

Abstract
The p53 tumor suppressor is controlled by an interactive network of factors that stimulate or inhibit its transcriptional activity. Within that network, Mdm2 functions as the major antagonist of p53 by promoting its ubiquitylation and degradation. Conversely, Tip60 activates p53 through direct association on target promoters as well as acetylation of p53 at lysine 120 (K120). This study examines the functional relationship between Mdm2 and Tip60 with a novel p53 regulator, NIAM (nuclear interactor of ARF and Mdm2). Previous work showed NIAM can suppress proliferation and activate p53 independently of ARF, indicating that other factors mediate those activities. Here, we demonstrate that NIAM is a chromatin-associated protein that binds Tip60. NIAM can promote p53 K120 acetylation, although that modification is not required for NIAM to inhibit proliferation or induce p53 transactivation of the p21 promoter. Notably, Tip60 silencing showed it contributes to but is not sufficient for NIAM-mediated p53 activation, suggesting other mechanisms are involved. Indeed, growth-inhibitory forms of NIAM also bind to Mdm2, and increased NIAM expression levels disrupt p53–Mdm2 association, inhibit p53 polyubiquitylation, and prevent Mdm2-mediated inhibition of p53 transcriptional activity. Importantly, loss of NIAM significantly impairs p53 activation. Together, these results show that NIAM activates p53 through multiple mechanisms involving Tip60 association and Mdm2 inhibition. Thus, NIAM regulates 2 critical pathways that control p53 function and are altered in human cancers, implying an important role for NIAM in tumorigenesis.
Keywords: NIAM, ARF, Mdm2, Tip60, p53, acetylation, ubiquitylation
Introduction
The p53 tumor suppressor is a key checkpoint regulator that induces cell death or arrest in response to numerous cellular insults, including DNA damage and oncogene activation.1-3 Ultimately, p53 protects the genome from stress-induced genetic mutations and chromosomal instability (CIN) and is commonly referred to as the “guardian of the genome”.4 The importance of p53 in preventing cancer is underscored by its functional inactivation through TP53 gene mutation or disruption of its regulators in the majority of human cancers.5-7
p53 is a transcription factor whose cellular expression and activity are controlled by many different mechanisms. The primary negative regulator of p53 is Mdm2 (mouse double minute-2), an E3 ubiquitin ligase that targets p53 for nuclear export or proteasomal degradation by mono- or polyubiquitylation, respectively.8-10 Mdm2 can also bind the p53 transactivation domain and directly block transcriptional initiation.11-13 Mdm2 itself is a transcriptional target of p53, creating a negative feedback loop that keeps p53 at low levels under normal conditions.14-16 Proper control of p53 stability and function by Mdm2 is critical in both development and cancer. Elevated p53 activity in Mdm2 knockout mice causes early embryonic lethality that is rescued by p53 deletion,17 whereas perpetual loss of p53 activity due to Mdm2 amplification or overexpression promotes tumorigenesis in sarcomas and many other human cancers.18-20
Numerous binding partners of Mdm2 and/or p53 influence their functional interaction and p53 activity.1,21 One important example is the alternative reading frame (ARF) tumor suppressor encoded by the INK4a/ARF locus.22 Oncogene activation and sustained DNA damage induce expression of ARF, which binds to Mdm2, inhibits its E3 ubiquitin ligase activity and restricts Mdm2–p53 association.22 In so doing, ARF stabilizes p53 and stimulates its transcriptional activation, thereby provoking apoptosis or cell senescence. While ubiquitylation plays a dominant role in p53 regulation, other post-translational modifications of p53, including acetylation, are also instrumental in controlling its activity.2,23-25 Tip60 (HIV-Tat1 interactive protein of 60 kDa), a MYST-family histone acetyltransferase, has particular relevance to ARF–Mdm2–p53 signaling. Tip60 is a potent activator of p53 required for its induction of apoptosis, autophagy, and cell cycle inhibition.26,28,29 It binds to p53, is recruited to p53 target promoters, and enhances expression of apoptosis and autophagy genes via acetylation of p53 lysine 120 (K120) in response to DNA damage.26,28-30 Tip60 also binds ARF and Mdm2, inhibits Mdm2-mediated neddylation of p53, and is negatively regulated by Mdm2-mediated ubiquitylation.31-33
We previously discovered NIAM (nuclear interactor of ARF and Mdm2), a novel growth inhibitor capable of activating p53.34 NIAM binds ARF and enhances its nuclear localization, while it is downregulated by Mdm2-mediated ubiquitylation and proteasome degradation. Those findings firmly placed NIAM within the ARF–Mdm2–p53 pathway, but how it activated p53 was unclear. ARF was not required, since NIAM caused a G1 phase arrest and stimulated p53-mediated expression of the p21 (also called CDKN1A/Cip1/WAF1) cell cycle inhibitor in ARF-null cells.34 In this study, we investigated mechanisms by which NIAM activates p53. Our findings reveal that NIAM controls p53 through at least 2 mechanisms, Tip60 association and Mdm2 inhibition.
Results
The NIAM N terminus is required and sufficient for p53 activation and inhibition of cell proliferation
NIAM acts through undefined mechanisms to activate p53, induce expression of p21, and suppress cell proliferation.34 To identify functional domains within NIAM responsible for those activities, we generated 2 deletion mutants. The N-terminus mutant (NT) contains a predicted nuclear/nucleolar localization signal domain (NLS) and a lysine-rich region (LYS-R) (Fig. 1A). The C-terminus mutant (CT) contains 2 phenylalanine and tyrosine-rich domains (FYRN and FYRC) that are thought to mediate protein–protein interactions.35,36 To assess their in vivo activities, each form of NIAM was HA-tagged and expressed in human U20S cells (p53 wild-type, ARF-null). In p53 reporter assays using a p21 promoter reporter construct, both wild-type (WT) and NT forms of NIAM activated p53 in a dose-dependent manner (Fig. 1B). By comparison, the CT mutant of NIAM was essentially inert. Likewise, in colony-formation assays, both WT and NT NIAM reduced proliferation relative to vector control (Vec), whereas the CT mutant had no growth-inhibitory activity (Fig. 1C). Similar results were obtained from transient growth analyses in mouse NIH 3T3 fibroblasts (p53 wild-type, ARF-null) (Fig. 1D). These data establish that NIAM’s N-terminal domains mediate its anti-proliferative and p53-stimulating activities.
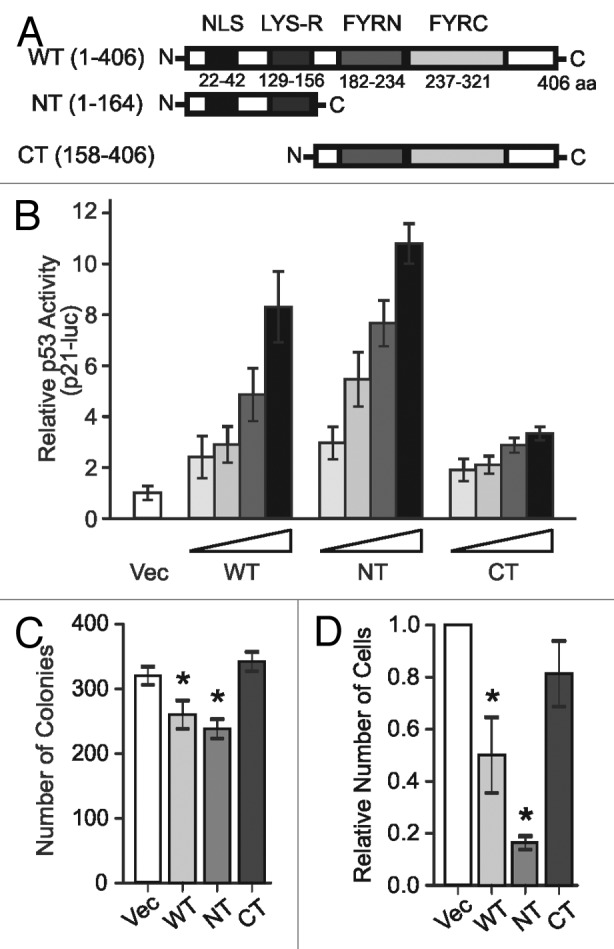
Figure 1. N terminus of NIAM is required and sufficient for activating p53 and inhibiting growth. (A) Schematic of wild-type (WT) mouse NIAM (1–406 aa) and deletion mutants NT (1–164) and CT (158–406). Locations of the predicted nuclear localization sequence (NLS), lysine-rich domain (LYS-R), and C-terminal “FY” rich regions are indicated. (B) p53-dependent luciferase activity in U2OS cells expressing increasing amounts of the indicated forms of NIAM (1, 2, 4, or 6 µg) relative to empty vector (Vec) control plus constant levels of the reporter constructs (80 ng Renilla plasmid and 800 ng of the p53-luc reporter plasmid). Mean and SD shown for 2 experiments with triplicate samples. (C) CFA in U2OS cells infected with vector (Vec) or indicated NIAM forms. Error bars equal the SD from mean for triplicate counts in a representative experiment. Asterisks denote statistical significance (P value < 0.05) as determined by an unpaired 2-tailed Student t test. (D) Growth inhibition was measured by a transient (6 d) growth assay in mouse NIH 3T3 fibroblasts infected with empty vector or NIAM mutants. Mean +/− SD of at least 3 different experiments is shown. Asterisks denote statistical significance (P < 0.006) relative to Vec as determined by a paired 2-tailed Student t test.
The predicted nuclear localization sequence of NIAM is contained within its N terminus. To assess the subcellular localization of WT and mutant NIAM, fibroblasts expressing each HA-tagged form were stained with HA antibodies and examined by confocal microscopy. The active WT and NT forms of NIAM both resided in nuclei, although NT accumulated more prominently within nucleoli (Fig. 2A). In contrast, the CT mutant was highly expressed throughout the cytoplasm and absent from the nucleus. Biochemical fractionation analyses yielded similar results and also showed that NIAM associates with chromatin. Specifically, western blotting of cytosolic, nucleoplasmic, and chromatin fractions from mouse and human cells showed that endogenous NIAM is almost exclusively bound to chromatin (Fig. 2B, top panels). The majority of exogenous WT and NT NIAM proteins likewise co-purified with chromatin, while the CT form was primarily cytosolic (Fig. 2B, bottom panels). Thus, the same N-terminal domains of NIAM are required to mediate its chromatin binding, activation of p53, and suppression of cell proliferation.
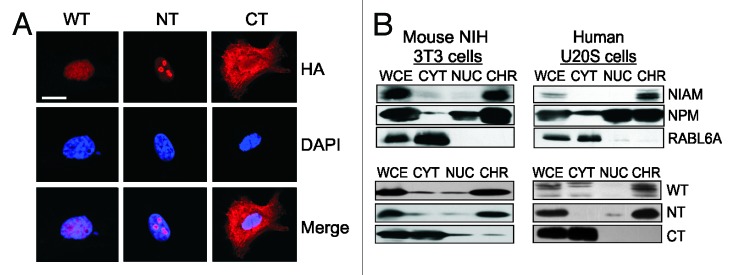
Figure 2. NIAM associates with chromatin via its N-terminal residues. The localization of NIAM wild-type (WT), N-terminal (NT), and C-terminal (CT) proteins was evaluated by immunofluorescence (A) and biochemical fractionation (B) of cells. (A) Representative confocal images of NIAM subcellular localization in ARF-null MEFs expressing the indicated HA-tagged forms of NIAM. NIAM proteins (red) were detected using HA antibodies and nuclei (blue) detected by DAPI staining. Scale bar, 10 μm. (B) Chromatin (CHR), nucleoplasmic (NUC), and cytoplasmic (CYT) fractions were isolated from mouse NIH 3T3 cells (left panel) or human U2OS cells (right panel), and protein localization in equivalently loaded fractions was examined by western blotting. WCE, whole-cell extracts. Top panels show endogenous NIAM localization to chromatin relative to NPM (chromatin associated nucleocytoplasmic shuttling protein) and RABL6A (cytoplasmic protein) controls. Bottom panels display localization patterns of HA-tagged WT, NT, and CT forms of NIAM, as detected using HA antibodies.
NIAM binds Tip60, promotes p53 acetylation, and requires Tip60 to fully activate p53
One important chromatin-bound regulator of p53 is the acetyltransferase Tip60.37 The ability of Tip60 to stimulate p53-mediated transcription is dependent on its association with p53 at target promoters, which does not require p53 acetylation at lysine 120 (K120).26,28,29 Besides their common ability to bind chromatin and activate p53, NIAM and Tip60 share some of the same binding partners, ARF and Mdm2.31,32,34 Such evidence led us to test if NIAM and Tip60 interact functionally to promote p53 activity. Initial assays evaluated NIAM and Tip60 association using tagged versions of each protein expressed in HEK293T cells. Interestingly, we found that Tip60 overexpression increased the levels of endogenous and exogenous NIAM, while NIAM overexpression decreased levels of Tip60, supporting the existence of a functional interplay between the 2 proteins (Fig. 3A, left panel). Immunoprecipitation (IP) with antibodies to either the Myc tag of NIAM or HA tag of Tip60 showed that NIAM and Tip60 can bind to each other in vivo (Fig. 3A, right panel). Both proteins are normally expressed at low levels in cells, and their detection is hindered by antibody limitations.28,29,34 Nonetheless, using p53-null mouse embryo fibroblasts, which express higher levels of NIAM and Tip60 due to the lack of Mdm2 expression,31,34 endogenous NIAM-Tip60 complexes were clearly detected by IP–western analyses (Fig. 3B).

Figure 3. NIAM is a novel interactor of Tip60. (A) Association between exogenous Myc-NIAM and HA-Tip60 was examined in transfected HEK293T cells (10 μg of each plasmid) by reciprocal IP-western analyses using HA, Myc, NIAM, and Tip60 antibodies (right). Immunoblots of whole-cell lysates revealed expression levels of each protein in the indicated lysates with GAPDH serving as loading control (left). *, Non-specific band; Endog, endogenous NIAM; Myc-tag, Myc-NIAM. (B) Endogenous NIAM-Tip60 complexes were detected in p53-null MEFs by immunoprecipitation with antibodies to NIAM or Tip60 followed by blotting with 2 different NIAM antibodies (polyclonal, pAb; monoclonal, mAb). (C) In vivo binding between Myc-tagged WT and NT NIAM, but not CT NIAM, with HA-tagged Tip60. HEK293T cells were transfected with 15 µg of each plasmid, complexes were pulled down using HA agarose (HA-IPs) and proteins detected with HA and Myc antibodies (upper panels). Relative levels of Myc-NIAM NT and CT protein expression in the cells were analyzed by immunoblotting of whole cell lysates (lower panels). (D) In vitro translated Tip60 can bind to GST-tagged WT and NT forms, but not the CT mutant. GST protein levels and Tip60 binding were measured by Coomassie staining and autoradiography, respectively.
To identify Tip60 interaction domains within NIAM, Myc-tagged forms of NIAM were co-expressed with HA-Tip60 in HEK293T cells. Tip60 complexes were isolated using HA-agarose and analyzed for NIAM association by immunoblotting, showing that Tip60 interacted specifically with the WT and NT forms of NIAM, but not with CT NIAM (Fig. 3C). Binding assays using in vitro-translated Tip60 and GST-tagged NIAM proteins yielded the same result, demonstrating that WT and NT NIAM, but not the CT mutant, were able to bind Tip60 directly (Fig. 3D). Together, these analyses reveal that the NIAM N-terminus binds Tip60, and that their mutual binding partners, ARF and Mdm2, are not required for that interaction.
We next tested if NIAM could promote p53–K120 acetylation. NIAM expression was sufficient to induce K120 acetylation of endogenous p53, suggesting NIAM stimulates Tip60 function toward p53 (Fig. 4A). Since the NIAM NT mutant effectively activates p53 and is required for Tip60 binding, we assessed its ability to induce K120 acetylation of p53. Surprisingly, the NT mutant was unable to promote that modification, similar to the CT mutant that lacked Tip60 binding capacity (Fig. 4B). This suggested that Tip60 association alone is insufficient for NIAM to promote p53–K120 acetylation, and it also raised a larger question about the importance of Tip60 to NIAM-mediated p53 activation. To address that issue, we measured NIAM’s ability to stimulate p53 transcriptional activity in cells following Tip60 depletion. Quantitative RT-PCR confirmed effective Tip60 knockdown in cells expressing 2 different Tip60 short hairpin RNAs (shRNAs, designated shT1 and shT2) compared with empty vector (EV) control cells (Fig. 4C). Loss of Tip60 expression significantly reduced the transcriptional activation of p53 by NIAM (Fig. 4D). However, p53 activation by NIAM was only partially reduced by Tip60 depletion, suggesting that NIAM acts through additional mechanisms to promote p53 activation.
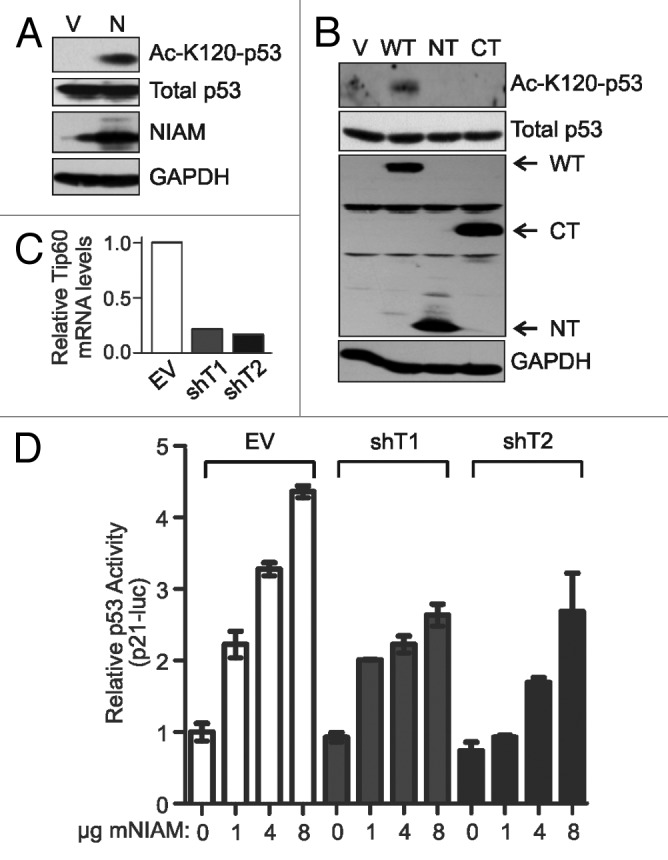
Figure 4. NIAM can promote acetylation of K120 on p53 and requires Tip60 to fully activate p53. (A) NIAM expression induces K120 acetylation of endogenous p53. U2OS cells expressing empty vector (V) (10 µg) or NIAM (N) (10 µg) were immunoblotted with antibodies specific to p53, HA-NIAM, GAPDH, and p53 K120 acetylation. (B) U2OS cells transfected with 5 to 15 µg of each indicated HA-tagged NIAM plasmid and lysates were immunoblotted with HA, K120 Ac-p53, p53, and GAPDH antibodies. Neither mutant of NIAM was capable of inducing acetylation of p53 at K120. (C) qRT-PCR demonstrates effective knockdown of Tip60 in H1299 cells expressing 2 separate shRNAs to Tip60 (shT1 and shT2) relative to empty vector (EV) control. (D) Cells expressing EV or Tip60 knockdown constructs were transfected with vector (0 µg) or increasing amounts of NIAM (1, 4, or 8 µg) along with the p53 cDNA (0.005 µg), p53-luciferase reporter construct (0.8 µg), and Renilla luciferase control (80 ng). Luciferase activity was measured in duplicate for each sample and results from a representative experiment are shown.
NIAM’s N-terminal residues mediate Mdm2 binding and confer protein instability
The primary antagonist of p53 is the Mdm2 E3 ubiquitin ligase, which destabilizes p53 and keeps it at low levels in cells.19,38 Mdm2 likewise binds and negatively regulates NIAM via polyubiquitylation and proteasomal degradation,34 although where it binds NIAM and if it does so directly or through shared partners is not known. To resolve those questions, in vitro binding assays were performed. In vitro-translated Mdm2 interacted with GST-tagged WT and NT NIAM, but not the CT mutant (Fig. 5A). Transient co-expression of both proteins in cells yielded identical binding results (Fig. 5B). Thus, NIAM and Mdm2 bind directly through amino acids 1–164 in the NIAM N terminus. Notably, stable expression of NIAM proteins in p53-positive cells showed reduced levels of WT and NT NIAM compared with the CT mutant (Fig. 5C, left panel), and half-life analyses demonstrated that the WT and NT proteins were short-lived (t1/2 = 2.2 to 2.4 h, respectively) relative to the CT protein (t1/2 > 48 h) (Fig. 5C, right panels). Thus, the N terminus of NIAM mediates Mdm2 binding and imparts protein instability.
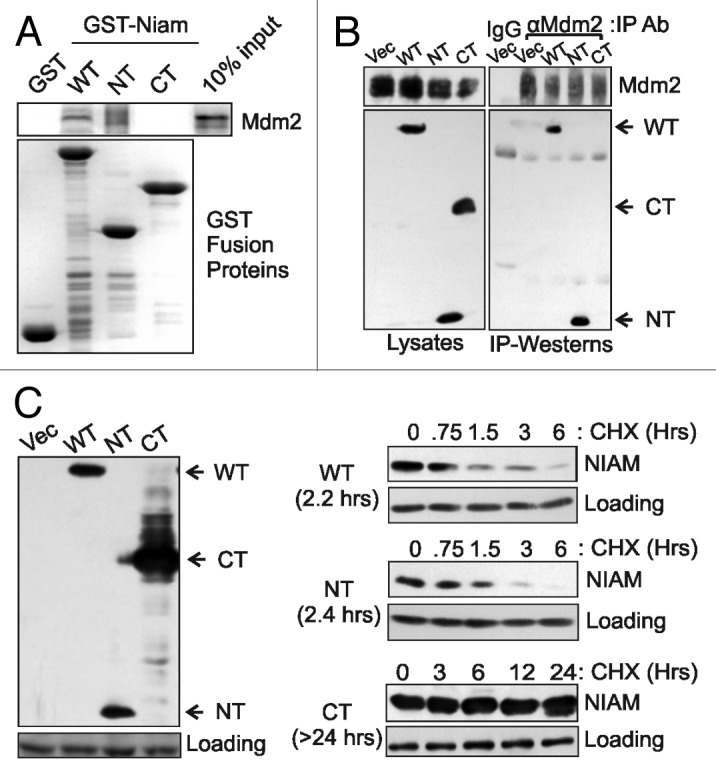
Figure 5. NIAM N-terminal sequences bind to Mdm2 and confer instability to the protein. (A) In vitro translated Mdm2 binds to GST-tagged WT and NT, but not CT, forms of NIAM. GST protein levels and Mdm2 binding were measured by Coomassie staining and autoradiography, respectively. (B) In vivo binding between the indicated HA-tagged NIAM proteins (10 µg each plasmid) and Mdm2 (2 µg plasmid) was assayed in transiently transfected U2OS cells. Whole-cell lysates were immunoprecipitated (IP) with a mixture of monoclonal and polyclonal antibodies to Mdm2 or control IgG. Mdm2 IPs were performed with roughly equivalent amounts of each NIAM form, followed by western blotting with Mdm2 and HA antibodies (right panel). Relative levels of Mdm2 and HA-NIAM protein expression in the cells were analyzed by immunoblotting of whole-cell lysates (left panel). (C) Mouse NIH 3T3 cells stably expressing each HA-tagged NIAM form or vector control were examined by western blotting with HA antibodies (left panel). The half-life of each NIAM protein was determined following treatment of the cells with cycloheximide (CHX) for the indicated times and blotting with HA antibodies. The average half-life calculated from at least 3 experiments is denoted in parentheses to the left of each blot (right panels).
NIAM interferes with Mdm2–p53 association and prevents p53 ubiquitylation
Both the WT and NT forms of NIAM activate p53 and bind Mdm2. Other Mdm2 binding proteins, such as ARF and nucleophosmin (NPM), promote p53 activation, at least in part, by limiting Mdm2–p53 association.39-42 Therefore, we examined whether NIAM could do the same. H1299 cells were transfected with a constant level of p53 plus increasing amounts of HA-tagged NIAM. Western blotting showed that higher levels of NIAM protein resulted in increased induction of the p53 targets, p21, and Mdm2 (Fig. 6A). Consistent with p53 activation, and despite the elevated expression of Mdm2, increasing levels of NIAM caused a concomitant and significant reduction in Mdm2–p53 association (Fig. 6B).

Figure 6. NIAM interferes with Mdm2-p53 association, prevents p53 polyubiquitylation, and promotes p53 transcriptional activity. (A) NIAM stimulates p53 transcriptional upregulation of p21 and Mdm2 in H1299 cells transfected with p53 expression plasmid (9 µg) and increasing amounts of an HA-NIAM construct (1, 5, or 10 μg). Direct lysates were blotted with antibodies to NIAM, p53, Mdm2, p21, and GAPDH. (B) IP–western analyses of p53-Mdm2 complexes in the same cells as (A) show reduced Mdm2 association with p53 in cells expressing higher levels of NIAM. (C) H1299 cells were transfected with a constant amount of p53 (2 µg) and His-ubiquitin (8 µg) plasmids plus increasing amounts of the HA-NIAM construct (5, 10, or 15 μg). His-Ub forms of p53 were pulled down on Ni2+-NTA− agarose, separated by SDS-PAGE, and blotted with antibodies to p53 (DO-1) (bottom panel). Total levels of p53 and NIAM in the samples were detected by western blotting of whole-cell lysates. Quantification of the total amount of ubiquitylation of p53 was calculated using ImageJ. (D) U2OS cells were transfected with increasing amounts of NIAM (3.75, 7.5, or 30 μg) and levels of endogenous p53, p21, and Mdm2 were assessed by immunoblotting. (E) U2OS cells were transfected with increasing amounts of HA-tagged NIAM (5, 10, 15, or 20 μg) and His-tagged ubiquitin (8 µg) plasmids. His-Ub forms of p53 were pulled down using Ni2+-NTA− agarose beads, separated by SDS-PAGE, and blotted with a p53 antibody (DO-1). Quantification of p53 ubiquitylation was done using ImageJ software.
Mdm2 primarily inhibits p53 by promoting its ubiquitylation, so we tested if NIAM could block that process. Cells were transfected with His-tagged ubiquitin along with a constant level of p53 and increasing amounts of NIAM. Ubiquitylated proteins were isolated on Ni2+-NTA-agarose under denaturing conditions, and polyubiquitylated p53 was detected by western blotting with p53 antibodies (Fig. 6C). Consistent with the ability of NIAM to impair Mdm2 binding to p53, NIAM expression significantly reduced p53 ubiquitylation. The same results were observed for endogenous p53, where NIAM reduced its polyubiquitylation and induced expression of its target genes in a dose-dependent manner (Figs. 6D and E). Together, these results show that NIAM can promote p53 activation by blocking its association with Mdm2 and inhibiting its polyubiquitylation.
NIAM abolishes Mdm2-mediated inhibition of p53 transcriptional activity and is required for maximal p53-dependent transactivation
The above data indicated that NIAM can interfere with the negative regulation of p53 by endogenous Mdm2. Many human cancers express abnormally high levels of Mdm2,18-20 so we tested the ability of NIAM to reactivate p53 in the presence of exogenous Mdm2. Relative p53 transcriptional activity was measured by p21–luciferase reporter assays and shown to be effectively reduced by Mdm2 overexpression (Fig. 7A). Both WT and NT forms of NIAM overcame the effects of Mdm2 and fully restored p53 activity to levels achieved in the absence of ectopic Mdm2. In contrast, the CT form of NIAM, which lacks Mdm2 binding, failed to inhibit Mdm2 and was unable to restore p53 activity. These data establish Mdm2 inhibition as a significant mechanism by which NIAM activates p53.
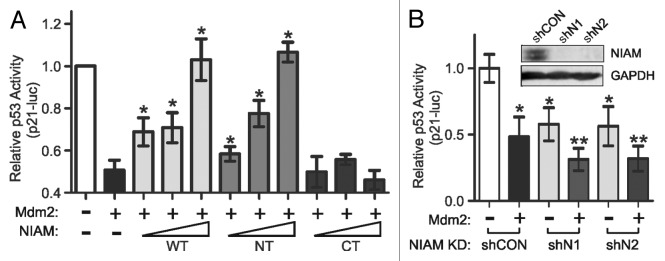
Figure 7. NIAM abolishes Mdm2-mediated inhibition of p53 transcriptional activity and is required for full p53 activation. (A) WT and NT forms of NIAM restored p53 activity after Mdm2-mediated inhibition of p53, whereas the NIAM CT mutant was inert. H1299 cells were transfected with constant levels of p53 (0.005 µg), Mdm2 (0.15 µg), p53 reporter (0.8 µg), and Renilla luciferase (80 ng) plasmids plus increasing amounts of HA-tagged forms of NIAM (1, 4, or 8 μg). The relative p53 activation of a p21–luciferase reporter was measured. Data are the mean +/− SD from 3 or more independent experiments (*, P < 0.01 relative to Mdm2 alone, as measured by an unpaired 2-tailed Student t test). (B) NIAM is required for full transcriptional activity of p53. NIAM was stably silenced in H1299 cells using 2 separate shRNAs (shN1 and shN2) vs. a scrambled shRNA control (shCON) and NIAM downregulation was confirmed by western blotting (inset). Cells were transfected with a p53 plasmid (0.005 µg) plus (+) or minus (−) the Mdm2 construct (0.15 µg), and p53 activation of the p21-luc reporter (0.8 µg of the reporter plus 80 ng Renilla luciferase) was measured. Mean +/− SD of data from at least 4 different experiments is shown. Statistical significance was determined by an unpaired 2-tailed Student t test. (*P < 0.0001 relative to shCON lacking Mdm2; **P < 0.0098 relative to Mdm2 alone or each NIAM knockdown in the absence of Mdm2).
Our cumulative findings suggest that NIAM is an important positive regulator of p53. In agreement with that conclusion, p53 transcriptional activity was significantly reduced by stable shRNA-mediated silencing of endogenous NIAM using 2 separate targeting shRNAs, designated shN1 and shN2 (Fig. 7B). The decrease in p53 activity caused by NIAM depletion was similar in magnitude to that caused by exogenous Mdm2 expression. Moreover, NIAM loss combined with Mdm2 overexpression resulted in an even greater impairment of p53 activity than either condition alone.
Discussion
The anti-cancer activities of p53 are governed by a network of cellular factors that bind and/or modify the protein through various post-translational modifications.1,2,24,25,43,44 NIAM was previously shown to induce p53 transcriptional activity and cell cycle arrest, although its mechanisms of action were undefined.34 Here, we explored how NIAM activates p53. Our findings reveal that NIAM is a chromatin-associated protein that indirectly promotes p53 activation through its interaction with 2 critical p53 regulators, Tip60 and Mdm2 (Fig. 8).
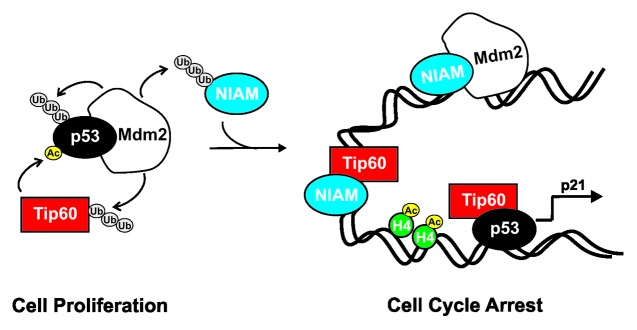
Figure 8. NIAM activates p53 by at least 2 different mechanisms. In proliferating cells, the Mdm2 E3 ubiquitin ligase polyubiquitylates p53, Tip60, and NIAM leading to their degradation at the proteasome and keeping their basal expression low. Tip60 is a histone acetyltransferase that acetylates histone H4 (inducing chromatin decondensation), promoting the p53-dependent recruitment of Tip60 to p53 gene targets, such as p21. Our data show NIAM is a chromatin bound protein capable of activating p53 independent of ARF through at least 2 mechanisms. First, NIAM can associate with Tip60 and induce p53 transactivation of p21, although it appears to do so without binding to p53 itself. Second, NIAM binds Mdm2 (which also appears exclusive of Tip60 and p53) and impairs Mdm2-p53 association, p53 polyubiquitylation, and Mdm2-mediated inhibition of p53 transcriptional activity. These effects of NIAM are associated with cell cycle arrest.
We have shown that Tip60, a major activator of p53, binds to NIAM and is required for NIAM to fully activate p53. Tip60 uses distinct mechanisms to differentially activate p53 target genes during a DNA damage response. For instance, Tip60 interacts with p53 and is recruited to high-affinity p53 target promoters, such as p21, where it promotes expression of genes responsible for cell cycle arrest.29 This step, which involves Tip60 acetylation of histone H4 but not p53, is thought to be important for cells to mount an effective DNA repair response.26,29 By comparison, Tip60 can also acetylate p53 at the evolutionarily conserved K120 residue45 to initiate an irreversible apoptotic response.28,29 Acetylation of K120 alters the DNA binding specificity of p53, favoring its accumulation on low-affinity apoptotic gene promoters, such as bax and puma, triggering cell death.28,29,46,47 We found that wild-type NIAM can promote p53 acetylation at K120. However, the N-terminal (NT) form lacks the ability to promote p53 acetylation despite retaining wild-type or greater abilities to bind Tip60, inhibit proliferation, and induce p53 transactivation of the p21 promoter. These findings suggest that NIAM activates p53 primarily thru Tip60 association, rather than p53 K120 acetylation, which is consistent with the fact that NIAM induces growth arrest and not cell death (ref. 34 and data herein).
The above conclusion raises several important questions. First, what is the significance of p53 K120 acetylation by WT NIAM? This acetylation is clearly dispensable to the NIAM functions described herein, yet we wonder if that modification could also be mediated by MOZ, a Tip60 relative that specifically promotes p21 upregulation and senescence.48 Of more immediate interest is determining how NIAM cooperates with Tip60 to activate p53 and induce p21. Does it do so by facilitating Tip60–p53 interaction on the p21 promoter, and is that associated with enhanced Tip60-mediated histone H4 acetylation and consequent relaxation of the chromatin? That is the clear prediction from our findings as well as earlier work by others regarding the control of p21 expression by Tip60-p53,26,29 and it is depicted in Figure 8. A related question is whether NIAM is part of a transcriptional complex containing p53 and/or Tip60 on p53 target promoters. To date, we have been unable to isolate cellular complexes containing NIAM and p53. Also the formation of NIAM–Tip60 complexes was observed in p53-null cells, supporting the conclusion that NIAM binds to Tip60 independently of p53. That heightens the probability that NIAM promotes Tip60-mediated activation of p53 by limiting their negative regulation by Mdm2 (as discussed below) and/or UHRF1 (ubiquitin-like with PHD and RING finger domains 1). UHRF1 is a newly identified suppressor of Tip60 and p53 whose depletion markedly increases p53/Tip60-dependent transactivation of both growth-inhibitory and apoptotic target genes.49 These questions are highly relevant to our understanding of p53 signaling and merit further investigation.
Since depletion of Tip60 only partially reduced p53 activation by NIAM, we examined other regulators and/or pathways by which NIAM controls p53. Our data establish Mdm2 inhibition as a central mechanism of NIAM-mediated p53 activation. This was supported by observations that NIAM binds to Mdm2, disrupts Mdm2–p53 association, and reduces p53 polyubiquitylation. Moreover, forms of NIAM (WT and NT) that lead to cell cycle arrest also interact directly with Mdm2 and completely abolish Mdm2-dependent transcriptional inhibition of p53. There are a few different mechanisms by which NIAM could inhibit Mdm2. The most obvious possibility is that NIAM could block Mdm2 ubiquitin ligase activity, similar to ARF.50 However, that seems improbable, since NIAM is effectively downregulated by Mdm2-mediated ubiquitylation.34 Our data are more consistent with the possibility that NIAM binds and sequesters Mdm2 away from p53 complexes, and we suggest those complexes reside on the DNA, since the vast majority of NIAM is chromatin bound. This mechanism would effectively impair any inhibitory modification of p53 by Mdm2, which includes not only ubiquitylation, but also neddylation51 and sumoylation.52 Future studies examining the effects of NIAM on those alternative lysine modifications of p53 are warranted. Notably, our data are not consistent with Tip60 binding to NIAM–Mdm2 complexes, since Tip60 and NIAM influence p53 stability through different mechanisms. Tip60 binds Mdm2 with p53 and relocalizes the complex to PML-like nuclear bodies without altering the ubiquitylation status of p53, presumably impairing p53 targeting to the proteasome.26,33 NIAM, on the other hand, substantially decreases p53 polyubiquitylation.
The association of NIAM with chromatin was predicted based on the presence of Phe/Tyr-rich regions (FYRN and FYRC), which are evolutionarily conserved among chromatin-associated proteins, such as the human mixed lineage leukemia (MLL) proteins.53 However, the role of those domains in chromatin binding is undefined. Our studies clearly showed that the NIAM C-terminus containing these domains is not essential to chromatin localization. Conversely, the NIAM NT form lacking FYRN/FYRC is exclusively nuclear and chromatin bound. It is possible that the positively charged Lys residues within NIAM’s NLS and LYS-R domains may mediate chromatin binding via electrostatic interactions with the DNA. Regardless of the mechanism of chromatin association, this study showed that NIAM N-terminal domains are required and sufficient for nearly all of NIAM’s observed activities, including association with Tip60 and Mdm2, p53 transcriptional activation, blocking Mdm2 inhibition of p53, and cell growth inhibition. Those same residues were also essential for chromatin association, implying that this association may be central to the function of NIAM. We tested if Tip60 and/or Mdm2 direct NIAM’s interaction with chromatin, since they also localize to that structure. But this hypothesis was not supported, since endogenous NIAM remains chromatin bound in Tip60-silenced cells, as well as in cells that lack functional p53 and express negligible levels of Mdm2 (Fig. S1). On the other hand, exogenous NIAM was dispersed throughout the nucleus and cytoplasm in cells lacking p53 and Mdm2, so it is possible that those factors (or other p53 regulated targets) can be quantitatively limiting for NIAM chromatin association.
In sum, this work reveals that NIAM is an important activator of p53. Depletion of NIAM in cells significantly impaired p53 transcriptional activity. NIAM acts through mechanisms involving Tip60 and Mdm2, 2 major regulators of p53 that are frequently downregulated (Tip60) or overexpressed (Mdm2) in human cancers.20,54-56 These observations strongly imply a tumor-suppressive role of NIAM in cancer, consistent with its downregulation at the mRNA level in various human tumors.34 Future studies evaluating the biological effects of NIAM loss on p53 function in response to genotoxic stresses and in tumor development in vivo will provide important insight into the physiological significance of NIAM in cancer.
Materials and Methods
Cell culture
Most cell types were maintained in standard Dulbecco modified Eagle medium containing 10% fetal bovine serum, 2 mM glutamine, and 100 µg/ml penicillin and streptomycin, including human HEK 293T (human embryonic kidney epithelial cells expressing SV40 large T antigen); human U2OS osteosarcoma cells; human MDA-MB-231 cells; and mouse NIH 3T3 fibroblasts. Primary mouse embryonic fibroblasts (MEFs; ARF-null provided by Martine Roussel, St. Jude’s Children Hospital and p53-null provided by Christine Eischen, Vanderbilt University) were grown in the same media supplemented with 0.1 mM nonessential amino acids and 55 µM 2-mercaptoethanol. Human H1299 lung cancer cells (American Type Culture Collection, CRL-5803) were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, and 100 µg/ml penicillin/streptomycin.
DNA constructs
Cloning of wild-type mouse NIAM into various expression vectors, including pXM-HA, pXM-Myc, pMSCV-IRES-GFP, and pGEX-4T-2, was described.34 NIAM deletion mutants were generated by PCR: 1–164 (NT) (forward primer: 5′ – CCATCGATAG CGTGTTGAGC GGCCTGGCC - 3′; reverse primer: 5′ – CCGAATTCCA GTTTTCGAGC ACCTCCCTC - 3′) and 158–406 (CT) (forward primer: 5′ - CCATCGATGA GGGAGGTGCT CGAAAACTG - 3′; reverse primer: 5′ – GGAATTCAAT CTGAAGACTG AATTG - 3′). PCR products were directly ligated into the pCRII-TOPO vector (Invitrogen), sequenced, and subcloned into the above expression vectors. Other expression plasmids used include pcDNA3.1-human p53,57 pCMV-human Mdm2 (Moshe Oren, Weizmann Institute of Science), pcDNA3-HA-Tip60 (Didier Trouche, Universite Paul Sabatier), and pRBG4-His-Myc-ubiquitin.34
Protein expression
Bacterial glutathione S-transferase (GST)-tagged fusion proteins were generated as described previously.34 For mammalian cell expression, cells were transfected using a modified calcium phosphate precipitation procedure.58 Production of ecotropic and amphotropic retroviruses in 293T cells and subsequent infections into MEFs, NIH 3T3, or H1299 cells were performed as described.59,60 Briefly, retroviral supernatants were collected 3 times at 12 h intervals between 36 and 60 h post-transfection, stored on ice, supplemented with 8 µg/mL polybrene, and passed through sterile 0.45-mm pore size filters. Cells (plated at 1.5–3 × 105 per 100 mm dish) were sequentially infected 2–3 times with 2.5 mL virus per round (8–16 h per round). Infection efficiency was monitored by flow cytometric measurement of GFP positivity using a FACScan (Becton Dickinson) and WinMDI analysis software.
Tip60 and NIAM silencing
To silence endogenous human Tip60, pLKO.1.puro-Tip60 shRNA constructs (Open Biosystems) were used with an empty vector included as control, whereas pSUPER-retro-neo/GFP viruses expressing 2 different shRNAs were used to silence human NIAM: shN1 (5′-GATCCCCGAA GGTACTTGCT AAAGAATTCA AGAGATTCTT TAGCAAGTAC CTTCTTTTTG GAAA-3′) and shN2 (5′-GATCCCCACT GGAAGTTCTG AAGAAATTCA AGAGATTTCT TCAGAACTTC CAGTTTTTTG GAAA-3′). A scrambled shRNA construct (5′-GATCCCCGGA ATCTCATTCG ATGCATACCT TCCTGTCAGT ATGCATCGAA TGAGATTCCT TTTTGGA AA-3′) was used as control. Viruses expressing both sets of shRNAs were generated and infected into cells exactly as described, including sterile cell sorting for GFP-positive cells in the NIAM silencing experiments.34,61
Knockdown of NIAM protein expression was confirmed by immunoblotting. Tip60 silencing was validated by quantitative real-time RT-PCR. Reverse transcription of Tip60 mRNA was performed with 2 μg of total RNA (isolated using RNeasy mini kit, Qiagen) using a High-Capacity cDNA Archive Kit (Applied Biosystems Inc). Subsequent PCR reactions consisted of 50 ng of cDNA added to 5 μl of SYBR Green PCR Master Mix (Applied Biosystems Inc), 1.0 μl (5 µM) of gene-specific Tip60 primers (forward primer 5′- CGAATTGTTT GGGCACTGAT G -3′; reverse primer 5′- GCTTCGATCA GACACCAGG -3′), and PCR-grade water to a total reaction volume of 10 μL. PCR was performed as follows: 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s, annealing of primers at 60 °C for 1 min, and extension at 72 °C for 1 min on a CFX96 Real-Time PCR detection system (BioRad). As a control, the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified using a forward primer (5′ -CCATGTTCGT CATGGGTGTG- 3′) and reverse primer (5′ - CAGGGGTGCT AAGCAGTTGG - 3′). Fold differences in Tip60 mRNA expression were calculated following normalization to GAPDH RNA expression and computed using BioRad CFX Manager 3.0 quantitation software (BioRad).
In vitro binding assays
Coupled in vitro transcription and translation (IVT) of plasmids containing Tip60 or Mdm2 were performed using the TNT kit (Promega Corp). 35S-labeled IVT products were incubated for 2 h at 4 °C with equivalent amounts of GST or GST-mouse NIAM proteins (WT, NT, or CT) that were previously bound to glutathione-sepharose, as described.34 Protein complexes were washed 4 times with NP-40 buffer (50 mM Tris [pH 8.0], 120 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, and 0.1 mM Na3VO4) supplemented with 1 mM NaF, protease inhibitor cocktail (Sigma Aldrich, P8340, 1:100 dilution), phosphatase inhibitor cocktail (Sigma Aldrich, P0044, 1:100 dilution), and 30 µM phenylmethylsulfonyl fluoride and resolved by SDS-PAGE. Gels were either fixed with 30% methanol and 10% acetic acid and stained with Coomassie Blue to confirm equivalent input of GST proteins, or proteins were transferred onto nitrocellulose membranes (Amersham), and autoradiography was used to detect protein interactions.
Proliferation assays
For colony-formation assays, U20S cells were infected with viruses containing empty vector (Vec), wild-type mouse NIAM (WT), or NIAM mutants (NT or CT). Cells were selected in puromycin for 3 d then replated in triplicate in a 6-well tissue culture dish. Dishes were cultured for 14 d before colonies were washed twice with phosphate-buffered saline fixed with methanol, and stained with Giemsa. For growth curves, NIH 3T3 cells expressing wild-type mouse NIAM or mutants were plated at 2 × 103 cells per well (6-well dishes) in triplicate. Cells were counted on day 6 with a hematocytometer.
p53 reporter assays
A p53 reporter construct containing 14 repeats of the p53-binding sequence (TGCCTGGACT TGCCTGG) in the p21 promoter element fused to the firefly luciferase gene (Stratagene) was co-transfected into U2OS cells with varying amounts of pXM-HA-NIAM forms plus empty pXM-HA or pCDNA3-Vec for a constant amount of DNA (12 µg) per transfection. For H1299 experiments, pCDNA3.1-p53 and/or pCMV-human MDM2 were also co-transfected into cells. Knockdown of Tip60 and NIAM was performed before plating for reporter assays. A pRL-SV40 construct containing Renilla luciferase (Promega, 80 ng) was included in all transfections to normalize for transfection efficiency. Luciferase activity was measured in duplicate or triplicate samples 48 h post-transfection using the Dual-Luciferase reporter assay system (Promega Corp.) and a Sirius V3.1 luminometer (Berthold Detection Systems).
Subcellular localization
For immunofluorescence studies, ARF-null MEFs were infected with retroviruses containing HA-NIAM forms or vector alone, and 2 d later cells (5 × 104) were replated on glass coverslips in a 6-well dishes. The next day, cells were fixed for 10 min with 4% paraformaldehyde and permeabilized for 10 min with 0.1% TritonX-100 in PBS. Exogenous HA-NIAM forms were detected by staining with an HA antibody (Roche Diagnostics, clone 3F10, 0.5 µg/mL) and HRP-coupled anti-rat IgG (GE Healthcare, NA935, 1:2000 dilution). Nuclei were visualized by staining with 4’, 6-diamidino-2-phenylindole (DAPI, Sigma) at 1 µg/mL for 1 min, and analyzed by confocal microscopy (Zeiss LSM 710).
To isolate chromatin, NIH 3T3, U2OS, MDA-MB-231, and H1299 cells were pelleted by centrifugation, washed one time in PBS, and biochemically fractionated, as previously described.62 Briefly, cells were lysed in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 1 mM NaF, protease inhibitor cocktail (Sigma Aldrich, P8340, 1:100 dilution), phosphatase inhibitor cocktail (Sigma Aldrich, P0044, 1:100 dilution), and 30 µM phenylmethylsulfonyl fluoride. Triton X-100 was added to a final concentration of 0.1%, and cells were incubated on ice for 8 min. After low-speed centrifugation (5 min, 1300 g, 4 °C), cytoplasmic proteins were harvested in the supernatant, and nuclei were pelleted. Buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, protease and phosphatase inhibitors) was then applied to the nuclear pellet for lysis. Nucleoplasmic proteins were isolated from the supernatant, and chromatin-bound proteins were pelleted during centrifugation (5 min, 1700 g, 4 °C). Identical cell equivalents per fraction (cytoplasmic, nucleoplasmic, and chromatin) were analyzed by SDS-PAGE and immunoblotting using antibodies to NIAM, nucleophosmin (NPM), histone H4, H2AX, and RABL6A (see details below).
Half-life analysis
NIH 3T3 cells expressing empty vector or various forms of HA-tagged mouse NIAM (WT, NT, or CT) were plated in 6-well dishes at 2 × 105 cells/well. Cells were treated with cycloheximide (Sigma Aldrich, C4859, 100 ug/mL) for different amounts of time (0, 0.75, 1.5, 3, 6, 12, or 24 h), harvested, and lysates resolved by SDS-PAGE. Immunoblotting was performed using antibodies (listed below) to NIAM, HA, GAPDH, and/or Mad1L.
Immunoprecipitation and western blot analyses
Frozen cell pellets were lysed on ice in Nonidet P-40 buffer and briefly vortexed, and lysates were incubated on ice for 45 min prior to sonication (2 × 5 s pulse) and clarification by centrifugation at 14 000 rpm for 10 min at 4 °C. Immunoprecipitations were performed using protein A/G-Sepharose plus antibodies against the hemagglutinin (HA) epitope (Roche Applied Science, rat monoclonal, clone 3F10, conjugated to agarose, 30 µl slurry), Myc (9E10 mouse monoclonal antibody, 150 µL of hybridoma supernatant), Tip60 (Santa Cruz, K17 sc-5727 and N-17 sc-5725, 3 µg each), p53 (Santa Cruz, FL393 sc-6243, rabbit polyclonal antibody, 5 µg), and Mdm2 (clone 2A10, mouse monoclonal antibody 300–400 µL; Calbiochem, Ab-1, OP-46 mouse monoclonal antibody, 5 µg). Species- and isotype-matched IgGs were used for immunoprecipitation controls. Immune complexes were washed 4 times with NP-40 buffer, separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes (Millipore), and analyzed by immunoblotting. For whole-cell analyses of protein expression levels, equivalent amounts of total cellular protein (50 –100 µg/lane) were loaded on gels. Proteins were detected on membranes by ECL (Amersham Biosciences) with antibodies against Mdm2 (Calbiochem, Ab-1, OP-46, mouse monoclonal, 1.0 µg/ml), p53 (Santa Cruz, DO-1, sc-126, 1 µg/mL), p21 (BD PharMingen, 554228, mouse monoclonal, clone 6B6, 2.5 µg/ml), GAPDH (Abcam, mouse monoclonal, ab8245, 1:10,000 dilution), HA (Roche Diagnostics, clone 3F10, 0.1 µg/mL), NPM (Zymed, 32–5200, 0.25 µg/mL), Mad1L (ProteinTech, 18322–1-AP, 0.24 ng/mL), RABL6A (rabbit polyclonal, 2 µg/mL),61,63 and NIAM (rabbit polyclonal antibody at 1.5 µg/ml34 and mouse monoclonal antibody [clone 11E12] at 1:5 dilution64).
p53 ubiquitylation assays
In vivo ubiquitylation was measured in H1299 cells expressing exogenous p53 and in U2OS cells with endogenous p53, as described.34 Cells were cotransfected with His-tagged ubiquitin and pXM-HA or pXM-HA-NIAM plus empty pcDNA3 or pXM-HA vector. Two days post-transfection, cells were harvested and lysed in 6 M guanidinium chloride, 0.1 M Na2HPO4/NaH2PO4, and 0.01 M TRIS-HCl (pH 8.0) and 10 mM β-mercaptoethanol. Ubiquitylated proteins were bound for 4 h at room temperature on 80–100 µl of nickel-nitrilotriacetic acid-agarose slurry (prewashed with the lysis buffer), washed extensively, and His-ubiquitin conjugated proteins eluted with 200 mM imidazole in 5% SDS, 0.15 M TRIS-HCl (pH 6.7), 30% glycerol, and 0.72 M β-mercaptoethanol for 20 min at room temperature.65,66 Eluates were resolved by SDS-PAGE and ubiquitylated p53 detected by immunoblotting with a p53 antibody (DO-1).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Drs Wei Gu, Moshe Oren, and Didier Trouche for sharing p53, Mdm2, and Tip60 reagents. We also thank Drs Martine Roussel and Christine Eischen for providing MEFs. Thanks to Meghan Naber and Kathryn Quelle for technical assistance as well as personnel in the DNA Core, Flow Cytometry and Central Microscopy facilities at the University of Iowa and Holden Comprehensive Cancer Center for their contributions. This work was supported by grants from the NIH (NCI R01 CA090367 to D.E.Q., NCI F30 8396126 to S.M.R., NCI P30CA086862 Cancer Center Support Grant, and a Pharmacological Sciences Training Grant T32 GM067795), and the Carver College of Medicine.
Glossary
Abbreviations:
- ARF
alternative reading frame protein
- FYRC
phenylalanine tyrosine-rich C-terminal domain
- FYRN
phenylalanine tyrosine-rich N-terminal domain
- GST
glutathione-S-transferase
- Mdm2
mouse double-minute 2
- MYST
(MOZ, Ybf2/Something about silencing protein 3 [Sas3], Something about silencing protein 2 [Sas2], Tip60/KAT5) lysine acetyltransferase family
- NIAM
nuclear interactor of ARF and Mdm2
- NLS
nuclear localization signal
- NPM
nucleophosmin/B23
- p21 (CDKN1A/WAF1/Cip1)
21-kDa inhibitor of cyclin-dependent kinases
- RABL6A
RAB-like isoform A
- Tip60
Tat-interacting protein of 60 kDa
References
- 1.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 3.Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–42. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 4.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–6. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 6.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senturk E, Manfredi JJ. Mdm2 and tumorigenesis: evolving theories and unsolved mysteries. Genes Cancer. 2012;3:192–8. doi: 10.1177/1947601912457368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 9.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–7. doi: 10.1016/S0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 10.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–14. doi: 10.1128/mcb.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–60. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 13.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi: 10.1016/0092-8674(92)90644-R. [DOI] [PubMed] [Google Scholar]
- 14.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–8. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7(7A):1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 16.Picksley SM, Lane DP. The p53-mdm2 autoregulatory feedback loop: a paradigm for the regulation of growth control by p53? Bioessays. 1993;15:689–90. doi: 10.1002/bies.950151008. [DOI] [PubMed] [Google Scholar]
- 17.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 18.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–3. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 19.Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res. 2003;1:993–1000. [PubMed] [Google Scholar]
- 20.Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets. 2005;5:27–41. doi: 10.2174/1568009053332636. [DOI] [PubMed] [Google Scholar]
- 21.Riley MF, Lozano G. The many faces of MDM2 binding partners. Genes cancer 2012; 3:226-39. [DOI] [PMC free article] [PubMed]
- 22.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 23.Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164–71. doi: 10.1016/S0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 24.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marouco D, Garabadgiu AV, Melino G, Barlev NA. Lysine-specific modifications of p53: a matter of life and death? Oncotarget. 2013;4:1556–71. doi: 10.18632/oncotarget.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Legube G, Linares LK, Tyteca S, Caron C, Scheffner M, Chevillard-Briet M, Trouche D. Role of the histone acetyl transferase Tip60 in the p53 pathway. J Biol Chem. 2004;279:44825–33. doi: 10.1074/jbc.M407478200. [DOI] [PubMed] [Google Scholar]
- 27.Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–7. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 28.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–51. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–39. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 30.Naidu SR, Lakhter AJ, Androphy EJ. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle. 2012;11:2717–28. doi: 10.4161/cc.21091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Legube G, Linares LK, Lemercier C, Scheffner M, Khochbin S, Trouche D. Tip60 is targeted to proteasome-mediated degradation by Mdm2 and accumulates after UV irradiation. EMBO J. 2002;21:1704–12. doi: 10.1093/emboj/21.7.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eymin B, Claverie P, Salon C, Leduc C, Col E, Brambilla E, Khochbin S, Gazzeri S. p14ARF activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol Cell Biol. 2006;26:4339–50. doi: 10.1128/MCB.02240-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dohmesen C, Koeppel M, Dobbelstein M. Specific inhibition of Mdm2-mediated neddylation by Tip60. Cell Cycle. 2008;7:222–31. doi: 10.4161/cc.7.2.5185. [DOI] [PubMed] [Google Scholar]
- 34.Tompkins VS, Hagen J, Frazier AA, Lushnikova T, Fitzgerald MP, di Tommaso A, Ladeveze V, Domann FE, Eischen CM, Quelle DE. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J Biol Chem. 2007;282:1322–33. doi: 10.1074/jbc.M609612200. [DOI] [PubMed] [Google Scholar]
- 35.García-Alai MM, Allen MD, Joerger AC, Bycroft M. The structure of the FYR domain of transforming growth factor beta regulator 1. Protein Sci. 2010;19:1432–8. doi: 10.1002/pro.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pless B, Oehm C, Knauer S, Stauber RH, Dingermann T, Marschalek R. The heterodimerization domains of MLL-FYRN and FYRC--are potential target structures in t(4;11) leukemia. Leukemia. 2011;25:663–70. doi: 10.1038/leu.2010.308. [DOI] [PubMed] [Google Scholar]
- 37.Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006;16:433–42. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 38.Love IM, Grossman SR. It takes 15 to tango: Making sense of the many ubiquitin ligases of p53. Genes Cancer. 2012;3:249–63. doi: 10.1177/1947601912455198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–6. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 40.Llanos S, Clark PA, Rowe J, Peters G. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat Cell Biol. 2001;3:445–52. doi: 10.1038/35074506. [DOI] [PubMed] [Google Scholar]
- 41.Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5:465–75. doi: 10.1016/S1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- 42.Korgaonkar C, Hagen J, Tompkins V, Frazier AA, Allamargot C, Quelle FW, Quelle DE. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol Cell Biol. 2005;25:1258–71. doi: 10.1128/MCB.25.4.1258-1271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu B, Zhu W-G. Surf the post-translational modification network of p53 regulation. Int J Biol Sci. 2012;8:672–84. doi: 10.7150/ijbs.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528–36. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lane DP, Madhumalar A, Lee AP, Tay B-H, Verma C, Brenner S, Venkatesh B. Conservation of all three p53 family members and Mdm2 and Mdm4 in the cartilaginous fish. Cell Cycle. 2011;10:4272–9. doi: 10.4161/cc.10.24.18567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arbely E, Natan E, Brandt T, Allen MD, Veprintsev DB, Robinson CV, Chin JW, Joerger AC, Fersht AR. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc Natl Acad Sci U S A. 2011;108:8251–6. doi: 10.1073/pnas.1105028108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu N, Wang J, Wang J, Wang R, Liu Z, Yu Y, Lu H. ING5 is a Tip60 cofactor that acetylates p53 in response to DNA damage. Cancer Res. 2013;73:3749–60. doi: 10.1158/0008-5472.CAN-12-3684. [DOI] [PubMed] [Google Scholar]
- 48.Rokudai S, Laptenko O, Arnal SM, Taya Y, Kitabayashi I, Prives C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc Natl Acad Sci U S A. 2013;110:3895–900. doi: 10.1073/pnas.1300490110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dai C, Shi D, Gu W. Negative regulation of the acetyltransferase TIP60-p53 interplay by UHRF1 (ubiquitin-like with PHD and RING finger domains 1) J Biol Chem. 2013;288:19581–92. doi: 10.1074/jbc.M113.476606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Honda R, Yasuda H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999;18:22–7. doi: 10.1093/emboj/18.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 52.Stindt MH, Carter S, Vigneron AM, Ryan KM, Vousden KH. MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle. 2011;10:3176–88. doi: 10.4161/cc.10.18.17436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 54.Sakuraba K, Yasuda T, Sakata M, Kitamura Y-H, Shirahata A, Goto T, Mizukami H, Saito M, Ishibashi K, Kigawa G, et al. Down-regulation of Tip60 gene as a potential marker for the malignancy of colorectal cancer. Anticancer Res. 2009;29:3953–5. [PubMed] [Google Scholar]
- 55.Sakuraba K, Yokomizo K, Shirahata A, Goto T, Saito M, Ishibashi K, Kigawa G, Nemoto H, Hibi K. TIP60 as a potential marker for the malignancy of gastric cancer. Anticancer Res. 2011;31:77–9. [PubMed] [Google Scholar]
- 56.Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature. 2007;448:1063–7. doi: 10.1038/nature06055. [DOI] [PubMed] [Google Scholar]
- 57.Zhao L, Samuels T, Winckler S, Korgaonkar C, Tompkins V, Horne MC, Quelle DE. Cyclin G1 has growth inhibitory activity linked to the ARF-Mdm2-p53 and pRb tumor suppressor pathways. Mol Cancer Res. 2003;1:195–206. [PubMed] [Google Scholar]
- 58.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–52. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 60.Modestou M, Puig-Antich V, Korgaonkar C, Eapen A, Quelle DE. The alternative reading frame tumor suppressor inhibits growth through p21-dependent and p21-independent pathways. Cancer Res. 2001;61:3145–50. [PubMed] [Google Scholar]
- 61.Muniz VP, Askeland RW, Zhang X, Reed SM, Tompkins VS, Hagen J, McDowell BD, Button A, Smith BJ, Weydert JA, et al. RABL6A promotes oxaliplatin resistance in tumor cells and is a new marker of survival for resected pancreatic ductal adenocarcinoma patients. Genes Cancer. 2013;4:273–84. doi: 10.1177/1947601913501074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Méndez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–12. doi: 10.1128/MCB.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tompkins V, Hagen J, Zediak VP, Quelle DE. Identification of novel ARF binding proteins by two-hybrid screening. Cell Cycle. 2006;5:641–6. doi: 10.4161/cc.5.6.2560. [DOI] [PubMed] [Google Scholar]
- 64.Hagen J, Tompkins V, Dudakovic A, Weydert JA, Quelle DE. Generation and characterization of monoclonal antibodies to NIAM: a nuclear interactor of ARF and Mdm2. Hybridoma (Larchmt) 2008;27:159–66. doi: 10.1089/hyb.2007.0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xirodimas DP, Chisholm J, Desterro JMS, Lane DP, Hay RT. P14ARF promotes accumulation of SUMO-1 conjugated (H)Mdm2. FEBS Lett. 2002;528:207–11. doi: 10.1016/S0014-5793(02)03310-0. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez MS, Desterro JMP, Lain S, Midgley CA, Lane DP, Hay RT. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999;18:6455–61. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.