of the histone H2AX (gamma-H2AX) is widely used as a DSB biomarker,22 but this marker is unsuitable for evaluating long-term radiation exposure, because it disappears when DSBs are repaired. Markers are therefore needed to trace radiation effects over long periods of time. In this study, we identified cyclin D1 as a candidate marker of long-term exposure to FR, because deregulated cyclin D1 localization persisted after FR exposures were stopped. Prolonged exposure to low-dose radiation irreversibly altered cyclin D1 localization in 31FR-31NR cells derived from normal human fibroblasts. Thus, cyclin D1 may serve as a stable marker of low-dose, long-term FR exposure. It has also been shown that senescent human fibroblasts express high levels of cyclin D1,23-26 and cell cycle deregulation often induces cellular senescence.27 Thus, cyclin D1 overexpression is considered a marker of cellular senescence. Importantly, senescent cells are negative for PCNA, because cell growth has been arrested. Long-term FR exposure resulted in PCNA/cyclin D1 double-positive cells, indicating that they are different from senescent cells.
Prevention of cyclin D1 overexpression
We previously reported that long-term FR exposure causes abnormal nuclear accumulation of cyclin D1 in human tumor cells, and that this accumulation results from AKT-mediated downregulation of cyclin D1 proteasomal degradation.7,28 API-2, which is an AKT inhibitor, therefore facilitates GSK3β-mediated cyclin D1 proteolysis in these cells. Our present data showed that API-2 abrogated nuclear cyclin D1 retention induced by long-term, low-dose FR exposure in normal human fibroblasts (Fig. 6). As we described previously, nuclear accumulation of cyclin D1 induces genomic instability. Reducing cyclin D1 levels, therefore, may promote genomic stability and mitigate the harmful effects of long-term FR. Cyclin D1 is considered a potential therapeutic target for treating cancer.29,30 Our results reveal that cyclin D1 is also a marker of long-term exposure to low-dose radiation in human cells.
In conclusion, we have described a unique response that normal cells exhibit to long-term, low-level radiation, namely the nuclear accumulation of cyclin D1. Abnormal localization of cyclin D1 persisted for at least 1 mo following the last dose of radiation. Thus, cyclin D1 may represent a useful marker for monitoring long-term effects of FR.
Materials and Methods
Cell culture conditions and drugs
Normal human diploid fetal lung fibroblasts (WI-38) were purchased from the American Type Culture Collection and grown in Dulbecco modified Eagle medium (DMEM)/F12 medium (Sigma) supplemented with 10% heat-inactivated fetal calf serum. Normal human diploid lung fetal fibroblasts (MRC5 and TIG3) were purchased from the Health Science Research Resources Bank and grown in minimum essential medium (Nacalai Tesque) supplemented with 10% heat-inactivated fetal calf serum. Serial passage experiments were started after 28.5, 33, or 23 population doublings for WI-38, MRC5, or TIG-3 cells, respectively. The AKT inhibitor API-2 was purchased from Calbiochem and dissolved in dimethyl sulfoxide. Cells were treated for 24 h with API-2 at a final concentration of 20 μM. Non-treated cells were used as controls.
Irradiation experiments
Cells were irradiated using a 150-kVp X-ray generator (Model MBR-1505R2, Hitachi) with a 0.5-mm Cu and 0.1-mm Al filter at a dose of 0.7 Gy/min. Low-dose X-ray fractions (0.01 or 0.05 Gy) were administrated twice a day and 5 d/wk. Other cells received 0.5-Gy, 1-Gy, or 2-Gy fractions once a day and 5 d/wk. These exposure regimens were chosen to constitutively activate DNA damage signaling. Total doses delivered over 31 d were 0.46 Gy, 2.3 Gy, 11.5 Gy, 23 Gy, or 46 Gy for cells exposed to FR of 0.01 Gy, 0.05 Gy, 0.5 Gy, 1 Gy, or 2 Gy, respectively.
Cell growth assay
Cells (1 × 105) were seeded into 25-cm2 flasks (Thermo Fisher Scientific), incubated overnight, and irradiated daily. Growth rates were monitored by counting cell numbers twice a wk. When the total cell number exceeded 1 × 105, cells were subcultured to 1 × 105 cells in a new flask.
Immunofluorescence
Immunofluorescence stainings were performed as described.17 Cells were seeded onto 18 × 18 mm coverslips and placed into 35-mm tissue culture dishes. Cells on coverslips were first treated with a hypotonic buffer (10 mM Tris–HCL, pH 7.4, 2.5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, and 0.5% NP-40) for 5 min, washed twice with phosphate buffered saline (PBS), and fixed with ice-cold acetone (5 min) and ice-cold methanol (5 min). Cells were then blocked in 5% ([w/v]) bovine serum albumin in PBS (PBS-BSA) for 15 min at room temperature. Antibodies against PCNA (Santa Cruz Biotechnology) and cyclin D1 (Nichirei Bioscience) were diluted with PBS-BSA and added to cells on coverslips. After a 1 h incubation, coverslips were washed 3 times with 0.1% Triton X-100 in PBS (PBST), and incubated with secondary antibodies conjugated with Alexa Fluor 488 (Molecular Probes) or Cy-3 (Jackson ImmunoResearch Laboratories) for 1 h. Cells were then washed 3 times with PBST and counterstained for DNA with Hoechst 33258 (4 µg/mL in Vectashield mounting medium; Vector Laboratories). Images were captured using a CCD camera attached to a fluorescence microscope (Keyence). For each data point, >50 cells were counted from at least 3 independent samples.
Western blot analyses
Western blotting was performed as described.7 To isolate nuclear fractions, cells were treated with a hypotonic buffer for 5 min on ice. Proteins were separated using sodium lauryl sulfate PAGE and electroblotted onto polyvinylidene fluoride membranes (Bio-Rad). Membranes were blocked with 5% (w/v) phospho-blocker (Cell Biolabs) for 1 h and then incubated with primary antibodies against β-actin (A2066, Sigma) and cyclin D1 (Nichirei Bioscience) for 1 h at room temperature or overnight at 4 °C. Next, membranes were incubated with either horseradish peroxidase-conjugated goat anti-rabbit (GE Healthcare) or anti-mouse IgG (R&D Systems) secondary antibodies. Protein bands were visualized using Chemi-Lumi One L western-blotting substrate (Nacalai Tesque), and band intensities were measured using Image Lab software (Bio-Rad).
Statistical analysis
Error bars represent standard deviations. All experiments were repeated at least 3 times using independent samples. Student t test was used for statistical analyses. Single and double asterisks indicate significant differences at P < 0.01 and P < 0.05, respectively.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the staff of the National Institute of Public Health for assistance with the research. This research was supported by a grant from the Japanese Ministry of Education and Science Kiban C (24510063). This work was performed at the Joint Usage/Research Center (RIRBM), Hiroshima University.
References
- 1.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–65. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 2.Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proc Natl Acad Sci U S A. 1998;95:1091–6. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J. 1997;326:61–8. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 5.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 6.DeGregori J. The Rb network. J Cell Sci. 2004;117:3411–3. doi: 10.1242/jcs.01189. [DOI] [PubMed] [Google Scholar]
- 7.Shimura T, Kakuda S, Ochiai Y, Nakagawa H, Kuwahara Y, Takai Y, Kobayashi J, Komatsu K, Fukumoto M. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene. 2010;29:4826–37. doi: 10.1038/onc.2010.238. [DOI] [PubMed] [Google Scholar]
- 8.Shimura T. Acquired radioresistance of cancer and the AKT/GSK3β/cyclin D1 overexpression cycle. J Radiat Res. 2011;52:539–44. doi: 10.1269/jrr.11098. [DOI] [PubMed] [Google Scholar]
- 9.Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000;102:55–66. doi: 10.1016/S0092-8674(00)00010-6. [DOI] [PubMed] [Google Scholar]
- 10.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 11.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan J, Geng L, Yazlovitskaya EM, Hallahan DE. Protein kinase B/Akt-dependent phosphorylation of glycogen synthase kinase-3beta in irradiated vascular endothelium. Cancer Res. 2006;66:2320–7. doi: 10.1158/0008-5472.CAN-05-2700. [DOI] [PubMed] [Google Scholar]
- 14.Tan J, Hallahan DE. Growth factor-independent activation of protein kinase B contributes to the inherent resistance of vascular endothelium to radiation-induced apoptotic response. Cancer Res. 2003;63:7663–7. [PubMed] [Google Scholar]
- 15.Shimura T, Ochiai Y, Noma N, Oikawa T, Sano Y, Fukumoto M. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle. 2013;12:773–82. doi: 10.4161/cc.23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimura T, Fukumoto M, Kunugita N. The role of cyclin D1 in response to long-term exposure to ionizing radiation. Cell Cycle. 2013;12:2738–43. doi: 10.4161/cc.25746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimura T, Toyoshima M, Adiga SK, Kunoh T, Nagai H, Shimizu N, Inoue M, Niwa O. Suppression of replication fork progression in low-dose-specific p53-dependent S-phase DNA damage checkpoint. Oncogene. 2006;25:5921–32. doi: 10.1038/sj.onc.1209624. [DOI] [PubMed] [Google Scholar]
- 18.Shimura T, Torres MJ, Martin MM, Rao VA, Pommier Y, Katsura M, Miyagawa K, Aladjem MI. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J Mol Biol. 2008;375:1152–64. doi: 10.1016/j.jmb.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292–6. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, Goradia A, Wasik MA, Klein-Szanto AJ, Rustgi AK, et al. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007;21:2908–22. doi: 10.1101/gad.1586007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-Zein R, Vral A, Etzel CJ. Cytokinesis-blocked micronucleus assay and cancer risk assessment. Mutagenesis. 2011;26:101–6. doi: 10.1093/mutage/geq071. [DOI] [PubMed] [Google Scholar]
- 22.Lassmann M, Hänscheid H, Gassen D, Biko J, Meineke V, Reiners C, Scherthan H. In vivo formation of gamma-H2AX and 53BP1 DNA repair foci in blood cells after radioiodine therapy of differentiated thyroid cancer. J Nucl Med. 2010;51:1318–25. doi: 10.2967/jnumed.109.071357. [DOI] [PubMed] [Google Scholar]
- 23.Dulić V, Drullinger LF, Lees E, Reed SI, Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci U S A. 1993;90:11034–8. doi: 10.1073/pnas.90.23.11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucibello FC, Sewing A, Brüsselbach S, Bürger C, Müller R. Deregulation of cyclins D1 and E and suppression of cdk2 and cdk4 in senescent human fibroblasts. J Cell Sci. 1993;105:123–33. doi: 10.1242/jcs.105.1.123. [DOI] [PubMed] [Google Scholar]
- 25.Atadja P, Wong H, Veillete C, Riabowol K. Overexpression of cyclin D1 blocks proliferation of normal diploid fibroblasts. Exp Cell Res. 1995;217:205–16. doi: 10.1006/excr.1995.1080. [DOI] [PubMed] [Google Scholar]
- 26.Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012;11:4642–9. doi: 10.4161/cc.22937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013;20:1241–9. doi: 10.1038/cdd.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimura T, Kakuda S, Ochiai Y, Kuwahara Y, Takai Y, Fukumoto M. Targeting the AKT/GSK3β/cyclin D1/Cdk4 survival signaling pathway for eradication of tumor radioresistance acquired by fractionated radiotherapy. Int J Radiat Oncol Biol Phys. 2011;80:540–8. doi: 10.1016/j.ijrobp.2010.12.065. [DOI] [PubMed] [Google Scholar]
- 29.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–72. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.