

Abstract
Parkinson’s disease (PD) is a multifocal degenerative disorder for which there is no cure. The majority of cases are sporadic with unknown etiology. Recent data indicate that untreated patients with de novo PD have increased colonic permeability and that both de novo and premotor patients have pathological expression of α-synuclein (α-syn) in their colon. Both endpoints potentially can serve as disease biomarkers and even may initiate PD events through gut-derived, lipopolysaccharide (LPS)-induced neuronal injury. Animal models could be ideal for interrogating the potential role of the intestines in the pathogenesis of PD; however, few current animal models of PD encompass these nonmotor features. We sought to establish a progressive model of PD that includes the gastrointestinal (GI) dysfunction present in human patients. C57/BL6 mice were systemically administered one dose of either LPS (2.5 mg/kg) or saline and were sacrificed in monthly intervals (n=5 mice for 5 months) to create a time-course. Small and large intestinal permeability was assessed by analyzing the urinary output of orally ingested sugar probes through capillary column gas chromatography. α-Syn expression was assessed by counting the number of mildly, moderately, and severely affected myenteric ganglia neurons throughout the GI tract, and the counts were validated by quantitative optical density measurements. Nigrostriatal integrity was assessed by tyrosine hydroxylase immunohistochemistry stereology and densitometry. LPS caused an immediate and progressive increase in α-syn expression in the large intestine but not in the small intestine. Intestinal permeability of the whole gut (large and small intestines) progressively increased between months 2 and 4 after LPS administration but returned to baseline levels at month 5. Selective measurements demonstrated that intestinal permeability in the small intestine remained largely intact, suggesting that gut leakiness was predominately in the large intestine. Phosphorylated serine 129-α-syn was identified in a subset of colonic myenteric neurons at months 4 and 5. Although these changes were observed in the absence of nigrostriatal degeneration, an abrupt but insignificant increase in brainstem α-syn was observed that paralleled the restoration of permeability. No changes were observed over time in controls. LPS, an endotoxin used to model PD, causes sequential increases in α-syn immunoreactivity, intestinal permeability, and pathological α-syn accumulation in the colon in a manner similar to that observed in patients with PD. These features are observed without nigrostriatal degeneration and incorporate PD features before the motor syndrome. This allows for the potential use of this model in testing neuroprotective and disease-modifying therapies, including intestinal-directed therapies to fortify intestinal barrier integrity.
Keywords: Parkinson, animal model, synuclein, enteric nervous system
Parkinson’s disease (PD) is synucleinopathy that classically has been defined as a movement disorder characterized by tremor, rigidity, akinesia, and postural instability due to degeneration of the nigrostriatal pathway. 1 The presence of Lewy bodies, the pathological hallmark of the disease, which are proteinaceous cytoplasmic aggregates comprised largely of the presynaptic terminal protein α-synuclein (α-syn), is required for a pathological diagnosis of the disease.2 Whereas the end-stage and postmortem parkinsonian profiles of PD are well characterized, much less is known about how the disease begins and progresses. Although this is due in part to the stage at which PD patients typically present motor symptoms, it is also due in part to our toxin animal models, which historically have consisted of rodents and nonhuman primates that are given acute and massive dopaminergic lesions.3 These models do not fully recapitulate the full spectrum of motor and nonmotor disease symptoms, especially those nonmotor symptoms that present in the extensive premotor phase.
Premotor symptoms such as constipation are present in the majority of PD patients and can occur years to decades before the onset of motor impairment.4–6 Several recent and converging lines of evidence indicate that α-syn is pathologically expressed in the intestines of patients with PD.7–14 This expression supports the Braak hypothesis,15 which postulates that PD pathogenesis spreads from the enteric nervous system to the brain via susceptible long, thin, unmyelinated axons before advancing rostrally through the central nervous system (CNS). Additional animal studies have begun to explore this provocative concept utilizing both toxin and genetic models of PD.16–25 Transgenic α-syn overexpression and missense mutations have produced slowed gastrointestinal (GI) motility,20 defecatory dysfunction and increased enteric α-syn expression,19 and enteric α-syn aggregates.24 Toxin models, especially rotenone, have displayed promising results, including delayed α-syn aggregate pathology and slightly decreased GI motility.16 A series of reports by Pan-Montojo and colleagues18,23 demonstrated that intragastrically administered rote-none induced Braak’s staging in mice23 and that this progressive pathology could be arrested by sympathetic and parasympathetic nerve resection.18 However, a recent report by Tasselli and colleagues17 showed that orally administered rotenone failed to reproduce enteric alterations while still inducing nigral degeneration. While animal models continue to be refined, we and our collaborators have demonstrated two critical pathological events in the early PD syndrome. First, we observed the pathological expression of colonic α-syn both in newly diagnosed de novo patients and in premotor patients years before their PD diagnosis.8,9,26 Second, the same cohort of newly diagnosed PD patients displayed abnormal intestinal permeability, which presumably was localized to the large intestine.26 Although both α-syn and increased intestinal permeability are well known events associated with inflammation,27–30 no animal models of PD have explored both of these early facets of disease pathogenesis over time.
Lipopolysaccharide (LPS) is a gut-derived, pro-inflammatory bacterial endotoxin that has demonstrated delayed and progressive nigral pathology when administered systemically and serves as a progressive model of PD.31,32 However, its use as model for premotor PD has not been explored. Therefore, in the present study, we hypothesized that systemically administered LPS would compromise GI permeability that, in turn, results in further leaks of LPS from the intestinal lumen into the intestinal submucosa and enteric neuronal tissues, promoting α-syn overexpression in neurons of the myenteric plexus of the GI tract over time.
Materials and Methods
Animals
Forty C57BL/6J male mice were used in the study and were housed five per cage under a 12-hour light/ dark cycle with food and water available ad libidum. The Rush University Institutional Animal Care and Use Committee approved all experiments.
Experimental Design
After a 1-week acclimation period, 7-week to 8-weekold C57/BL6 mice were systemically administered one intraperitoneal dose of either LPS (2.5 mg/kg; n=25) or saline (n=5). This dose was chosen because it is not sufficient to induce nigrostriatal degeneration.31 Baseline controls were sacrificed initially (n=5) at age 2 months, and LPS animals were sacrificed in monthly intervals for 5 months to create a time course (n=5 per month). An additional 10 control C57BL/6J male mice ages 2 months (n=5) and 7 months (n=5) were sacrificed at the end of the study to control for the effects of time. One week before each sacrifice, intestinal permeability was assessed in the small and large intestines using poorly absorbed sugar markers (lactulose, mannitol, and sucralose), as described below. The expression of α-syn was assessed post-mortem by counting the number of mildly, moderately, and severely affected myenteric ganglia neurons and was validated by measuring optical density. Stereological assessments of tyrosine hydroxylase (TH)-positive nigral neurons in the substantia nigra and of α-syn–positive neurons in the dorsal motor nucleus of the vagus nerve (DMNX) also were performed, as we have previously described.33
Intestinal Permeability Analyses
A stock solution of lactulose (1.605 g), sucrose (0.225 g), sucralose (0.225 g), and mannitol (0.450 g) in 100 mL distilled H2O was created and aliquoted for use throughout the study. Mice were deprived of food for 12 hours before the experiment. Each mouse received a 0.2-mL sugar bolus by oral gavage followed by 2 mL of lactated Ringer’s solution subcutaneously immediately after sugar gavage to facilitate urine output. Mice were then individually placed in metabolic chambers for 5 hours to collect urine before being returned to cages. Urinary volume was measured and stored at −80°C. Urinary sugars were measured using capillary column gas chromatography.26 Lactulose and mannitol are reported as a ratio (the L/M ratio), and sucralose is reported as the percentage excretion of oral dose (Fig. 1), as we have previously described.26,34–37
FIG. 1.

(A, B) Intestinal permeability was assessed in untreated mice at baseline (white bars) and in lipopolysaccharide (LPS)-treated mice (black bars) over time. (C, D) Additional control C57BL/6J male mice ages 2 months (n=5) and 7 months (n=5) were sacrificed at the end of the study to control for the effects of time. NS indicates nonsignificant. Gas chromatography was used to measure the urinary output of orally ingested sugar probes (lactulose, mannitol, and sucralose) as described in the text (see Materials and Methods). The ratio of mannitol to lactulose (L/M ratio) reflects small intestine permeability, whereas sucralose reflects small and large intestinal permeability. General linear model main effects were observed for the L/M ratio and sucralose; however, (B) the effect of LPS treatment was observed only for sucralose permeability with subsequent significant Dunnett’s pairwise comparisons at 2 months, 3 months, and 4 months post-LPS. (C, D) No differences were observed in control animals between the initial and final time points. In A through D, each bar represents n=5 mice, and data are presented as the mean percentage of oral dose±standard error of the mean.
Tissue Processing
At each sacrifice time point, mice were deeply anesthetized with sodium pentobarbital and transcardially perfused with saline followed by 4% paraformaldehyde. Brains were removed and transferred to 30% sucrose, in which they remained for 72 hours until equilibrium; coronally sliced at 40 microns on a freezing microtome; and stored in an ethylene glycol-based cryoprotectant before immunohistochemistry. The intestines were removed, extensively flushed with phosphate-buffered saline, and stored in 4% paraformaldehyde for 24 hours at 4°C. One-centimeter coronal samples from the duodenum, jejunum, ascending colon, transverse colon, and descending colon were systematically retrieved and vertically embedded in Tissue-Tek, snap-frozen, and sliced at 20 microns in a cryostat.
Immunohistochemistry
Brains were stained as free-floating sections, whereas intestinal sections were stained on the slide. Initially, the brains were rinsed of cryoprotectant, and the slides were rinsed of Tissue-Tek before they were quenched with endogenous peroxidases using a 20-minute incubation in a 0.1 M sodium periodate solution. Non-specific background staining was blocked for 1 hour in 3% goat serum and 2% bovine serum albumin. Sections were incubated with primary antibody (rabbit anti-TH, 1:1,000 dilution [AB152; Millipore Corporation, Billerica, MA]; rabbit anti-α-syn, 1:1,000 dilution [2642; Cell Signaling Technology, Beverly, MA]; or rabbit anti-α-syn/phosphorylated serine 129 [ab59264; Abcam, Cambridge, UK]), 1% bovine serum albumin, 1% serum, and 0.4% Triton-X at 4°C for 18 hours. The sections were then washed, incubated with appropriate secondary antibodies (biotinylated goat anti-rabbit, 1:200 dilution; BA-1000; Vector Laboratories, Burlingame, CA) for 1 hour, washed again, and incubated with avidin-biotin complex (Vector Laboratories) for 2 hours. Tissues were then incubated in imidazole-acetate buffer, pH 7.3, for 30 minutes before they were visualized with 3-3′-diaminobenzidine tetrahydrochloride in 0.01% hydrogen peroxide with (α-syn) or without (TH and phosphorylated serine 129-α-syn) nickel enhancement. The sections were allowed to dry overnight, dehydrated through increasing alcohol concentrations and xylenes, and coverslipped with cytoseal (23244257; Fisher Scientific International, Hampton, NH).
Quantification and Assessment of TH and α-Syn Immunoreactivity
Stereological assessments of TH-positive neurons in the substantia nigra pars compacta (SNpc) and α-syn–positive neurons in the DMNX were performed using an optical fractionator unbiased sampling protocol. Counting of α-syn and visualization of phosphorylated serine 129-α-syn–immunoreactive myenteric neurons was performed by an investigator who was blinded to GI region and time point. Two sections were analyzed per GI region (duodenum, jejunum, ascending colon, transverse colon, and descending colon) for each animal at every time point. Under ×20 magnification, myenteric plexus ganglia were identified between the outer longitudinal and inner circular muscle layers. At least 20 ganglia were identified per section. Coronal sectioning allowed for the evaluation of all myenteric ganglia neurons around the entire GI section perimeter. Neurons were labeled as mild, moderate, or severe according to their α-syn–immunoreactive intensity. Mild neurons contained faint α-syn staining largely localized to the cell periphery, moderate neurons displayed more intense α-syn immunoreactivity in the periphery and cell body, and severe neurons exhibited profound α-syn immunoreactivity throughout the entire cell body (Fig. 2). The optical density of individual neurons was used to establish quantitative differences between these categorizations (mild, 26.08±9.3; moderate, 94.24±12.4; severe, 136.3±3.3) to validate this rating scheme and was assessed using Kruskal-Wallis and Dunn’s multiple comparison tests. Densitometric measurements, expressed as gray levels, were performed using an Olympus microscope (Olympus, Tokyo, Japan) equipped with a × 20 objective connected to a Scion image analysis system (Scion Corporation, Frederick, MD), as previously described.38 Because the total number of neurons in a coronal section will vary depending on the number of ganglia observed, data are expressed as the average number of mild, moderate, and severe neurons per ganglia±standard error of the mean (Fig. 3).
FIG. 2.

These microphotographs show (A–H) α-synuclein (α-syn) staining and (I–P) phosphorylated serine 129-α-syn staining in the myenteric plexus of (A–D, I–L) the small intestines and (E–H, M–P) the large intestines of saline-treated control mice (A, E, I, M) at baseline and at (B, F, J, N) 1 month, (C, G) 3 months, (K, O) 4 months, and (D, H, L, P) 5 months after lipopolysaccharide (LPS) treatment in LPS-treated mice. Note that in, neurons with mild α-syn staining (black arrows), α-syn immunoreactivity is light and is restricted to the cell periphery. Neurons with moderate α-syn staining (white arrows) display more intense α-syn immunoreactivity in the periphery and soma, whereas severely stained neurons (black arrowheads) exhibit pro-found immunoreactivity throughout the cell body. Over time, the number of severe neurons per ganglia increased in LPS-treated mice, whereas the number of mild neurons per ganglia decreased in the large intestine. No significant changes were detected in the small intestine over time in LPS-treated mice. (O) Phosphorylated serine 129-α-syn was observed in a subset of myenteric neurons beginning at 4 months post-LPS that persisted (P) through 5 months post-LPS in the large intestine. Black arrows indicate neurons that were devoid of staining, and black arrowheads indicate neurons that were positive for phosphorylated serine 129-α-syn. Scale bars=40 μm in H (also applies to A–G) and P (also applies to I–O).
FIG. 3.

The average numbers of mild (light gray), moderate (dark gray), and severe (black) α-synuclein–immunoreactive myenteric neurons per ganglia were assessed in (A) the small intestine and (B) the large intestine. This classification scheme was validated through optical density measurements, as described in the text (see Materials and Methods), which are presented above each neuronal category data set. No differences were observed in control animals between the initial and final time points in any neuronal category in (C) the small intestine or (D) the large intestine. Bars represent n=5 mice, and data are presented as the average number of neurons per ganglia±standard error of the mean.
Statistical Analyses
Statistical comparisons were made using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA). For intestinal permeability, stereological assessments of the DMNX and SNpc, and striatal densitometry, we used a general linear model with Dunnett’s contrasts to compare individual LPS time points versus saline baseline. For immunohistochemistry assessments, myenteric neurons were classified as mild, moderate, or severe based on α-syn staining intensity. Quantitative differences between these categorizations were assessed using Kruskal-Wallis and Dunn’s multiple comparison tests. Linear regression analyses were used to detect changes in neuronal categorizations in LPS-treated animals over time. Mann-Whitney tests were used to compare control animals at 2 months and 7 months. In all tests, we adopted a 5% significance level.
Results
Intestinal permeability tests were performed 1 week before sacrifice (Fig. 1A–D). One animal was not included in the intestinal permeability assessments because partial regurgitation of gavaged sugar probes was noted during testing. The widely used 5-hour urinary L/M ratio26,27 represents small bowel permeability, whereas sucralose represents whole gut permeability. Taken together, L/M ratio and sucralose measurements are used to assess large intestine (colon) permeability.26 Mannitol is absorbed passively via paracellular and transcellular transport, whereas lactulose is absorbed via a paracellular route throughout the small intestine. Therefore, an increased L/M ratio represents increased small intestinal permeability. Lactulose and mannitol can also be absorbed passively in the colon; however, because colonic bacteria rapidly metabolize these sugars, the changes in urinary lactulose and mannitol are not reliable indices of colonic permeability.
In contrast, sucralose is absorbed passively via a paracellular route throughout the small and large intestines and is not fermented by bacteria; thus, its urinary levels represent total gut (small and large bowel) permeability. Whereas the overall general linear model (GLM) was significant for the L/M ratio (F[5,23]=6.88; P=0.001), no effect was observed for LPS treatment (P=0.533) (Fig. 1A). For sucralose assessments, the overall GLM was significant (F[5,23]=3.95; P<0.01), and we observed an effect for LPS treatment (P=0.004) (Fig. 1B). Pairwise comparisons demonstrated significant increases in sucralose permeability at 2 months, 3 months, and 4 months post-LPS compared with saline controls before returning to baseline levels at 5 months post-LPS. An additional group of controls at the initial (n=5) and final (5 months later; n=5) time points demonstrated no differences in the L/M ratio (Fig. 1C) or in sucralose (Fig. 1D) permeability.
CNS assessments included α-syn stereology in the DMNX, TH stereology in the SNpc, and TH striatal densitometry (Fig. 4A–F). The overall GLM was not significant for α-syn in the DMNX (F[5,21]=1.81; P=0.155) (Fig. 4A). Surprisingly, the increase in brainstem α-syn at 5 months post-LPS paralleled the abrupt restoration of intestinal permeability (Fig. 1B). Although no changes were observed in the SNpc (F[5,24]=2.46; P=0.062) (Fig. 4B), the overall GLM was significant for striatal densitometric assessments (F[5,20]=2.85; P=.042) (Fig. 4C). However, no effect for striatal LPS treatment was observed (P=0.233) (Fig. 4C). The additional group of controls at the initial (n=5) and final (5 months later; n=5) time points indicated no differences in DMNX α-syn stereology (Fig. 4D), SNpc TH stereology (Fig. 4E), or striatal TH densitometry (Fig. 4F).
FIG. 4.
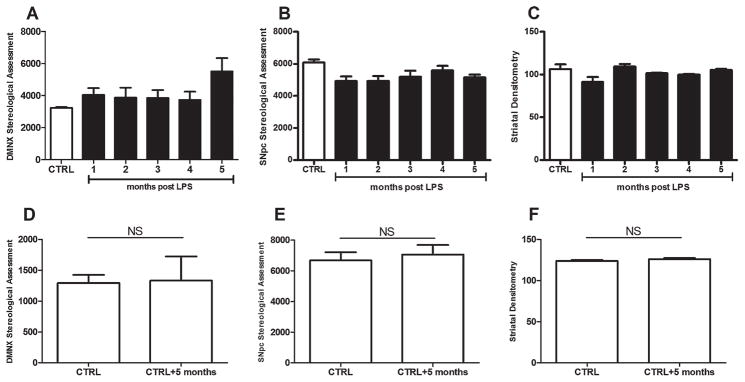
Central nervous system assessments included α-synuclein (α-syn) stereology in (A) the dorsal motor nucleus of the vagus nerve (DMNX), (B) tyrosine hydroxylase (TH) stereology in the substantia nigra pars compacta (SNpc), and (C) TH striatal densitometry. Each bar represents n=5 mice. Although the overall general linear model was not significant (NS) for α-syn in the DMNX, an abrupt but insignificant increase in α-syn–positive neurons was observed 5 months after lipopolysaccharide (LPS) treatment. (D) This increase occurred in the absence of stereological differences between control (CTRL) groups at the initial and final time points. Although no changes were observed (B) in the SNpc, (C) the overall general linear model was significant for striatal densitometric assessments; however, no effect of striatal LPS treatment was observed. The additional group of controls at the initial time point (n=5 mice) and at the final time point (5 months later; n=5 mice) indicated no differences in (E) SNpc TH stereology or (F) or striatal TH densitometry.
One week after intestinal permeability tests were performed, animals were sacrificed, and coronal GI samples were processed as described above (see Materials and Methods). Myenteric neurons were visualized (Fig. 2) and classified as mild, moderate, or severe based on α-syn staining intensity. This categorization scheme was validated by the significant intensification of differences in optical density (Fig. 3) assessed by the Kruskal-Wallis test (data not shown; P<0.001) and subsequent Dunn’s multiple comparisons tests (data not shown; all P<0.05). Neurons that expressed α-syn-immunoreactivity were detected in the myenteric ganglia of control and LPS-treated animals in all GI regions across all time points. In saline baseline controls, the observed α-syn immunoreactivity was predominately mild in both the small and large intestines. After LPS treatment, the distribution of mild, moderate, and severe neurons changed considerably in the large intestine (Fig. 3B), but not in the small intestine (Fig. 3A). The additional group of vehicle-injected controls at the initial (n=5) and final (5 months later; n=5) time points demonstrated no differences in the average numbers of mild, moderate, or severe myenteric neurons in the small (Fig. 3C) or large (Fig. 3D) intestines. Phosphorylated serine 129-α-syn staining in the gut demonstrated selective staining in the large intestine beginning at 4 months and continuing into 5 months post-LPS in a subset of myenteric neurons. No phosphorylated serine 129-α-syn was detected in the CNS.
When explored over time, linear regression indicated that LPS did not affect the average number of neurons per ganglia in the small intestine in any category (R2<0.226; P>0.419) (Table 1, Fig. 5). However, in the large intestine, the average number of severe neurons per ganglia increased (R2=0.840; P=0.029), whereas the number of mild neurons decreased (R2=0.869; P=0.021) in LPS-treated animals over time. The average number of moderate neurons per ganglia remained consistent in the large intestine (R2=0.292; P=0.347).
TABLE 1.
Tabular results of linear regression analysesa
| α-Synuclein staining intensityb | |||
|---|---|---|---|
| Location | Mild | Moderate | Severe |
| Small intestine | |||
| R2 | 0.226 | 0.007 | 0.127 |
| P | 0.419 | 0.894 | 0.556 |
| Large intestine | |||
| R2 | 0.869 | 0.292 | 0.840 |
| P | 0.021 | 0.347 | 0.029 |
Although lipopolysaccharide did not affect the average number of neurons with mild, moderate, or severe α-synuclein immunoreactivity in the small intestine, in the large intestine, the average number of severely immunoreactive neurons per ganglia increased over time, whereas the number of mildly reactive neurons per ganglia decreased over time (graphical data are presented in Fig. 3).
Large R2 values with significantly non-zero slopes are depicted in bold.
FIG. 5.
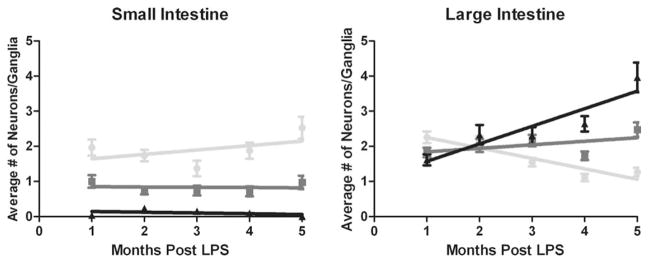
Charts illustrate linear regression analyses of mild (light gray), moderate (dark gray), and severe (black) α-synuclein-immunoreactive myenteric neurons per ganglia in lipopolysaccharide (LPS)-treated mice over time. Although LPS did not affect the average number of mild, moderate, or severe neurons in the small intestine, over time, the average number of severe neurons increased whereas the number of mild neurons decreased in the large intestine (numerical data are presented in Table 1).
Discussion
LPS is an endogenous, pro-inflammatory endotoxin that has been used to model characteristics of PD, including extensive microglial activation, increased αsyn accumulation, and selective degeneration of the nigrostriatal pathway.31,32,39–43 To date, no studies have reported on GI involvement in LPS models of PD. In this study, we demonstrated that systemic exposure to low-dose LPS causes progressive increases in α-syn expression, followed by gut leakiness, and these increases are preferentially localized to the large intestine in mice. Both of these events preceded phosphorylated serine 129-α-syn expression in a subset of colonic myenteric neurons at 4 and 5 months post-LPS. Whereas previous reports demonstrated that higher doses of systemic LPS produced progressive lesions of the nigrostriatal pathway,31,32 the current dosing paradigm was intended to fall below the lesion threshold, leaving nigrostriatal dopamine neurons structurally intact. Although similar findings were observed by Drolet and colleagues in 2009.16 using a low-dose rotenone model of PD, including delayed myenteric plexus α-syn accumulation in the absence of nigrostriatal damage, both studies require replication.
Enteric nervous system susceptibility to inflammation is intuitive given its contact with luminal GI contents; indeed, the magnitude and diversity of the intestinal microbiota are capable of inducing inflammatory and oxidative pathways.26,44 Under physiological conditions, the intestinal barrier is semipermeable and allows low levels of antigens to pass the mucosa to facilitate interactions with the immune system.27 Although transient compromises in barrier integrity may result in mucosal damage, under normal physiological conditions, damage is minimized once the initiating insult is resolved. However, under pathological conditions, such as patients with inflammatory bowel disease or PD, prolonged permeability dysfunction may lead to chronic inflammation and neuronal damage. 28,30 It is important to note that the intestinal permeability data observed in this study support what was observed in the human PD condition: namely, increased sucralose permeability and a normal L/M ratio.26 Collectively, these data strongly implicate the large intestine as the site of intestinal hyperpermeability. Whereas sucralose permeability increased over time, a surprising finding in the current data set was that, at the final sucralose time point, it abruptly returned to baseline levels. Although the overall GLM examining the expression of α-syn within the DMNX by LPS across all time points was not significant and prevented a statistical probing of individual data points, an obvious increase in α-syn expression was seen within this structure at the 5-month time point. Whether the return to baseline for intestinal permeability at 5 months is associated in any way with the increased α-syn expression in the DMNX at 5 months remains to be determined. It is intriguing to speculate that further refinements to this model, such as increased dosing regimens and extended time points, could offer insights into the temporal and regional specificities of α-syn and the role of the enteric nervous system in the development and spreading of PD. Although the delayed observations of (1) apparent spontaneous recovery of intestinal permeability and (2) up-regulated brainstem α-syn point to delayed GI coping mechanism(s) and potential CNS vulnerability in the DMNX, respectively, additional studies are necessary to validate and understand the underlying mechanism(s) responsible for these observations.
Whereas previous laboratories have utilized qualitative and semi-quantitative rating scales to assess enteric α-syn pathology,16,45–47 this is the first report to quantify subdivisions of α-syn–immunoreactive myenteric neurons differentiated by densitometry at multiple sites in the small and large intestines. These data sets were expressed as the number of neurons per ganglia, because the total neuronal number will vary in coronal sections, depending on the number of ganglia observed. Whereas our data demonstrated a robust, inverse correlation between mild and severe neurons/ganglia in the large intestine over time, no changes were observed in the small intestine over time in LPS-treated mice. Additional controls that were examined in parallel at the initial and final time points indicated no differences in any dependent variable, suggesting that changes in LPS groups were due to treatment rather than time. These data suggest that the large intestine is preferentially susceptible to systemic endotoxin exposure and support the observed localized functional changes in permeability. In addition, the gradual increase in large intestine permeability observed in the current study appeared secondary to the abrupt increases in myenteric α-syn expression, suggesting that increased cellular α-syn expression precedes functional hyperpermeability dysfunction. Conversely, phosphorylated serine 129-α-syn was not observed until the 4-month and 5-month time points. This finding suggests that, although up-regulated α-syn may precede hyperpermeability, both phenomena precede pathological α-syn accumulation in the colon. Collectively, these data suggest that, whereas functional damage to the GI tract may be restored with time, cellular α-syn pathology persists. Additional studies will be necessary to test whether reductions in chronic hyperpermeability can similarly reduce cellular α-syn burden.
Although the initial reports on enteric Lewy pathology 48,49 were published nearly 30 years ago, several reports have recently emerged evaluating GI α-syn pathology in human PD samples7–9,14,50–55 and animal models.16–25 Whereas attempts have been made to determine whether frank myenteric neuronal degeneration occurs or whether specific neurochemical phenotypes are preferentially susceptible, these issues require further clarification. Moving forward, the establishment of universal methods will be helpful, because incongruent protocols surrounding sample retrieval and processing may easily provide conflicting data sets. Nonetheless, one common observation surrounds observed α-syn pathology and the decreasing rostrocaudal gradient of vagal innervation throughout the GI tract. While α-syn pathology has been identified throughout the GI tract in PD, many reports claim that α-syn burden follows a decreasing rostro-caudal gradient.10,14,51 In a recent critical quantitative analysis of myenteric plexus in PD, the authors demonstrate no loss of myenteric neurons and a decrease in the percentage of Lewy body-positive ganglia between the stomach and the duodenum.52 It is noteworthy that this percentage of Lewy body-positive ganglia remained consistent throughout all intestinal regions examined (duodenum, ileum, colon, and rectum) and, thus, deviated from the predictable decreasing intestinal vagal innervation pattern. In rodent studies, Noorian et al.20 recently demonstrated in transgenic A53T mice that the percentage of α-syn–labeled myenteric ganglia followed decreasing vagal innervation patterns nearly identical to those in early innervation reports.56 Although it is becoming clear that virtually all vagal efferent terminals in the GI tract are immunopositive for α-syn, it will be important to distinguish between α-syn–positive terminals and myenteric neurons in future analyses. Indeed, a rodent study by Phillips et al.25 illustrated this point: those authors observed very low densities (≈3%) of myenteric α-syn neurons in the stomach. It is noteworthy that the proportion of α-syn–immunopositive neurons in their report actually followed an increasing rostro-caudal gradient in the proximal GI tract. These findings suggest that, if the Braak15 hypothesis of an unbroken chain of α-syn neurons transferring a pathogen via transynaptic retrograde transport is correct, then the number of susceptible neurons follows an increasing rostrocaudal gradient in the proximal GI tract.25 In the current data sets, the distal GI tract, which possesses the least amount of vagal innervation, was most susceptible to inflammation-induced PD pathology. Future studies might employ strategic enteric lesions to further elucidate regional GI vulnerabilities.
Given the proportionally high density of α-syn–immunoreactive vagal efferent terminals in the stomach and duodenum, as well as the presence of α-synimmunoreactive neurons, it stands to reason that the proximal GI tract is the most susceptible site for PD initiation. However, if pathology is initiated by compromised GI barrier integrity, leading to chronic inflammation, then the site with the most luminal bacteria (the distal GI tract) presumably would be more susceptible. Although these hypotheses are not necessarily mutually exclusive, available human and animal data suggest that, functionally, the large intestine is preferentially affected in PD.57–61 Symptomatically, constipation and defecatory dysfunction are the most prevalent premotor GI complaints among patients with PD,51 supporting the notion that the large intestine is the dominant segment of the GI tract implicated in the disease. However, less frequent and often late appearing symptoms, including dysphagia, sialorrhea, postprandial fullness, and early satiety, suggest the involvement of proximal GI segments, such as the esophagus, stomach, and small bowel, leading to the gastroparesis and intestinal dysmotility responsible for these symptoms during the course of PD. Increased permeability and α-syn pathology selectively in the large intestine, as demonstrated in the current animal data set, also are features of patients with newly diagnosed, pharmacologically untreated PD.26 Our recent human data and current animal data strongly suggest that the large intestine is the initial site of GI injury in α-syn neurodegenerative disorders like PD. Therefore, if α-syn pathological burden truly follows a decreasing rostrocaudal gradient throughout the GI tract in PD, then there is an apparent disconnect between α-syn pathology and functional impairment of the affected region.
It is becoming increasingly evident that non-motor symptoms like GI dysfunction play a critical role in parkinsonian etiology, because they can manifest years to decades before the emergence of cardinal motor symptoms. As premotor biomarkers, such as colonic submucosal biopsies, are validated, developing animal models that encompass premotor phenomena, including GI pathology, will be critical to ultimately test disease-modifying strategies. Whereas the mechanisms surrounding disease initiation and propagation are incompletely understood, we now report on a robust animal model to potentially interrogate the role of the intestines and the enteric nervous system in PD-like pathogenesis. Fully elucidating and modeling the early parkinsonian spectrum will allow the testing of gut-directed interventions in the disease course. Examples of these potential interventions to prevent and/or treat PD are prebiotics, probiotics, and dietary modification, which could impact intestinal barrier integrity and colonic milieu.
Acknowledgments
Funding agencies: This work was supported by an internal grant from Rush University.
We are most appreciative for the assistance and guidance of Drs. Chris Forsyth and Celeste Napier as well as statistical input by Dr. Glenn Stebbins and Mr. Mario Moric.
Footnotes
Relevant conflicts of interest/financial disclosures: Nothing to report.
Full financial disclosures and author roles may be found in the online version of this article.
References
- 1.Litvan I, Bhatia KP, Burn DJ, et al. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinson disorders. Mov Disord. 2003;18:467–486. doi: 10.1002/mds.10459. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:2045–2047. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Bove J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx. 2005;2:484–494. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2003;2:107–116. doi: 10.1016/s1474-4422(03)00307-7. [DOI] [PubMed] [Google Scholar]
- 5.White L, Petrovitch H, Ross GW, et al. Prevalence of dementia in older Japanese-American men in Hawaii: the Honolulu-Asia Aging Study. JAMA. 1996;276:955–960. [PubMed] [Google Scholar]
- 6.Ross GW, Abbott RD, Petrovitch H, Tanner CM, White LR. Pre-motor features of Parkinson’s disease: the Honolulu-Asia Aging Study experience. Parkinsonism Relat Disord. 2012;18(suppl 1):S199–S202. doi: 10.1016/S1353-8020(11)70062-1. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396:67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 8.Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord. 2012;27:716–719. doi: 10.1002/mds.25020. [DOI] [PubMed] [Google Scholar]
- 9.Shannon KM, Keshavarzian A, Mutlu E, et al. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord. 2012;27:709–715. doi: 10.1002/mds.23838. [DOI] [PubMed] [Google Scholar]
- 10.Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F. Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 1988;76:217–221. doi: 10.1007/BF00687767. [DOI] [PubMed] [Google Scholar]
- 11.Wakabayashi K, Takahashi H, Ohama E, Ikuta F. Parkinson’s disease: an immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol. 1990;79:581–583. doi: 10.1007/BF00294234. [DOI] [PubMed] [Google Scholar]
- 12.Takeda S, Yamazaki K, Miyakawa T, Arai H. Parkinson’s disease with involvement of the parasympathetic ganglia. Acta Neuropathol. 1993;86:397–398. doi: 10.1007/BF00369454. [DOI] [PubMed] [Google Scholar]
- 13.Wakabayashi K, Takahashi H. Neuropathology of autonomic nervous system in Parkinson’s disease. Eur Neurol. 1997;38(suppl 2):2–7. doi: 10.1159/000113469. [DOI] [PubMed] [Google Scholar]
- 14.Beach TG, Adler CH, Sue LI, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 16.Drolet RE, Cannon JR, Montero L, Greenamyre JT. Chronic rote-none exposure reproduces Parkinson’s disease gastrointestinal neuropathology. Neurobiol Dis. 2009;36:96–102. doi: 10.1016/j.nbd.2009.06.017. [DOI] [PubMed] [Google Scholar]
- 17.Tasselli M, Chaumette T, Paillusson S, et al. Effects of oral administration of rotenone on gastrointestinal functions in mice [serial online] Neurogastroenterol Motil. 2013;25:e183–e193. doi: 10.1111/nmo.12070. [DOI] [PubMed] [Google Scholar]
- 18.Pan-Montojo FJ, Schwarz M, Winkler C, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice [serial online] Sci Rep. 2012;2:898. doi: 10.1038/srep00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Magen I, Yuan PQ, Subramaniam SR, Richter F, Chesselet MF, Tache Y. Mice overexpressing wild-type human alpha-synuclein display alterations in colonic myenteric ganglia and defecation [serial online] Neurogastroenterol Motil. 2012;24:e425–e436. doi: 10.1111/j.1365-2982.2012.01974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noorian AR, Rha J, Annerino DM, Bernhard D, Taylor GM, Greene JG. Alpha-synuclein transgenic mice display age-related slowing of gastrointestinal motility associated with transgene expression in the vagal system. Neurobiol Dis. 2012;48:9–19. doi: 10.1016/j.nbd.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Hallett PJ, McLean JR, Kartunen A, Langston JW, Isacson O. Alpha-synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol Dis. 2012;47:258–267. doi: 10.1016/j.nbd.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HJ, Suk JE, Lee KW, Park SH, Blumbergs PC, Gai P, Lee SP. Transmission of synucleinopathies in the enteric nervous system of A53T alpha-synuclein transgenic mice. Exp Neurobiol. 2011;20:181–188. doi: 10.5607/en.2011.20.4.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan-Montojo F, Anichtchik O, Dening Y. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice [serial online] PLoS One. 2010;5:e8762. doi: 10.1371/journal.pone.0008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuo YM, Li Z, Jiao Y, et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum Mol Genet. 2010;19:1633–1650. doi: 10.1093/hmg/ddq038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips RJ, Walter GC, Wilder SL, Baronowsky EA, Powley TL. Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson’s disease? Neuroscience. 2008;153:733–750. doi: 10.1016/j.neuroscience.2008.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forsyth CB, Shannon KM, Kordower JH, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease [serial online] PLOS One. 2011;6:e28032. doi: 10.1371/journal.pone.0028032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bjarnason I, MacPherson A, Hollander D. Intestinal permeability: an overview. Gastroenterology. 1995;108:1566–1581. doi: 10.1016/0016-5085(95)90708-4. [DOI] [PubMed] [Google Scholar]
- 28.Keita AV, Soderholm JD. The intestinal barrier and its regulation by neuroimmune factors. Neurogastroenterol Motil. 2010;22:718–733. doi: 10.1111/j.1365-2982.2010.01498.x. [DOI] [PubMed] [Google Scholar]
- 29.Lema Tome CM, Tyson T, Rey NL, Grathwol S, Britschqi M, Brudin P. Inflammation and alpha-synuclein’s prion-like behavior in Parkinson’s disease—is there a link? Mol Neurobiol. 2012;47:561–574. doi: 10.1007/s12035-012-8267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farhadi A, Banan A, Fields J, Keshavarzian A. Intestinal barrier: an interface between health and disease. J Gastroenterol Hepatol. 2003;18:479–497. doi: 10.1046/j.1440-1746.2003.03032.x. [DOI] [PubMed] [Google Scholar]
- 31.Qin L, Wu X, Block ML, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Qin L, Wilson B, Wu X, Qian L, Granholm AC, Crews FT, Hong JS. Endotoxin induces a delayed loss of TH-IR neurons in substantia nigra and motor behavioral deficits. Neurotoxicology. 2008;29:864–870. doi: 10.1016/j.neuro.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu Y, Kordower JH. Age-associated increases of α-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol Disease. 2007;25:134–149. doi: 10.1016/j.nbd.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 34.Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol. 2009;43:163–172. doi: 10.1016/j.alcohol.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keshavarzian A, Farhadi A, Forsyth CB, et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol. 2009;50:538–547. doi: 10.1016/j.jhep.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, Keshavarian A. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008;28:1026–1033. doi: 10.1111/j.1478-3231.2008.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farhadi A, Keshavarzian A, Fields JZ, Sheikh M, Banan A. Resolution of common dietary sugars from probe sugars for test of intestinal permeability using capillary column gas chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;836:63–68. doi: 10.1016/j.jchromb.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 38.Ma SY, Ciliax BJ, Stebbins G, et al. Dopamine transporter-immunoreactive neurons decrease with age in the human substantia nigra. J Comp Neurol. 1999;409:25–37. doi: 10.1002/(sici)1096-9861(19990621)409:1<25::aid-cne3>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 39.Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. 1998;70:1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- 40.Bing G, Lu X, Zheng NA, Jin L, Qi Y, Kim H-C. Microglia mediated dopaminergic cell death in the substantia nigra: a new animal model for Parkinson’s disease [abstract] Neurosci Abstracts. 1998;24:44. [Google Scholar]
- 41.Herrera AJ, Castano A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. 2000;7:429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Stanton DM, Nguyen XV, Liu M, Zhang Z, Gash D, Bing G. Intrapallidal lipopolysaccharide injection increases iron and ferritin levels in glia of the rat substantia nigra and induces locomotor deficits. Neuroscience. 2005;135:829–838. doi: 10.1016/j.neuroscience.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 43.Choi DY, Liu M, Hunter RL, et al. Striatal neuroinflammation promotes parkinsonism in rats [serial online] PLoS One. 2009;4:e5482. doi: 10.1371/journal.pone.0005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gill SR, Pop M, DeBoy RT, et al. Metagenomic analysis of the human distal gut micobiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 2010;119:703–713. doi: 10.1007/s00401-010-0665-2. [DOI] [PubMed] [Google Scholar]
- 46.Bloch A, Probst A, Bissig H, Adams H, Tolnay M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subject. Neuropathol Appl Neurobiol. 2006;32:284–295. doi: 10.1111/j.1365-2990.2006.00727.x. [DOI] [PubMed] [Google Scholar]
- 47.Bottner M, Zorenkov D, Hellwig I, et al. Expression pattern and localization of alpha-synuclein in the human enteric nervous system. Neurobiol Dis. 2012;48:474–480. doi: 10.1016/j.nbd.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 48.Kupsky WJ, Grimes MM, Sweeting J, Bertsch R, Cote LJ. Parkinson’s disease and megacolon: concentric hyaline inclusions (Lewy bodies) in enteric ganglion cells. Neurology. 1987;37:1253–1255. doi: 10.1212/wnl.37.7.1253. [DOI] [PubMed] [Google Scholar]
- 49.Qualman SJ, Haupt HM, Yang P, Hamilton SR. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson’s Gastroenterology. 1984;87:848–856. [PubMed] [Google Scholar]
- 50.Devos D, Lebouvier T, Lardeux B, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–48. doi: 10.1016/j.nbd.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 51.Cersosimo MG, Raina GB, Pecci C, et al. Gastrointestinal manifestations in Parkinson’s disease: prevalence and occurrence before motor symptoms. J Neurol. 2013;260:1332–1338. doi: 10.1007/s00415-012-6801-2. [DOI] [PubMed] [Google Scholar]
- 52.Annerino DM, Arshad S, Taylor GM, Alder CH, Beach TG, Greene JG. Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathol. 2012;124:665–680. doi: 10.1007/s00401-012-1040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pouclet H, Lebouvier T, Coron E, et al. A comparison between rectal and colonic biopsies to detect Lewy pathology in Parkinson’s disease. Neurobiol Dis. 2012;45:305–309. doi: 10.1016/j.nbd.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 54.Lebouvier T, Neunlist M, Bruley des Varannes S, et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms[serial online] PLOS One. 2010;5:e12728. doi: 10.1371/journal.pone.0012728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lebouvier T, Chaumette T, Damier P, et al. Pathological lesions in colonic biopsies during Parkinson’s disease. Gut. 2008;57:1741–1743. doi: 10.1136/gut.2008.162503. [DOI] [PubMed] [Google Scholar]
- 56.Berthoud HR, Carlson NR, Powley TL. Topography of efferent vagal innervation of the rat gastrointestinal tract. Am J Physiol. 1991;260:200–207. doi: 10.1152/ajpregu.1991.260.1.R200. [DOI] [PubMed] [Google Scholar]
- 57.Abbott RD, Petrovitch H, White LR, et al. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57:456–462. doi: 10.1212/wnl.57.3.456. [DOI] [PubMed] [Google Scholar]
- 58.Abbott RD, Ross GW, Petrovitch H, et al. Bowel movement frequency in late-life and incidental Lewy bodies. Mov Disord. 2007;22:1581–1586. doi: 10.1002/mds.21560. [DOI] [PubMed] [Google Scholar]
- 59.Savica R, Carlin JM, Grossardt BR, et al. Medical records documentation of constipation preceding Parkinson disease: a casecontrol study. Neurology. 2009;73:1752–1758. doi: 10.1212/WNL.0b013e3181c34af5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao X, Chen H, Schwarzschild MA, Ascherio A. A prospective study of bowel movement frequency and risk of Parkinson’s disease. Am J Epidemiol. 2011;174:546–551. doi: 10.1093/aje/kwr119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ross GW, Abbott RD, Petrovitch H, Tanner CM, White LR. Premotor features of Parkinson’s disease: the Honolulu-Asia Aging Study experience. Parkinsonism Relat Disord. 2012;18(suppl 1):S199–S202. doi: 10.1016/S1353-8020(11)70062-1. [DOI] [PubMed] [Google Scholar]