

Abstract
Next-generation sequencing (NGS) technologies have made high-throughput sequencing available to medium- and small-size laboratories, culminating in a tidal wave of genomic information. The quantity of sequenced bacterial genomes has not only brought excitement to the field of genomics but also heightened expectations that NGS would boost antibacterial discovery and vaccine development. Although many possible drug and vaccine targets have been discovered, the success rate of genome-based analysis has remained below expectations. Furthermore, NGS has had consequences for genome quality, resulting in an exponential increase in draft (partial data) genome deposits in public databases. If no further interests are expressed for a particular bacterial genome, it is more likely that the sequencing of its genome will be limited to a draft stage, and the painstaking tasks of completing the sequencing of its genome and annotation will not be undertaken. It is important to know what is lost when we settle for a draft genome and to determine the “scientific value” of a newly sequenced genome. This review addresses the expected impact of newly sequenced genomes on antibacterial discovery and vaccinology. Also, it discusses the factors that could be leading to the increase in the number of draft deposits and the consequent loss of relevant biological information.
Keywords: Next-generation sequencing, Drafts, Prokaryotic genomes, Computational tools, Omics
Core tip: Next-generation sequencing (NGS) technologies have made high-throughput sequencing available to medium- and small-size laboratories, culminating in a tidal wave of genomic information. The quantity of bacterial genomes has not only brought excitement to the field of genomics, it has also heightened expectations that NGS would boost antibacterial discovery and vaccine development. Although many possible drug and vaccine targets have been discovered, the success rate of genome-based analysis has remained below expectations. Furthermore, NGS has consequences for genome quality, resulting in an exponential increase in draft genome deposits in public databases. This review will address the expected impact of newly sequenced genomes on antibacterial discovery and vaccinology, as well as the impact of NGS on draft bacterial genomes.
INTRODUCTION
Since its release in 2005, next-generation sequencing (NGS) has been responsible for a drastic reduction in the price of genome sequencing and for a tidal wave of genetic information[1]. NGS technologies have made high-throughput sequencing available to medium- and small-size laboratories. The new possibility of generating a large number of sequenced bacterial genomes not only brought excitement to the field of genomics but also heightened expectations that the development of vaccines and the search for new antibacterial targets would be boosted. Nevertheless, these expectations were shown to be naïve. The complexity of host-bacteria interactions and the large diversity of bacterial genetic products have been shown to play greater roles in vaccine development and antibacterial discovery[2-4].
Additionally, as with any methodology, NGS presents its own drawbacks. Among the new sequencing technologies the most consolidated in the market are the 454 GS FLX platform (Roche), Illumina (Genome Analyzer) and SOLiD (Life Technologies)[5,6]. These devices are capable of generating millions of reads, providing high coverage genomic but with a drawback, reads are considerably smaller than the ones produced by Sanger methodology[7,8]. While Sanger methodology produces reads ranging from 800 to 1000 bases, NGS platforms produces reads ranging from 50 (SOLiD V3) to 2 × 150 bases (Illumina)[9]. The small amount of information contained in each read makes it difficult to completely assemble a genome using exclusively computational tools[10,11]. Therefore small reads made the genome assembly process a quite more laborious task.
In recent years, approaches that use hybrid assemblies were developed to facilitate the assembly process. They take advantage of high read quality of second generation sequencers, i.e., Illumina (Genome Analyzer), and longer read lengths from third generation sequencers, i.e., SMRT sequencers (Pacific Biosciences) and Ion Torrent PGM[12,13]. Although empirically logical, this kind of approach wasn’t facilitated due to the lack of integration between sequencers.
In order to improving and verifying quality genome is essential to know which combination of sequencing data, computer algorithms, and parameters can produce the highest quality assembly[14,15]. Also, it is necessary to know the more likely type of error data a sequencer platform will present. For instance, Illumina and SOLiD are more likely to present nucleotide substitution, while 454 GS FLX and Ion Torrent are more likely to present indels[16]. Nearly none bioinformatic system has been developed to integrate reads from different sequencers into a single assembly[12,17]. This new developed approaches aim to reduce the manual intervention in finishing genomes, since repetitive regions may be solved using an hybrid approach.
Although NGS is directly responsible for considerable growth in the size of genomic databases, it has also been indirectly responsible for a decrease in genome quality[1,10]. The number of draft genome (partial data) deposits in public databases has grown exponentially since 2005 (Figure 1). In general, if no further studies will be developed using a particular organism’s genome, it is more likely to be deposited as a draft genome. Otherwise, the painstaking tasks of improving and finishing the genome (complete data) must be undertaken[18].
Figure 1.
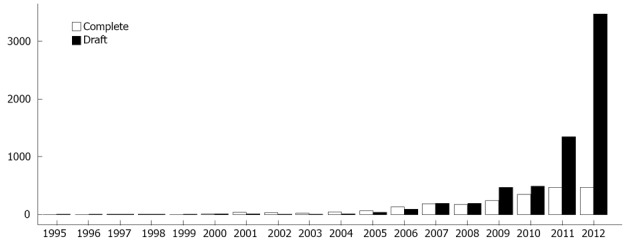
Number of complete genome and draft genome (partial data) deposits in public databases.
This review will address the “scientific value” of a newly sequenced genome and the amount of insight it can provide. We will address the factors that could be leading to the increase in the number of draft deposits and the consequent loss of relevant biological information. Additionally, we will summarize the expectations created by NGS technologies regarding vaccine development and antibacterial discovery.
OVERVIEW OF SEQUENCING AND ASSEMBLY
For 30 years, sequencing technologies based on Sanger chemistry dominated the market. Although sequencing had undergone numerous improvements over the years, gene cloning techniques were still necessary to obtain genomic DNA sequences. Therefore, the time and cost required to obtain a complete genome sequence remained high. Moreover, the capacity of parallel sequencing was quite limited[19-21]. NGS platforms made it possible to sequence complete prokaryotic genomes using massively parallel sequencing more rapidly and at a lower cost[20,22].
Although NGS has facilitated sequencing processes, its relatively smaller reads make the assembly process a computational challenge[10,11]. The main limitation of short-read assembly methods is their inability to resolve repetitive regions of the genome without paired libraries[11]. The assembly of repetitive regions was an important issue even before the introduction of NGS platforms; shorter reads only made the problem worse.
In 2001, Kececioglu et al[23] argued about the impossibility of correctly assembling regions of the genome that contain identical copies of a sequence. Usually, long DNA repeats are not exact copies. They contain small differences that could, in principle, permit their correct assembly. Nevertheless, a major difficulty arises from sequencing errors. Assembly software must accept imperfect sequencing alignments to avoid missing genuine connections between sequences[22]. With the small amount of information within each read adding to the inherent sequencing error, it is difficult to separate true differences within repeated sequences from sequencing errors.
A study by Phillippy et al[24] revealed that the majority of contig ends in draft genomes were associated with repeated regions. They concluded that it was possible to categorize the majority of mis-assembly events into two general classes: (1) repeat collapse or expansion; and (2) sequence rearrangement and inversion. Each of these classes exhibits specific mis-assembly signatures: the first class is the result of incorrect assembly in repetitive regions, including fewer or additional copies; the second class is the result of the rearrangement of multiple repeated copies, which is caused by the insertion of a read between them. The second class may be considered more influential because, if not fixed, it might be interpreted as a real biological rearrangement event[25,26]. If the assembler cannot resolve the region between two genomic fragments, a gap is formed. Gaps may occur due to: (1) an intrinsic characteristic of the sequencing platform that leads to incomplete or incorrect information; or (2) the inability of an assembly algorithm to handle regions of low complexity or repeated DNA[18,27,28]. The process of identifying and closing these gaps is quite laborious and requires additional manual intervention.
Gap closure processes usually involve the design of primers flanking the gap region to perform semi-automated sequencing of the unrepresented parts of the genome[28]. Several bioinformatics methodologies have been developed to facilitate gap closure. IMAGE is a tool that uses de Bruijn methodology to fill gaps with short reads that are aligned with flanking regions of the gap and were not used in the assembly[28]. In 2011, Cerdeira et al[29] generated a similar strategy by using CLC Genomics Workbench for the recursive alignment of unused short reads from the SOLiD platform. GapFiller is another tool that uses local alignment; its main advantage is the use of paired reads to estimate gap size and allows define the type of paired library: reverse-reverse, forward-forward, reverse-forward and forward-reverse[30].
From a purely practical standpoint, assembly tools are not required to produce a perfectly finished genome as an output. Their main function is to reduce the sequencing reads to a manageable number of contigs[26]. The process of finishing a genome, ensuring that gaps are closed and the gene order is correct, requires human decision-making. Therefore, the lack of fully automated processes constitutes a bottleneck in generating complete genomes.
“SCIENTIFIC VALUE” OF A NEWLY SEQUENCED GENOME
The value of a newly sequenced genome can be assessed using many different metrics. If publications are considered the main “currency” within the scientific community, there has been a considerable decrease in the value of new sequences over the last four decades.
The introduction of Sanger methodology in 1977 was one of the main landmarks in the early stages of the genomic era[31]. During the first years of using Sanger sequencing, a sequence of no more than 1000 nucleotides was sufficient for a work to be accepted in a journal such as Cell (current impact factor: 32.40) or Nature (current impact factor: 36.28)[32-34]. In 1980, the shotgun DNA sequencing methodology was introduced, enabling the sequencing of longer DNA fragments[35]. Complete bacterial operons were sequenced and published in journals such as Molecular Microbiology (current impact factor: 5.01) and Proceedings of the National Academy of Sciences (PNAS - current impact factor: 9.68)[36-38].
A combination of DNA sequencing improvements and the newly developed TIGR Assembler[39] culminated in the publication of the first complete bacterial genomes in 1995. Papers containing the complete nucleotide sequences of Haemophilus influenzae Rd (1830137 base pairs) and Mycoplasma genitalium (580070 base pairs) were both published in Science (current impact factor: 31.20)[40,41]. Almost 20 years later, a paper containing the sequence of a prokaryotic genome alone may be published in the Genome Announcement section of the Journal of Bacteriology (current impact factor: 3.82) or in Standards in Genomic Sciences (SIGS - has not been published sufficiently long to receive an impact factor). A recent article by Smith even refers to the not-so-distant “death” of the “genome paper”, noting that the space for genome publication may come to an end soon[42].
The publication impact of newly sequenced genomes decreased following DNA sequencing improvements, and the reason is no mystery. High-impact journals only publish groundbreaking original scientific research or results of outstanding scientific importance. To produce a higher-impact publication, more information must be extracted from genomes. For instance, several genomes may be examined in a comparative genomic analysis or pangenomic study[43,44], or an analysis may focus on the presence or absence of specific markers or on small differences between DNA sequences[26,45]. In this context, the genome becomes a stepping stone to the main goal, the comparative analysis. As the basis of the analysis, the genome sequence remains important. Nevertheless, it may not be of sufficient importance for one to undertake the painstaking task of completing the genome sequence.
WHAT IS LOST WHEN WE OPT FOR A DRAFT GENOME?
Over the years, arguments have been presented in favor both of complete genomes[41,46] and of the superior “tradeoff” that a draft genome represents[47]. The discussion has been centered around two main points: (1) to provide the greatest amount of useful data, sequences must be as complete as possible; and (2) draft genomes (partial data) are sufficient for most scientific contexts. The issue at stake is the extra money and manpower necessary to finish a genome. Is the additional information contained in a finished genome worth the investment? To answer this question, one must identify the information that is lost from a draft and analyze the quality of data that is generated using drafts. Furthermore, it is necessary to understand the limits of draft genome use.
The first issue to consider is whether it is possible to properly identify all of an organism’s genes in a draft genome. Gene characterization consists of the following: (1) gene prediction with the identification of an open reading frame (ORF); and (2) the functional annotation of the gene product. The main gene identification problems in drafts are associated with the partial or complete loss of ORFs[10]. Such errors may lead either to over-annotation, due to the annotation of multiple fragments originating from the same ORF, or to under-annotation, possibly due to the absence of partial or entire domains from the ORF[10]. These problems affect genomic analyses, causing errors due to missing ORFs that are not annotated or due to multiple fragments that belong to the same ORF but are annotated separately. In other words, the mere absence of a gene from a draft cannot be considered definitive proof of its absence from the organism’s genome[10,41].
The pangenomic approach is one type of analysis that may be impaired by reliance on draft genomes, because many genes in a draft may be misidentified due to fragmentation. Pangenomic projects attempt to characterize the gene pool of a bacterial species as the genes that are present in all strains (the “core genome”) and the genes that are present in only a few species (the “dispensable genome”)[43]. Horizontal gene transfer (HGT) analysis is another approach that cannot be performed using drafts. HGT is one of the main sources of variability among bacteria because it allows the acquisition of several new genes[36,37]. There is evidence that most gaps in genomic sequences are associated with transposases, insertion sequences and integrases, structures that usually flank a genomic island[48]. Another approach that may be impaired by reliance on drafts is phylogenomics, which aims to reconstruct both the vertical and lateral gene transfer processes of a bacterial species using a whole-genome analysis[49].
Although not strictly related to drafts, the functional annotation of genes is another feature that is usually neglected when we opt for a draft genome (Figure 2). Complete genomes may also present this problem because the quality of functional annotation is related to the amount of effort dedicated to a genome. DNA sequence is being generated much more rapidly than it can be analyzed; thus, a large proportion of the sequence information in databases has been annotated solely by automatic algorithms[50]. It is disturbing that although automatic annotation algorithms have improved over the years, misannotation has increased over time[50]. The misannotation of a reference strain is particularly harmful because the error will likely be propagated to other genomes. In our attempts to exploit the full potential of NGS, we risk having databases filled with incomplete and/or incorrect genomic data.
Figure 2.
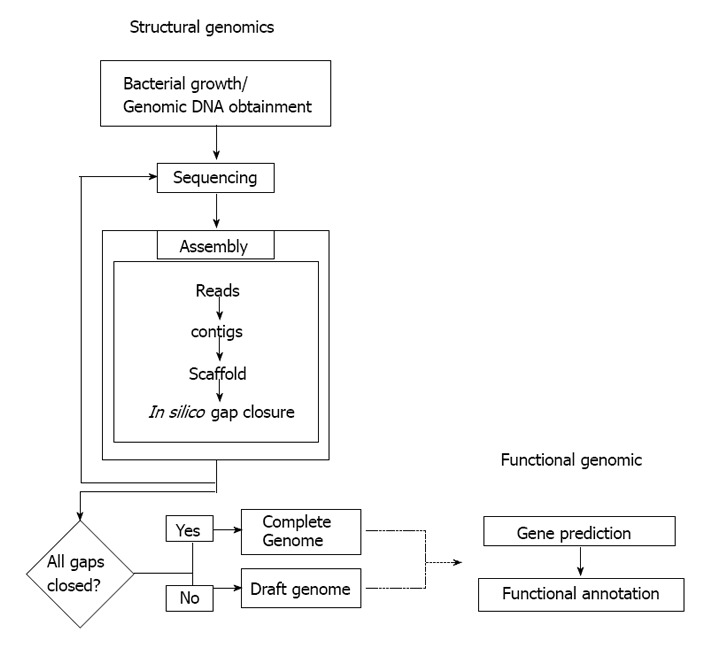
General workflow during sequencing process a bacterial genome.
Because the purpose of many sequencing projects is to identify a small number of differences between a newly sequenced genome and the sequence of a closely related species, a large number of genomes are left as drafts[26]. Considering the constant evolution of organisms, a sequenced genome represents a snapshot in the biological history of a species. Therefore, a single finished genome might be useful for decades of future studies. By opting for draft genomes, we may be shutting down the full gamut of future scientific analysis.
VACCINE DEVELOPMENT
Genomic information was expected to boost vaccine discovery. In an attempt to measure the impact of genomic information on this field, Prachi et al[2] analyzed all the patent applications that contained genomic information. They observed that there was an enormous increase in such applications shortly after the first complete genomes were released, but since 2002, there has been a continuous decrease. The authors attributed this decrease to more stringent legal requirements, which call for empirical evidence to complement in silico data.
The initial increase in patent applications containing genomic information was related to the development of a new paradigm in vaccine development. In 2000, Rappuoli[51] described the “reverse vaccinology” (RV) concept, in which he proposed inverting the traditional process of antigen identification. Instead of identifying the antigenic components of a pathogenic organism using serological or biochemical methods, RV uses the organism’s genome to predict all of its protein antigens. RV approaches mainly focus on secreted proteins because they are more likely to induce immune responses. Secreted proteins are involved in several processes that modulate the host-pathogen relationship, such as cell adhesion and invasion, as well as resistance to stress conditions[52-54]. Over the years, several methodologies have been developed to predict secreted proteins and to evaluate their potential immunological properties.
In 2010, Vaxign was released as the first vaccine design tool with a web interface (http://www.violinet.org/vaxign/). Vaxign allows users to submit their own sequences to perform vaccine target predictions. The Vaxign predictions have been consistent with existing reports for organisms such as Mycobacterium tuberculosis and Neisseria meningitides[55]. Another vaccine design tool is MED (Mature Epitope Density - http://med.mmci.uni-saarland.de/). MED attempts to select the more promising vaccine targets by identifying proteins with higher concentrations of epitopes[56]. There are also tools exclusively for protein epitope prediction, such as Immune Epitope Analysis (http://tools.immuneepitope.org/main/) and Vaxitope (http://www.violinet.org/vaxign/vaxitop/index.php).
Because a large number of bacterial genomes are already available, reverse vaccinology is quite accessible and inexpensive. Nevertheless, as has been previously discussed[57,58], the expectations for reverse vaccinology techniques do not correspond to reality, given the small number of vaccines have been developed using the bacterial genome sequences available[59]. This occurs because there are also several factors that are involved in the host response during infection, for example, the production of antibodies by the immune system.
ANTIBACTERIAL DISCOVERY
The period between the 1930s and the 1960s is known as the “golden age” of antibiotic discovery[11,60]. During this period, most of the known classes of antibiotics were discovered. These discoveries involved screening natural products regardless of their mechanisms of action. After most of the low-hanging fruits were harvested, the rate of antibacterial discovery decreased, culminating in a slowdown beginning in the 1990s[61].
Hopes for turning this void into a rapid acceleration accompanied the completion of the first bacterial genome sequences. The goal was to use comparative genomic analysis to identify potential targets present in a desirable spectrum (e.g., the bacteria responsible for upper respiratory tract infections)[3,4,62]. It was naive to assume that having the genome sequences would be sufficient for this level of discovery; a possible drug target must undergo numerous stages, from discovery through human clinical tests, and it is not possible to develop drugs for all potential targets[3,62]. Nevertheless, the prospect of exploring hundreds of potential targets revived the interest of pharmaceutical companies.
After some years of trials, several companies ended their target-based programs because of a lack of productivity. Despite reports of multi-resistant bacterial strains, the efforts to discover new antibacterial targets were again reduced[63,64]. Although genomics has not been able to reverse the lack of new antibiotic development, it has significantly improved screening methodologies. Genomics has facilitated high-throughput drug campaigns, which are being used to determine the mechanisms of action of antibacterial compounds and bacterial resistance mechanisms[4].
CONCLUSION
Several next-generation platforms have been developed in recent decades, as well as bioinformatics programs to an enhancement of performance and optimization omics techniques. Is not yet possible to integrate reads from different sequencers into a single assembly[17,23]. This newly developed approach aims to reduce the amount of manual intervention needed to complete a genome sequence by using a hybrid approach to resolve repetitive regions.
Improvements are expected not only in sequencing platforms but also in assemblers. Recently, two groups assessed the quality of the currently available assemblers. The 2011 Assemblathon was the first competition among assemblers[65]. For this competition, simulated data were generated and groups of assemblers were asked to blindly assemble it. The use of simulated data poses a problem in determining the applicability of the results to other data sets. The 2012 GAGE (Genome Assembly Gold-Standard Evaluations) competition for assembling real data resulted in the following conclusions: (1) the data quality has a greater influence on the final outcome than the assembler itself; and (2) the results do not support the current measures of correctness (related to contiguity)[26].
There is a large gap between the availability of genomic sequences in databases and the commercial production of vaccines and antibiotics in recent years, especially in the fields of investment and success (“expected return”). Drug development for all potential targets and effective vaccines has produced limited success. In contrast, there has been an acceleration in the discovery of new targets due to the refinement of bioinformatics tools for this purpose, such as epitope mapping and searching for secreted proteins. However, the major problems facing vaccine and antibiotic development, such as resistance mechanisms and host immune responses, remain unsolved.
Genome analysis constitutes a strategy for the expansion and diversification of the pharmacology and vaccinology sectors. This methodology can be used to explore a large number of targets and to reduce the costs of molecular and immunological tests. Finally, to improve the production of antibiotics and vaccines, it is necessary to know more about bacterial regulatory pathways. New interactome and microbiome studies must be implemented to assist this search.
ACKNOWLEDGEMENTS
This work involved the collaboration of various institutions, including the Genomics and Proteomics Network of the State of Pará of the Federal University of Pará (Rede Paraense de Genômica e Proteômica da Universidade Federal do Pará), the Amazon Research Foundation (Fundação Amazônia Paraense - FAPESPA), the National Council for Scientific and Technological Development (Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq), the Brazilian Federal Agency for the Support and Evaluation of Graduate Education (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES) and the Minas Gerais Research Foundation (Fundação de Amparo à Pesquisa do estado de Minas Gerais).
Footnotes
Supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) in Brazil, processes BEX 12954-12-8 and 11517-12-3, to Barbosa EGV and Aburjaile FF
P- Reviewers: Bhattacharya SK, Faik A S- Editor: Ma YJ L- Editor: A E- Editor: Lu YJ
References
- 1.Zhang J, Chiodini R, Badr A, Zhang G. The impact of next-generation sequencing on genomics. J Genet Genomics. 2011;38:95–109. doi: 10.1016/j.jgg.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prachi P, Donati C, Masciopinto F, Rappuoli R, Bagnoli F. Deep sequencing in pre- and clinical vaccine research. Public Health Genomics. 2013;16:62–68. doi: 10.1159/000345611. [DOI] [PubMed] [Google Scholar]
- 3.Pucci MJ. Use of genomics to select antibacterial targets. Biochem Pharmacol. 2006;71:1066–1072. doi: 10.1016/j.bcp.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Mills SD. When will the genomics investment pay off for antibacterial discovery? Biochem Pharmacol. 2006;71:1096–1102. doi: 10.1016/j.bcp.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 5.Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet. 2011;52:413–435. doi: 10.1007/s13353-011-0057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metzker ML. Sequencing technologies-the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 8.Magi A, Benelli M, Gozzini A, Girolami F, Torricelli F, Brandi ML. Bioinformatics for Next Generation Sequencing Data. Genes. 2010;1:294–307. doi: 10.3390/genes1020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 10.Klassen JL, Currie CR. Gene fragmentation in bacterial draft genomes: extent, consequences and mitigation. BMC Genomics. 2012;13:14. doi: 10.1186/1471-2164-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95:315–327. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bashir A, Klammer AA, Robins WP, Chin CS, Webster D, Paxinos E, Hsu D, Ashby M, Wang S, Peluso P, et al. A hybrid approach for the automated finishing of bacterial genomes. Nat Biotechnol. 2012;30:701–707. doi: 10.1038/nbt.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribeiro FJ, Przybylski D, Yin S, Sharpe T, Gnerre S, Abouelleil A, Berlin AM, Montmayeur A, Shea TP, Walker BJ, et al. Finished bacterial genomes from shotgun sequence data. Genome Res. 2012;22:2270–2277. doi: 10.1101/gr.141515.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker M. De novo genome assembly: what every biologist should know. Nature Methods. 2012;9:333–337. [Google Scholar]
- 15.Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012;22:557–567. doi: 10.1101/gr.131383.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faircloth BC, Glenn TC. Not all sequence tags are created equal: designing and validating sequence identification tags robust to indels. PLoS One. 2012;7:e42543. doi: 10.1371/journal.pone.0042543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diguistini S, Liao NY, Platt D, Robertson G, Seidel M, Chan SK, Docking TR, Birol I, Holt RA, Hirst M, et al. De novo genome sequence assembly of a filamentous fungus using Sanger, 454 and Illumina sequence data. Genome Biol. 2009;10:R94. doi: 10.1186/gb-2009-10-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chain PS, Grafham DV, Fulton RS, Fitzgerald MG, Hostetler J, Muzny D, Ali J, Birren B, Bruce DC, Buhay C, et al. Genomics. Genome project standards in a new era of sequencing. Science. 2009;326:236–237. doi: 10.1126/science.1180614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shendure J, Mitra RD, Varma C, Church GM. Advanced sequencing technologies: methods and goals. Nat Rev Genet. 2004;5:335–344. doi: 10.1038/nrg1325. [DOI] [PubMed] [Google Scholar]
- 20.Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, Wang MD, Zhang K, Mitra RD, Church GM. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 21.Richardson P. Special Issue: Next Generation DNA Sequencing. Genes. 2010;1:385–387. [Google Scholar]
- 22.Munroe DJ, Harris TJ. Third-generation sequencing fireworks at Marco Island. Nat Biotechnol. 2010;28:426–428. doi: 10.1038/nbt0510-426. [DOI] [PubMed] [Google Scholar]
- 23.Kececioglu J, Ju J. Separating repeats in DNA sequence assembly. Proceedings of the 5th International Conference on Computational Biology, 2001. New York: ACM; 2001. pp. 176–183. [Google Scholar]
- 24.Phillippy AM, Schatz MC, Pop M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biol. 2008;9:R55. doi: 10.1186/gb-2008-9-3-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soares SC, Abreu VA, Ramos RT, Cerdeira L, Silva A, Baumbach J, Trost E, Tauch A, Hirata R, Mattos-Guaraldi AL, et al. PIPS: pathogenicity island prediction software. PLoS One. 2012;7:e30848. doi: 10.1371/journal.pone.0030848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ricker N, Qian H, Fulthorpe RR. The limitations of draft assemblies for understanding prokaryotic adaptation and evolution. Genomics. 2012;100:167–175. doi: 10.1016/j.ygeno.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Pop M. Genome assembly reborn: recent computational challenges. Brief Bioinform. 2009;10:354–366. doi: 10.1093/bib/bbp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11:R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerdeira LT, Carneiro AR, Ramos RT, de Almeida SS, D’Afonseca V, Schneider MP, Baumbach J, Tauch A, McCulloch JA, Azevedo VA, et al. Rapid hybrid de novo assembly of a microbial genome using only short reads: Corynebacterium pseudotuberculosis I19 as a case study. J Microbiol Methods. 2011;86:218–223. doi: 10.1016/j.mimet.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 30.Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13:R56. doi: 10.1186/gb-2012-13-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Boer HA, Gilbert SF, Nomura M. DNA sequences of promoter regions for rRNA operons rrnE and rrnA in E. coli. Cell. 1979;17:201–209. doi: 10.1016/0092-8674(79)90308-8. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura K, Inouye M. DNA sequence of the gene for the outer membrane lipoprotein of E. coli: an extremely AT-rich promoter. Cell. 1979;18:1109–1117. doi: 10.1016/0092-8674(79)90224-1. [DOI] [PubMed] [Google Scholar]
- 34.Porter AG, Barber C, Carey NH, Hallewell RA, Threlfall G, Emtage JS. Complete nucleotide sequence of an influenza virus haemagglutinin gene from cloned DNA. Nature. 1979;282:471–477. doi: 10.1038/282471a0. [DOI] [PubMed] [Google Scholar]
- 35.Messing J, Crea R, Seeburg PH. A system for shotgun DNA sequencing. Nucleic Acids Res. 1981;9:309–321. doi: 10.1093/nar/9.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown NL, Misra TK, Winnie JN, Schmidt A, Seiff M, Silver S. The nucleotide sequence of the mercuric resistance operons of plasmid R100 and transposon Tn501: further evidence for mer genes which enhance the activity of the mercuric ion detoxification system. Mol Gen Genet. 1986;202:143–151. doi: 10.1007/BF00330531. [DOI] [PubMed] [Google Scholar]
- 37.Postle K, Good RF. DNA sequence of the Escherichia coli tonB gene. Proc Natl Acad Sci USA. 1983;80:5235–5239. doi: 10.1073/pnas.80.17.5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Overduin P, Boos W, Tommassen J. Nucleotide sequence of the ugp genes of Escherichia coli K-12: homology to the maltose system. Mol Microbiol. 1988;2:767–775. doi: 10.1111/j.1365-2958.1988.tb00088.x. [DOI] [PubMed] [Google Scholar]
- 39.Sutton GG, White O, Adams MD and Kerlavage A. TIGR Assembler: A new tool for assembling large shotgun sequencing projects. Genome Sci Technol. 1995;1 Suppl 1:S9–S19. [Google Scholar]
- 40.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 41.Fraser CM, Eisen JA, Nelson KE, Paulsen IT, Salzberg SL. The value of complete microbial genome sequencing (you get what you pay for) J Bacteriol. 2002;184:6403–6405; discusion 6405. doi: 10.1128/JB.184.23.6403-6405.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith DR. Death of the genome paper. Front Genet. 2013;4:72. doi: 10.3389/fgene.2013.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Soares SC, Silva A, Trost E, Blom J, Ramos R, Carneiro A, Ali A, Santos AR, Pinto AC, Diniz C, et al. The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PLoS One. 2013;8:e53818. doi: 10.1371/journal.pone.0053818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jakobsen TH, Hansen MA, Jensen PØ, Hansen L, Riber L, Cockburn A, Kolpen M, Rønne Hansen C, Ridderberg W, Eickhardt S, et al. Complete genome sequence of the cystic fibrosis pathogen Achromobacter xylosoxidans NH44784-1996 complies with important pathogenic phenotypes. PLoS One. 2013;8:e68484. doi: 10.1371/journal.pone.0068484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parkhill J. In defense of complete genomes. Nat Biotechnol. 2000;18:493–494. doi: 10.1038/75346. [DOI] [PubMed] [Google Scholar]
- 47.Branscomb E, Predki P. On the high value of low standards. J Bacteriol. 2002;184:6406–6409; discussion 6409. doi: 10.1128/JB.184.23.6406-6409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kingsford C, Schatz MC, Pop M. Assembly complexity of prokaryotic genomes using short reads. BMC Bioinformatics. 2010;11:21. doi: 10.1186/1471-2105-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dagan T. Phylogenomic networks. Trends Microbiol. 2011;19:483–491. doi: 10.1016/j.tim.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 50.Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3:445–450. doi: 10.1016/s1369-5274(00)00119-3. [DOI] [PubMed] [Google Scholar]
- 52.Wooldridge K. Bacterial secreted proteins: secretory mechanisms and role in pathogenesis. United States: Caister Academic Press; 2009. pp. 300–315. [Google Scholar]
- 53.Simeone R, Bottai D, Brosch R. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr Opin Microbiol. 2009;12:4–10. doi: 10.1016/j.mib.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 54.Stavrinides J, McCann HC, Guttman DS. Host-pathogen interplay and the evolution of bacterial effectors. Cell Microbiol. 2008;10:285–292. doi: 10.1111/j.1462-5822.2007.01078.x. [DOI] [PubMed] [Google Scholar]
- 55.He Y, Xiang Z, Mobley HL. Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol. 2010;2010:297505. doi: 10.1155/2010/297505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santos AR, Pereira VB, Barbosa E, Baumbach J, Pauling J, Röttger R, Turk MZ, Silva A, Miyoshi A, Azevedo V. Mature Epitope Density--a strategy for target selection based on immunoinformatics and exported prokaryotic proteins. BMC Genomics. 2013;14 Suppl 6:S4. doi: 10.1186/1471-2164-14-S6-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seib KL, Zhao X, Rappuoli R. Developing vaccines in the era of genomics: a decade of reverse vaccinology. Clin Microbiol Infect. 2012;18 Suppl 5:109–116. doi: 10.1111/j.1469-0691.2012.03939.x. [DOI] [PubMed] [Google Scholar]
- 58.Tettelin H. The bacterial pan-genome and reverse vaccinology. Genome Dyn. 2009;6:35–47. doi: 10.1159/000235761. [DOI] [PubMed] [Google Scholar]
- 59.Donati C, Rappuoli R. Reverse vaccinology in the 21st century: improvements over the original design. Ann N Y Acad Sci. 2013;1285:115–132. doi: 10.1111/nyas.12046. [DOI] [PubMed] [Google Scholar]
- 60.Walsh C. Where will new antibiotics come from? Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 61.Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 63.Projan SJ. Why is big Pharma getting out of antibacterial drug discovery? Curr Opin Microbiol. 2003;6:427–430. doi: 10.1016/j.mib.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 64.Bush K, Pucci MJ. New antimicrobial agents on the horizon. Biochem Pharmacol. 2011;82:1528–1539. doi: 10.1016/j.bcp.2011.07.077. [DOI] [PubMed] [Google Scholar]
- 65.Earl D, Bradnam K, St John J, Darling A, Lin D, Fass J, Yu HO, Buffalo V, Zerbino DR, Diekhans M, et al. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res. 2011;21:2224–2241. doi: 10.1101/gr.126599.111. [DOI] [PMC free article] [PubMed] [Google Scholar]