

Abstract
Endoglin, also known as cluster of differentiation CD105, was originally identified 25 years ago as a novel marker of endothelial cells. Later it was shown that endoglin is also expressed in pro-fibrogenic cells including mesangial cells, cardiac and scleroderma fibroblasts, and hepatic stellate cells. It is an integral membrane-bound disulfide-linked 180 kDa homodimeric receptor that acts as a transforming growth factor-β (TGF-β) auxiliary co-receptor. In humans, several hundreds of mutations of the endoglin gene are known that give rise to an autosomal dominant bleeding disorder that is characterized by localized angiodysplasia and arteriovenous malformation. This disease is termed hereditary hemorrhagic telangiectasia type I and induces various vascular lesions, mainly on the face, lips, hands and gastrointestinal mucosa. Two variants of endoglin (i.e., S- and L-endoglin) are formed by alternative splicing that distinguishes from each other in the length of their cytoplasmic tails. Moreover, a soluble form of endoglin, i.e., sol-Eng, is shedded by the matrix metalloprotease-14 that cleaves within the extracellular juxtamembrane region. Endoglin interacts with the TGF-β signaling receptors and influences Smad-dependent and -independent effects. Recent work has demonstrated that endoglin is a crucial mediator during liver fibrogenesis that critically controls the activity of the different Smad branches. In the present review, we summarize the present knowledge of endoglin expression and function, its involvement in fibrogenic Smad signaling, current models to investigate endoglin function, and the diagnostic value of endoglin in liver disease.
Keywords: Telangiectasia, Signalling, Transforming growth factor-β, Disease, Bleeding disorders
Core tip: Endoglin is an accessory receptor for transforming growth factor-β impacting various aspects of its signaling and biological functions. Endoglin mutations are inherited as autosomal dominant disorders and may cause severe defects in different organs, including brain, lung and liver. In the present review, we will highlight the pathogenesis of several of these disorders and give an overview about the important role of endoglin dysfunction in the pathology of liver fibrosis.
INTRODUCTION
Endoglin (OMIM 131195) was originally identified 25 years ago by immunofluorescence staining of vascular endothelium with a monoclonal antibody (mAb 44G4) that was produced against a human pre-B leukemia cell line[1]. It is composed as a homodimer of two subunits with an apparent molecular weight of 95 kDa that are linked by disulfide bonds[1]. Two years later, cDNA clones were isolated from an endothelial cell λgt11 expression library using a rabbit antibody prepared against endoglin purified from placenta[2]. Subsequent screening with an endoglin-specific cDNA probe resulted in the isolation of a different splice variant in which the encoded cytoplasmic tail contains only 14 amino acids (aa) as opposed to the stretch of 47 residues that was published previously[3]. The ENG gene was mapped to the long arm of human chromosome 9 (9q34→qter) by Southern blot analysis of DNA isolated from human-hamster somatic cell hybrids and by fluorescent in situ hybridization coupled with DAPI banding on human chromosomes[4]. The detailed chromosomal assignment was subsequently predicted from the fact that the mouse homolog is located on chromosome 2 directly in the close proximity of the adenylate kinase-1 gene that is syntenic to human chromosome subband 9q34.1[5,6].
Mutations within endoglin were first brought into context of hereditary hemorrhagic telangiectasia type I (HHT-1) in three affected individuals in whom nucleotide substitutions or deletions gave rise to premature termination codons[7]. Since that, several hundred independent mutations or variations have been identified in the ENG gene that most often show regional distribution[8-12]. The different mutations show different phenotype-genotype correlation with the severity of HHT-1[13]. Moreover, it has been shown that soluble endoglin (sol-Eng) is an anti-angiogenesis factor that contributes to the pathogenesis of pre-eclampsia that is associated with hypertension, proteinuria, premature labor, hemolysis, liver abnormalities, thrombocytopenia, seizures and death[14,15]. Increased levels of sol-Eng in vascular surgical specimens were also brought into context with the pathogenesis of arteriovenous malformations (AVM) of the brain and aberrant cerebral vascular remodelling[16]. Other reports propose sol-Eng as a marker in diabetic patients[17] for estimating progression or treatment efficacy of the atherosclerotic process[18,19], systemic lupus erythematosus[20], non-small cell lung cancer patients[21], hypertension[22], disturbed angiogenesis in systemic sclerosis[23], Alzheimer’s disease[24], breast cancer[25], premalignant lesions of the colon mucosa[26], outcome of biliary atresia[27] and cystic fibrosis associated liver disease[28], unexplained fetal death[29], malaria pathogenesis[30], prostate cancer[31] and many other diseases. In addition, endoglin expression was found to be related to tumor size, aggressiveness and metastatic potential in patients with gastroenteropancreatic neuroendocrine tumors[32].
A similar phenotype, i.e., HHT type 2, is observed when the activin-like kinase (ALK)-1 receptor is functionally altered[33]. Likewise, mutations in the gene encoding Smad4 (MADH4) can cause a syndrome called Juvenile Polyposis/Hereditary Hemorrhagic Telangiectasia Syndrome (JPHT), consisting of both juvenile polyposis and hereditary hemorrhagic telangiectasia phenotypes[34]. Also, the mutations of other yet unidentified genes on the long arm of chromosome 5[35] and on the short arm of chromosome 7[36] were linked to the formation of other HHT types.
Endoglin expression and dysregulation has been shown in a number of cell types, including mesangial cells, cardiac and scleroderma fibroblasts, and hepatic stellate cells (HSC), suggesting some important function in cell and organ homeostasis and disease formation[37-40]. In particular, many independent findings demonstrate that endoglin is a critical factor that orchestrates transforming growth factor-β (TGF-β) signaling in wound healing in the pathogenesis of fibrosis. In regard to hepatic fibrogenesis, it was shown that endoglin is expressed in HSC[41] representing the most pro-fibrogenic cell type within the liver. Interestingly, endoglin expression is up-regulated during liver damage and transiently induced in HSC by TGF-β1[40]. In this hepatic subpopulation, endoglin binds to the TGF-β type II receptor (TβRII), becomes phosphorylated by the activity of the TβRII, and shows highest expression during maximal cell activation with a transdifferentiation-dependent cellular localisation and ligand affinity[40]. Interestingly, transient overexpression of endoglin results in a stronger activation of the Smad1/Smad5 signaling cascade and a prominent increase of α-smooth muscle actin expression, thereby promoting cellular activation and transdifferentiation[40], while contrarily the activity of the TGF-β1/Smad3 pathway is inhibited[42]. All these findings demonstrate that endoglin is one of the central switches controlling fibrotic and anti-fibrotic activities by producing different variant forms, adjusting ligand affinity, amending expression levels, and interacting with a versatile receptor network, thereby modulating the specific outcome of TGF-β-dependent and -independent pathways.
In the present review, we will summarize the actual knowledge of endoglin function and discuss the impact of this receptor on disease formation, hepatic fibrogenesis and its diagnostic value in initiation, progression and prognosis of various liver diseases.
MOLECULAR AND BIOCHEMICAL CHARACTERISTICS OF ENDOGLIN
The human endoglin gene contains 15 exons numbered 1 to 14, where exon 9 is split into 9a and 9b (Figure 1)[7]. Beside the full length endoglin (FL-Eng), a splice variant has been identified, i.e., short-endoglin (S-Eng), that is characterized by the retention of intron 14 in the mature mRNA[3,40,43]. The expression of S-Eng is increased in senescent endothelial cells and alternative splicing is most likely performed by the alternative splicing factor or splicing factor-2 (ASF/SF2)[44,45]. However, the orthologous S-Eng mRNAs of men and mice give rise to different proteins (Figure 1) with either shortened and in part alternate C-termini[2,43] or a full length endoglin with a peptide insertion in rat[40]. Although the C-terminal domain of FL-Eng does not possess catalytic activity, it is substrate for different kinases and comprises several protein-protein interaction domains (Figure 1)[46]. Therefore, structural alterations imposed by differential splicing of the mRNA that encodes the intracellular domain results in functional consequences for Eng in signaling (see below). In addition to splicing, Northern blot analysis of mouse and rat transcripts revealed two mRNA species differing in molecular weight more than the size of the retained intron 14 of S-Eng. Analysis of the corresponding cDNA with 3’-RACE and inspection of the rat genomic DNA sequence confirmed a variation in the non-coding region of the mRNA and the presence of a second polyadenylation signal in the genomic DNA[40]. Whether this differential polyadenylation modulates mRNA stability or other features of the mRNA is currently not known. Since it has been realized that endoglin mutations are causative for HHT-1[7], a wealth of different mutations in the endoglin gene which lead to altered expression or formation of aberrant protein products has been identified (see below). Nevertheless, mutations are not spread randomly in the genomic sequence. A bias for mutations is found in the orphan domain and the N-terminal zona pellucida (ZP-N) subdomain in which three highly conserved cysteines (Cys363, Cys382 and Cys412) are exceptionally prone to mutations[47].
Figure 1.
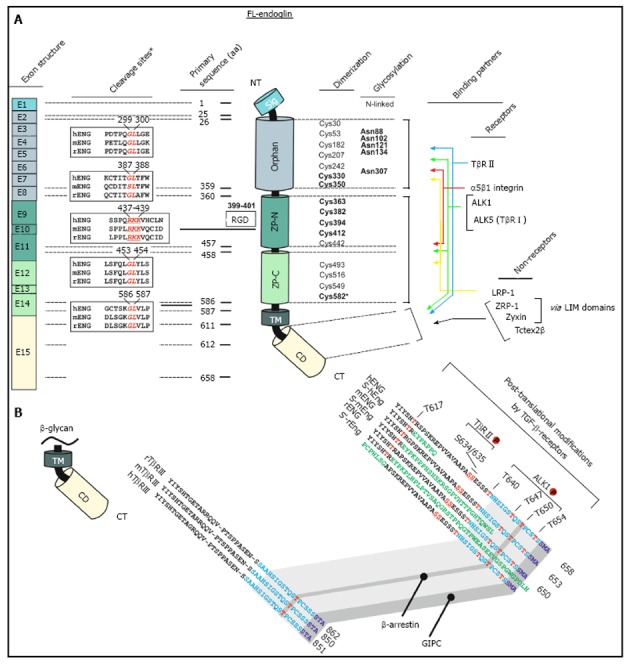
Schematic representation of the structural and functional modules of human endoglin. A: Left, first panel, Exon structure: Structure of the endoglin gene and assignment of respective protein modules. Left, second panel, cleavage sites: Predicted consensus proteolytical cleavage sites deduced from the primary sequence and experimentally confirmed matrix metalloproteinases-14 cleavage site between aa positions 586 and 587[59]. Middle panel, primary sequence positions: aa boundaries of the structural domains of human endoglin and location of the Arginine-Glycin-Aspartic acid (RGD) sequence that is only present in human endoglin. Right, first panel, dimerization: The cysteine residues of the extracellular domain of endoglin are depicted including the 8 highly conserved residues within the ZP-domain[47]. Cysteines that are involved in dimerization are shown in bold. Cysteine 582 that is involved in human endoglin dimerization is not present in the mouse and rat homologues[56]. Right, second panel, glycosylation: This figure part displays verified N-linked glycosylation sites within human endoglin. There is experimental evidence for O-linked glycosylation, but respective sites are not shown[1]. Right, third panel, binding partners: Depicted are interaction partners of endoglin as either receptor proteins (upper) or cytosolic non receptors proteins (lower). Colored arrows indicate interacting domains of endoglin with its binding partners. Zyxin and ZRP-1 bind to endoglin via their LIM-domains; B: Displayed is an aa alignment of betaglycan (left) and endoglin (right) of human, mouse and rat. Receptor kinase substrates (serine and threonines) are shown in red. Threonine 650 is essential for binding to β-arrestin2[66]. The C-termini of betaglycan and endoglin that are highly conserved are indicated in light blue. The PDZ-I domain which binds to GIPC is depicted in dark blue[67]. The alternative C-termini which results from differential splicing are shown in green.
Biochemical characteristics
Endoglin, a type I transmembrane glycoprotein, is expressed as a disulfide-bound dimer at the cell surface[48]. Endoglin belongs structurally to the zona pellucida (ZP) family of sperm receptors sharing a ZP domain of approximately 260 aa in their extracellular part[49,50]. This domain is localized between Lys362-Asp561 (Figure 1) and contains eight highly conserved cysteine residues[47]. Common characteristics of ZP domain proteins are that they are: (1) shed to generate a soluble form; (2) membrane proteins with a hydrophobic region at their C-termini; (3) strongly glycosylated; and (4) finally highly expressed in the corresponding tissues in which they occur[50].
Among TGF-β-family receptors, endoglin and betaglycan constitute the TGF-β type III receptor family. Both receptors share a high degree of similarity, especially in their intracellular domain (Figure 2) that is also the most conserved region between endoglin from different species (Figure 3), implying that this region has an important function, although lacking enzymatic activity[40].
Figure 2.

Sequence alignment of rat endoglin and betaglycan. The protein sequences of rat endoglin and betaglycan were aligned using the ClustalW2 algorithm. Respective sequences of rat endoglin (AAS67893) and betalycan (AAA42236.1) were taken from the GenBank. Please note the high degree of similarity of both proteins at their C-termini. Fully conserved aa in endoglin are marked by asterisk (*), positions that carry aa with strongly similar properties by a colon (:) and positions with weakly similar properties by a period (.), respectively. Please note that the highest degree of homology is found at the C-terminal regions that encompass the cytosolic part of endoglin
Figure 3.
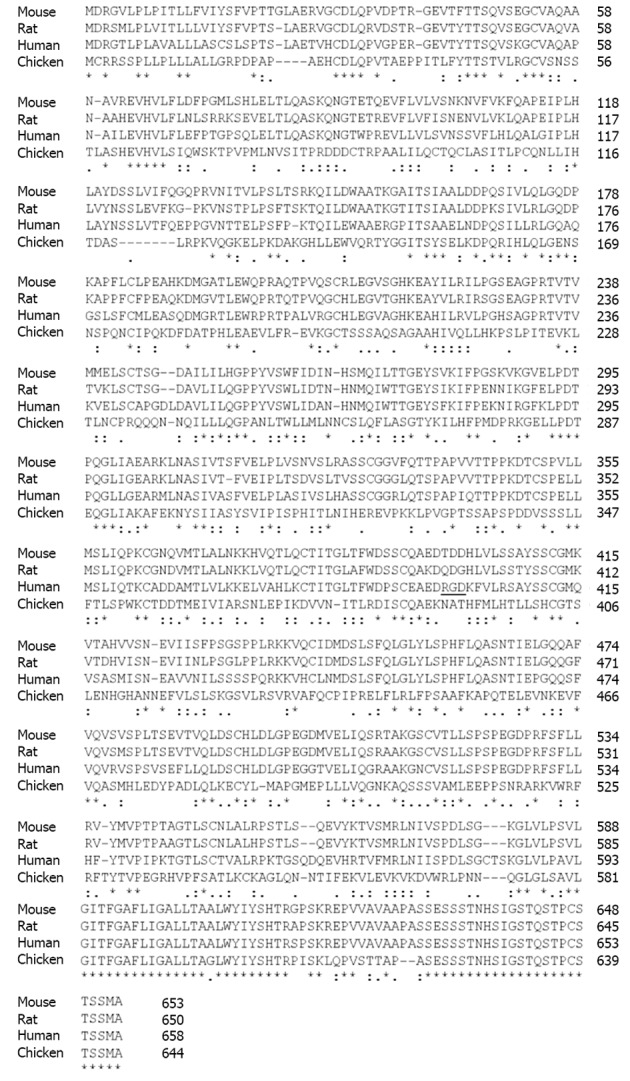
Sequence alignment of endoglin from different species. The protein sequences of rat, mouse, human, and chicken endoglin were aligned using the ClustalW2 tool (http://www.expasy.org/genomics/sequence_alignment). Sequences of mouse (NP_031958), rat (AAS67893), human (NP_001108225) and chicken (AAT84715) were taken from the GenBank (http://www.ncbi.nlm.nih.gov/). The Arginine-Glycin-Aspartic acid sequence in human endoglin (aa 399-aa 401) is underlined. Fully conserved aa in endoglin are marked by asterisk (*), positions that carry aa with strongly similar properties by a colon (:) and positions with weakly similar properties by a period (.), respectively. Please note that the highest degree of homology is found at the C-terminal regions that encompass the cytosolic part of endoglin.
In line, the signaling specificity of endoglin compared to betaglycan is at least for some specific functions determined by the extracellular domain (ECD)[51]. Since both of these receptors possess no enzymatic activity in their short C-terminal domain and are not obligatory for general signaling, they have been assigned an accessory/modulating function in signaling[52]. The primary sequence of FL-Eng comprises 658 aa in human[2,3], 650 aa in rat[41], and 653 aa in mouse (Figure 3)[53]. The ECD of human Eng harbors a Arginine-Glycin-Aspartic acid (RGD) peptide representing a potent binding site for integrins which is not present in the rat and mouse homologues[2,41,53]. Along with the FL-Eng, a splice variant designated S-Eng has been identified. The longer mRNA is due to the retained intron 14 (see above) and codes for a protein with a shortened C-terminus of 14 aa in human and 35 aa in mouse because of an in-frame stop codon present in the intron which is not found in rat resulting in a protein that contains a 49 aa insertion[3,41,53]. As outlined below, the shortening of the C-terminal domain of the splice variant in human and mouse have structural and functional consequences because specific modules are missing. The mentioned insertion in rat FL-Eng only causes minor effects which may be due to sterical alterations in the C-terminal domain[40]. In addition to these splice variants, two transcripts in mouse and rat occur which differ in the 3’-non-coding part and which arise from differential polyadenylation[40]. With respect to post-translational modifications, the primary Eng sequence contains several potential N- and O-dependent glycosylation sites. Initial enzymatic de-glycosylation studies confirmed the usage of both N- and O-dependent glycosylation consensus motives[48]. In a more detailed study, single N-dependent glycosylation sites (Asn88, Asn102, Asn121, Asn134 and Asn307) have been identified by mutational analysis[54]. Although the corresponding N-glycosylation sites seem to influence the stability of the corresponding domain, e.g., Asn102 and Asn307[54], the removal of carbohydrates by peptide N-glycosidase F (PNGase F) was shown to be exiguous for function of the ECD[55].
In general, FL-Eng has a tripartide structure comprising a short intracellular region (47 aa), a single transmembranal portion (25 aa), a large ECD (561 aa) and a predicted signal peptide (25 aa)[3]. Preceding the ZP domain there is an orphan domain (Glu26-Ile359), sharing no similarity to other protein families/domains[47]. The ZP domain (Gln360-Gly586) is further subdivided in a ZP-N (Gln360-Ser457) and ZP-C (Pro458-Gly586) subdomain (Figure 1). Deletion and substitution studies revealed that at least Cys582 in human FL-Eng in the ECD is involved in intermolecular disulfide binding[56]. Additional work revealed that the six cysteines between Cys330 and Cys412 are necessary to mediate receptor dimerization[57], allowing the receptor to be expressed as a dimer at the cell surface, or in case of the soluble form as a secreted dimer[58]. A high resolution structure established for the ECD of endoglin revealed information about the sterical arrangement of the 3-dimensional protein fold[47]. These studies confirmed the three-modular-structure (orphan domain, ZP-N and ZP-C domain) and further raised the hypothesis of the occurrence of a putative cleavage site for a sheddase with specificity for the linker region between the folded domains of ZP-N and ZP-C at position Arg437-Lys438-Lys439 (RKK)[47]. However, the biochemical elucidation showed that the cleavage site is located closer to the membrane at position Gly586-Leu587. The executing enzyme was shown to be matrix metalloprotease-14 (MMP-14 or MT1-MMP)[59,60] promoting a shedding process that is similar to that described for betaglycan before[61]. On a functional level, endoglin is able to interact with the TGF-β signaling receptors (cf. Figure 1, Figure 4)[62] as well as other regulatory proteins[63-67]. These interactions are mediated by the different subdomains (or combinations). In general, FL-Eng is able to interact with ALK5 and TβRII independent of ligand and the activation state of the signaling receptors[56]. In more detail it was shown that TβRII interacts with the region 437-558 (mainly ZP-C domain) of the endoglin ECD. In contrast to TβRII, ALK5 contacts two regions, spanning aa 26-437 and aa 437-558 (orphan and ZP-C domains)[56]. Similarly, the second type I receptor ALK1 was shown to interact with the region Glu26-Gly586 of the ECD of endoglin[68].
Figure 4.
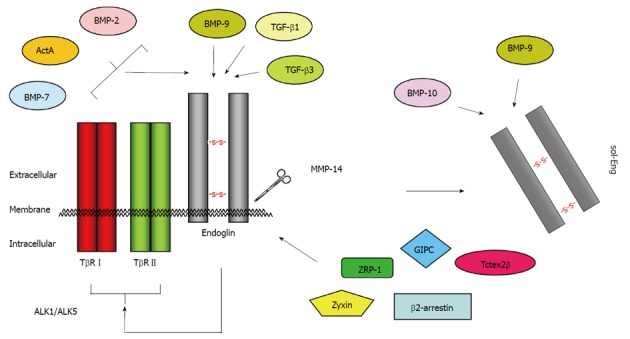
Binding partners of endoglin. Endoglin physically interacts via its extracellular domain with TGF-β1, TGF-β3 and BMP-9[110]. The short cytoplasmic domain has affinity for ZRP-1[63], Zyxin[64], GIPC[67], β-arrestin-2[66], and Tctex2β[65]. In conjunction with TβRI and TβRII, the binding spectrum is extended to BMP-2, BMP-7 and ActA[111]. After proteolytic cleavage (shedding) by MMP-14 (also known as membrane-type matrix metalloproteinase MT1-MMP), the soluble form of endoglin (sol-Eng) is released[59]. This form has capacity to bind BMP-9 and BMP-10[113]. TGF: Transforming growth factor; BMP: Bone morphogenetic protein.
Since the soluble variant of endoglin comprises all these ECD, it should in principal also be capable of mediating the same receptor interactions. Nevertheless, the binding of the soluble ECD to membrane bound endoglin could not be shown[57].
Whereas the binding to the ECD of FL-endoglin is independent of the signaling receptor activity, interaction of TβRII, ALK5 and ALK1 with the intracellular domain of endoglin is regulated by the activation state of the signaling receptors since binding of the constitutive active ALK5/ALK1 could not be detected, while the binding of kinase dead and wild type ALK5/ALK1 could be demonstrated[56,68]. In line, the association of endoglin with the inactive form (kinase dead) of TβRII was reported to be stronger when compared to wild type TβRII[56].
It is known that FL-Eng is phosphorylated at serine and threonine residues[69,70] and both ALK5 and TβRII use the C-terminus of endoglin as a substrate[56,70,71]. In turn, FL-Eng inhibits autophosphorylation of TβRII but enhances phosphorylation of ALK5 by TβRII leading to a stronger Smad2 transcriptional activity (see below)[56]. Aside from ALK5, ALK1 is also able to phosphorylate the FL-Eng C-terminus, but in contrast to ALK5, primarily on threonine residues[70]. Threonine phosphorylation by ALK1 (Thr654) necessitates serine phosphorylation by TβRII which is enforced by removal of the C-terminal PDZ domain[70]. Moreover, ALK1 phosphorylation and binding of endoglin was observed only in the presence of TGF-β1 and this phosphorylation leads to loss of FL-Eng from focal adhesions (see below)[70]. This modulates proliferative and adhesive properties of endothelial cells. In more detail, it was shown that the exponentiation activity of endoglin on ALK1 signaling and Smad1 activity is located between residues 26-558 within the ECD of endoglin[68]. Another interaction with the ECD of endoglin is mediated by integrin α5β1, which contacts not only the RGD-peptide but several parts of the ECD of endoglin. Clustering of α5β1/endoglin/ALK1 leads to an enhancement of TGF-β1-mediated Smad1/Smad5 activation and signaling[72]. Recently, leucine-rich α2-glycoprotein 1 (Lrg1) has been further shown to interact with the ECD of endoglin. This protein is a regulator of endothelial functions during angiogenesis. In addition to endoglin, it interacts with ALK5 and TβRII directly and facilitates recruitment of ALK1 into the receptor complex thereby promoting Smad1/Smad5-signaling[73].
In contrast to the signaling type I and type II receptors, the type III receptors betaglycan and endoglin do not possess a kinase activity in their short intracellular domains[3,53,41]. Nevertheless, respective domains have important functional implications for the interaction with the signaling receptors as described above. Although the C-termini of betaglycan and endoglin are very homologous to each other (Figure 2), several residues used as substrates by the signaling receptors are unique to endoglin[70].
Phosphorylation by a respective receptor serves as a switch to regulate the interaction with a certain receptor. Besides receptor interactions, other regulatory proteins have been identified which specifically bind to the C-terminal domain of FL-Eng. Using the two hybrid method, zyxin and zyxin-related protein-1 (ZRP-1) were found to specifically and exclusively, with respect to type III receptors, interact with FL-Eng[63,64]. Association with FL-Eng redirects these proteins from focal adhesions to actin stress fibers and leads to endoglin dependent inhibition of cell migration[63,64]. Another protein identified in the yeast system is the dynein light chain member Tctex2β. In addition to FL-Eng, Tctex2β also interacts with TβRII and betaglycan and it inhibits TGF-β signaling[65].
However, it has to be mentioned here that all these interaction screens have been solely performed using protein baits of the endoglin intracellular domain which have not been posttranslationally modified, e.g., phosphorylated. The interaction of at least zyxin with endoglin is stronger with the so called -ΔSMA deletion mutant that lacks the 3'-carboxyl-terminal protein part harbouring the PDZ-domain[64]. In line, removal of this domain causes an increase in endoglin phosphorylation[70] implying that this modification (phosphorylation) most likely modulates/regulates protein-protein interaction with the carboxyterminal domain (CD) of endoglin. Therefore, it is most likely that the group of proteins able to interact with endoglin is currently somewhat underestimated.
Based upon the high homology of the CD of endoglin and betaglycan, it is not surprising that both β-arrestin2 and GIPC were found to associate with both proteins[66,67,74,75]. The interaction of β-arrestin2 and endoglin is lost in the absence of threonine 650 and increases when co-expressed with TβRII and ALK1[66]. Whether the latter receptor regulates this interaction via phosphorylation is unclear since Thr650 is not a prominent ALK1 substrate[70]. On a functional level, β-arrestin2 causes endocytosis of the receptor complex, including endoglin, TβRII and ALK1, and impacts MAPK-signaling in an endoglin-dependent manner[66]. In contrast to β-arrestin-2, the C terminus of the G alpha interacting protein (GAIP)-interacting protein (GIPC) binds to the C terminus of endoglin in a manner that is restricted to the endoglin class I PDZ-motif. This leads to a stabilization of endoglin at the plasma membrane and changes in Smad1/Smad5 activation and endothelial cell migration (see below)[67]. Moreover, GIPC mediates the interaction of endoglin and phosphatidylinositol 3-kinase in a TGF-β1 dependent manner to regulate endothelial cell sprouting and capillary tube stability[76].
ENDOGLIN FUNCTION AND IMPACT ON TGF-β SIGNAL TRANSDUCTION
Endoglin is an accessory receptor for TGF-β impacting various aspects of its signaling and biological functions. Special features for the full length, soluble and short forms of endoglin have been reported. In the following, we provide a brief overview about TGF-β signaling and the impact of the different endoglin protein variants. Functional aspects of FL-endoglin are summarized in Figure 5.
Figure 5.
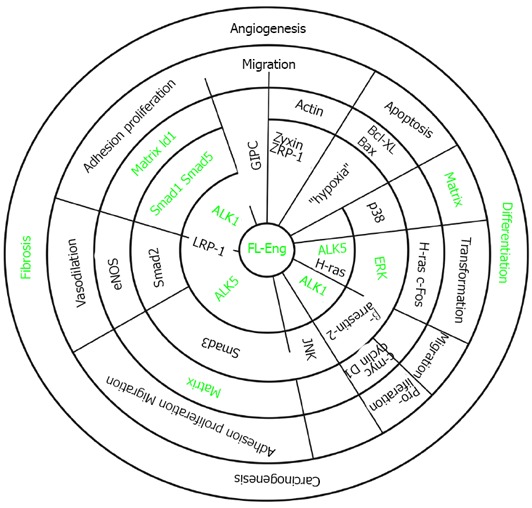
Association of endoglin with different signaling cascades. The concentrical circles display the hierarchy of signaling. Signaling starts at the membrane with receptors and adaptors (inner two circles). The next circle represents activated intermediates and adaptors. Thereafter, target genes are indicated. These are involved in shaping a cellular response which is part of a complex process (last circle). Partially open radial lines indicate that the corresponding molecules interact or interaction of molecules is mediated by the protein displayed on the radial line (LRP-1). The green font indicates items addressed in liver cells which are modulated by endoglin.
Brief overview of TGF-β signaling
Signaling by ligands of the TGF-β superfamily is initiated by binding of the ligand to a heterooligomeric membrane receptor complex. Binding of TGF-β1 is mediated by a homodimer of the TGF-β type II receptor which in turn recruits and phosphorylates a type I receptor (ALK5 or ALK1) homodimer into the complex. After ligand binding, the receptor complexes are internalized in general via two different pathways. Endocytosis mediated by clathrin-coated vesicles, enriched for Smad anchor for receptor activation (SARA), leads to active signaling. Depending on the type I receptor involved, the signal is propagated to two different Smad protein subfamilies, with the specificities of ALK5 phosphorylating Smad2/Smad3 or ALK1 in triggering phosphorylation of Smad1/Smad5. Phosphorylated Smads bind to the common Smad4, translocate into the nucleus and regulate transcription of target genes. Of these, the I-Smads, i.e., Smad6 and Smad7, are important regulators since they are direct target genes and shut off the signaling cascade at diverse points in a negative feedback loop. If internalization occurs via the lipid-rafts-caveolae-1, the receptors are bound to I-Smad/Smurf complexes targeting the receptor for ubiquitination and degradation[77].
Since this simple “core” of TGF-β signaling is involved in the regulation of a wide array of different target genes and control of diverse cellular responses, cells are endowed with a plethora of switches to adjust this cascade for their needs. Such cell type specific regulators for example are the type III receptors, i.e., betaglycan and endoglin, which are engaged in TGF-β receptor-complex formation and modulation of downstream signaling.
In the liver and especially in HSC, it has been assumed that the key operating TGF-β1 pathway is the ALK5/Smad3 branche that regulates proliferation, activation and profibrogenic responses of these cells. However, it has been anticipated that other signaling modalities like the ALK1/ALK5/Smad1/Smad5/Id1 axis is also engaged by TGF-β1 in regulating HSC physiology under normal and pathological conditions[40,78,79].
Impact of full length endoglin on TGF-β1-signaling
Analysis regarding the role of endoglin in signaling was primarily based on TGF-β-signaling and Smad-activation in monocytes and myoblasts[51,80]. Since it is known that endoglin is the candidate gene affected in HHT-1, detailed experimental work has been done using different endothelial cells[7,81]. So far the functional data regarding the involvement of endoglin in HSC are rather sparse. Endoglin is expressed in quiescent HSC and transdifferentiated myofibroblasts (MFB) and is transiently upregulated during cellular activation[40,41]. Upregulation of endoglin during activation/differentiation of cells is also seen in endothelial cells and monocytes[82,83]. Similar to other cell types, endoglin is not only affecting TGF-β1-signaling but is itself regulated by this ligand on the transcriptional level, most likely involving the Sp1 transcription factor[40,84-86]. As a mutual prerequisite, endoglin is membrane localized and interacts with and is phosphorylated by TβRII in HSC[41,40]. Overexpression of endoglin causes an increased phosphorylation of Smad1/Smad5 in HSC of rat and mouse origin[40,79]. In line with the HSC data, it was previously found that endoglin enhances ALK1/Smad1/5 signaling in endothelial cells and other cell types[87], leading to increased proliferation and migration (characteristics of the activation phase of angiogenesis), responses which are negatively affected upon endoglin reduction[72,88,89]. However, other laboratories claimed that endoglin causes reduced activation of ALK1/Smad1/5 as well as reduced migration and proliferation[90] or even having no impact on Smad-signaling at all[66]. These differences might be explained in part by the experimental set up (method used to modulate the endoglin expression, i.e., siRNA vs. knockout, concentration of the ligand, time scale of stimulation, cell type analyzed) and by the expression level of the two corresponding type I receptors, i.e., ALK1 and ALK5, both of which are expressed in HSC[78]. On the other hand, ALK5/Smad3 signaling that inhibits proliferation and migration (characteristics of the resolution phase of angiogenesis) is blocked by endoglin[67,88,91]. Interestingly, in contrast to ALK5/Smad3 which is downregulated, the signaling via ALK5/Smad2 leading to increased eNOS expression/activity is promoted in endothelial cells[56,92]. This effect is in part due to a stabilization of the Smad2 protein[92].
Although collagen type I expression is reduced, the overexpression of endoglin has no significant impact on ALK5/Smad3/Smad2 activation in mouse and rat HSC cell lines[40,79]. An inhibitory role of endoglin in collagen type I expression has been well documented in diverse kinds of cells, including mesangial cells, fibroblast of different origins and myoblasts[87,93-95] and was attributed to a reduced Smad3 activation[87,94]. A contribution of MAPK in the endoglin dependent modulation of collagen expression and Smad3 phosphorylation was postulated for JNK1 and ERK1/2[94,96].
In HSC, endoglin causes an increase in TGF-β1 dependent ERK1/2 activation[79]. A positive effect of endoglin on ERK1/2 activation was also observed in human T cells upon crosslinking of endoglin[97]. In line with an enhancement of ERK1/2 phosphorylation, TGF-β1 mediated expression of the connective tissue growth factor (CTGF) is promoted by endoglin in HSC[79]. There are several other reports showing an ERK1/2 dependent expression of CTGF, once more underscoring these results[98,99]. Nevertheless, the activation of ERK1/2 and increased expression of CTGF by endoglin is most likely cell type specific. In endothelial cells and epidermal cells it was shown that endoglin, in association with β-arrestin2, leads to suppression of ERK1/2 activation and a change in the cellular distribution[66,100]. On the contrary, in myoblasts in which TGF-β1 and endoglin have only a minor effect on ERK1/2 activation, CTGF is reduced in the presence of endoglin[87,95]. A negative impact of endoglin on CTGF expression was also found in scleroderma fibroblasts by some groups[39,101]. However, in a subset of scleroderma fibroblasts it was shown that the TGF-β1/ALK1/Smad1 pathway mediates fibrogenic responses, e.g., collagen I and CTGF expression, and that endoglin promotes this ALK1 pathway[102,103]. Finally, it was shown that ERK1/2 and Smad1 activation are functionally linked[102]. If endoglin-dependent up-regulation of ERK1/2 phosphorylation in HSC is directly linked to Smad1 activation and CTGF expression, and if ALK1 is involved in these responses is currently under investigation. Moreover, if the co-expressed betaglycan is involved in the up-regulation of CTGF is actually only speculative[101]. In addition, the basis of the forced expression of α-smooth muscle actin (α-SMA) in endoglin overexpressing cells needs to be analyzed[40,79]. One comprehensible option is a direct promoting effect on TGF-β1 signaling mediating α-SMA expression, which was shown to rely not exclusively on Smad3[104], or alternatively endoglin may cause a general shift in the transdifferentiation process leading finally to up-regulation of α-SMA.
Role of short (S-) endoglin on TGF-β1-signaling
Similar to FL-Eng, the S-Eng splice variant, although missing a large part of the C-terminal tail, binds to TGF-β1[3] and interacts with the signaling type II receptor[40] and both type I receptors (ALK5 and ALK1)[44]. FL-Eng was shown to be phosphorylated at serine residues by TβRII receptor[70] that fortuitously can be detected by a phospho-specific NF-κB antibody[105]. TβRII-mediated phosphorylation of both isoforms of rat endoglin could be detected in HSC using this antibody[40], implying a functional association of endoglin and the TGF-β-signaling receptors in HSC. Both splice variants are co-expressed in endothelial cells and HSC and can form heteromeric L-/S-Endoglin dimers[40,43]. Nevertheless, S-Eng is unable to substitute for FL-Eng since animals that carry an S-Eng transgene on an Eng null background are not viable, implying that S-Eng alone is inappropriate to rescue the lethal phenotype[43]. Using the afore mentioned S-Eng overexpressing animals in a model for tumor angiogenesis and metastatic infiltration by injecting Lewis lung carcinoma (3LL) cells, it was found that tumor growth is retarded when compared to control mice[43]. Even more, in a model of chemically induced skin tumors, overexpression of S-Eng in the vascular endothelium reduces benign tumor formation[43]. Nevertheless, functional data obtained in the rat system for the specific S-Eng variant yielded similar results when compared to FL-Eng[40]. Whether these results can be transferred to the mouse or human system is questionable due to the completely different C-termini.
Soluble endoglin: more than just a disease marker
As described above, endoglin can be shedded by MT1-MMP (MMP-14) from the cell surface to generate a soluble extracellular domain (sol-Eng) which reduces spontaneous and VEGF-induced endothelial sprouting[59]. In addition, the occurrence of sol-Eng has been observed in the serum/plasma of patients suffering from diverse tumors[106]. In pre-eclamptic women, the elevation sol-Eng precedes the onset of the disease, correlates with the severity of the disease and therefore its detection is of prognostic value[107]. Increased serum levels of sol-Eng have been found in cystic fibrosis associated liver disease (CFLD) patients, with the highest levels in patients suffering from HCV coupled with cirrhosis[28]. Significantly elevated sol-Eng levels are also observed in patients with hepatocellular carcinoma [Hepatocellular carcinoma (HCC)] combined with cirrhosis[108]. However, the role of sol-Eng in TGF-β1 signaling is presently controversial. Initially it was shown that the soluble domain is able to reduce TGF-β1-mediated reporter-gene activity and eNOS activation in endothelial cells[14]. In line with a ligand sequestering function, complexes of sol-Eng and TGF-β1 have been detected in serum of breast cancer patients using ELISA and co-immunoprecipitation[58]. Nevertheless, although part of the TGF-β1 ligand binding complex, a direct binding of TGF-β1 to endoglin is questionable[109,110]. If the signaling receptor type I and type II are present/co-expressed, endoglin can be precipitated together with labelled ligand. If endoglin on the other hand is overexpressed in cells lacking type I and type II receptor, there is no binding of TGF-β1 to endoglin[110]. The increase of the sol-Eng concentration in pre-eclamptic women and a few studies with a focus on sol-Eng function, using overexpression systems and luciferase assays, suggest that sol-Eng indeed has a functional role in TGF-β1 signaling[14,59]. In addition, we could show by co-immunoprecipitation that heterologous expressed sol-Eng is able to bind to TGF-β1 directly (SKM unpublished data) but experimental data suggest that it is unlikely for soluble endoglin to simply interfere with TGF-β1 signaling by competing with membrane bound type II receptor for TGF-β1. Using a BIACore facility, the measured dissociation constants are 5 pM for TβRII/TGF-β1[111] and in the micromolar range for sol-Eng/TGF-β1[112], underscoring the higher affinity of TβRII for TGF-β1 compared to the soluble endoglin counterpart. On the other hand, Van Le et al found that CHO-overexpressed and purified soluble endoglin increased TGF-β1 mediated p3TP-lux activity in U937 monocytic cells[55] in which L-endoglin was shown to antagonize several TGF-β1-responses[80]. Nevertheless, direct ligand binding and functional mechanisms used by sol-Eng to affect cellular responses have to be analyzed in more detail in the future. There are currently no data focussing on functional aspects of sol-Eng, especially in the liver.
ENDOGLIN IN DISEASE
As outlined above, mutations that affect human endoglin function are inherited as autosomal dominant disorders and may cause AVM in different organs, including brain, lung and liver (Figure 6). In the following paragraphs we will highlight the pathogenesis of several of these disorders and associated diseases and give an overview about the important role of endoglin dysfunction in the pathology of liver fibrosis.
Figure 6.
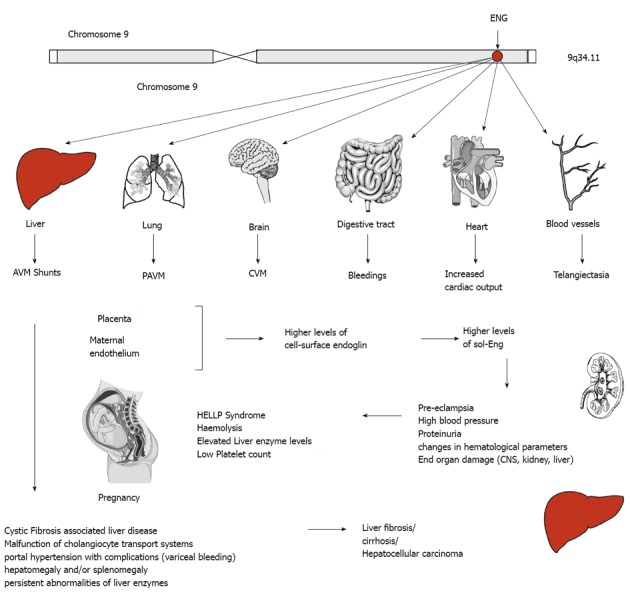
Endoglin and disease. The human endoglin gene (ENG) is located on the long arm of human chromosome 9. Mutations are inherited in an autosomal dominant manner and affect several organs. In liver, abnormal connection formed between blood vessels, arteriovenous malformations (AVM), malfunction of the cholangiocyte transport system gives rise to liver damage indicated by portal hypertension, persistent abnormalities of liver enzymes, hepatomegaly and/or splenomegaly, fibrosis, cirrhosis or even hepatocellular carcinoma. Intrahepatic connection between arteries and veins results in a large amount of blood bypasses for which the heart compensates by increasing the cardiac output resulting on long term in heart insufficiency. Similar arteriovenous (pulmonary AVM, cerebral AVM) are found in lung and brain. In the digestive tract bleedings occur and telangiectasias of blood vessels are found on the skin of the hands, face and mouth. During pregnancy, the placenta and the maternal endothelium produce higher levels of cell-surface endoglin that is shedded and leads to higher systemic concentration of soluble endoglin (sol-Eng) that leads to an imbalance of the antiangiogenic factors resulting in life-threatening obstetric complication (e.g., pre-eclampsia, HELLP syndrome).
Hereditary hemorrhagic telangiectasia
Hereditary hemorrhagic telangiectasia (HHT, Osler-Weber-Rendu syndrome) is an autosomal dominant inherited vascular disorder with a variety of clinical manifestations. Common symptoms of this disease occur due to the forming of AVM in small and large blood vessels. This leads to epistaxis, gastrointestinal bleeding and microcytic anemia due to iron deficiency, along with characteristic mucocutaneous telangiectasia[113]. AVM are found in pulmonary, hepatic and cerebral vascular tissue (Figure 6). The diagnosis of HHT is based on these clinical features, which are summarized in consensus criteria known as the “Curaçao criteria”[114]. Rupture of AVM contributes to significant morbidity.
Mutations in at least five genes result in manifestation of hereditary hemorrhagic telangiectasia. However, about 85% of the cases develop due to mutations of the ENG gene (coding for endoglin) and ACVRL1 (activin A receptor type II-like 1 kinase 1, ALK1)[115]. This disease is usually autosomal dominantly inherited, varying in penetrance and expression. Juvenile Polyposis/Hereditary Hemorrhagic Telangiectasia (JPHT) is a rare juvenile form of HHT which is associated with polyposis and occurs due to mutations in the MADH4 gene coding for Smad4[116]. In gene linkage analyses, two other loci have been shown to be in a disequilibrium with HHT symptoms; one on chromosome 5, defining HHT-3[35], the other on chromosome 7[13], defining HHT-4. However specific genes on these chromosomes involved in disease formation remain to be identified. Mice deficient for endoglin or ALK1 expression show clinical features of HHT[117]. Eng knockout (null) mice are embryonically lethal, dying at day 10.5 p.c. due to impaired extraembryonic vascular development and several cardiac defects (see below). Heterozygous animals show clinical symptoms of HHT-1 with variable penetrance. Human patients with HHT-1 exhibit less endoglin expression in peripheral blood monocytes and newborn umbilical vein endothelial cells[118].
To prevent fatal clinical events like stroke, high-output heart failure, pulmonary hypertension and hemorrhage, the embolization of visceral AVM is a valuable course of treatment. Furthermore, symptomatical treatment approaches with antiangiogenic or antihormonal agents have been investigated. In some patients, the use of antiangiogenic therapies known from cancer therapy, such as thalidomide[119], lenalidomide[120] and bevacizumab[121], reduces the incidence of nasal and gastrointestinal bleeding. The β-receptor blocker propanolol, usually used for prophylaxis of esophageal variceal bleeding in patients with liver cirrhosis or the treatment in infantile haemangiomas, was able to decrease cellular migration and tube formation, concomitantly with reduced RNA and protein levels of ENG and ALK1 in cell culture[122]. Other studies showed that tamoxifen, an estrogen receptor antagonist, and the selective estrogen receptor modulator, raloxifene, can reduce episodes of epistaxis and transfusion requirements in patients suffering from nasal vascular malformations[123,124]. However, limited controlled studies, severe side effects of those drugs and the need for life long treatment limits the applicability for most patients.
Pre-eclampsia
Pre-eclampsia is a disease of high incidence (about 3%) in pregnant women with an onset after 20 wk of gestation. It complicates pregnancy and can lead to death of mother and baby. The disease is characterized by new-onset hypertension (140 mmHg or diastolic blood pressure 90 mmHg) and proteinuria (excess of protein in the urine of at least 0.3 g of protein/d)[125]. Eclampsia is characterized by additionally occurring grand mal seizures[126]. Typical complications for the pregnant woman are the involvement of the central nervous system, acute renal or liver failure, and changes in hematological parameters. Women with pre-eclampsia are prone to higher lifetime cardiovascular morbidity, including hypertension and ischemic heart disease. Effects on the fetus can be severe and include prematurity, fetal growth restriction, oligohydramnios and placental abruption. A family history of pre-eclampsia, advanced maternal age, obesity or pregestational diabetes increases the mothers risk to develop this condition[127].
The pathophysiology of pre-eclampsia is still poorly understood. Prior to the development of clinical symptoms, cells migrating to the placenta lack the expression of endothelial surface adhesion markers. This leads to incomplete invasion of maternal arteries by the developing trophoblast, resulting in placental ischemia and the release of antiangiogenic factors, including sol-Eng and soluble fms-like tyrosine kinase (sFlt1)[128]. Vascular endothelial growth factor (VEGF) and placental growth factor are antagonized by soluble fms-like tyrosine kinase-1 (sFlt1 or sVEGFR-1) and sol-Eng antagonizes TGF-β1 and TGF-β3 activity[129]. These effects on vascular homeostasis promote changes in placental circulation. Numerous studies show the effect of VEGF and TGF-β signaling pathways on circulation and angiogenesis. These pathways directly influence the development of pre-eclampsia. By regulating endothelial cell proliferation, migration, vascular permeability and secretion, VEGF-A is an important ligand for angiogenesis. It binds to two tyrosine kinase receptors, VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1). The soluble receptor VEGFR-1 (sFlt1) acts as an endogenous VEGF inhibitor. In patients with pre-eclampsia, sFlt1 is overexpressed in the maternal circulation[130], which corresponds to a decrease of VEGF and placental growth factor expression in the placenta of pre-eclampsia patients[131]. This leads to the development of major symptoms of the disease due to abnormal trapping of VEGFs. The role of sFlt1 is underlined by studies in which pregnant rats were treated with exogenous sFlt1, inducing severe pre-eclampsia. Immunoprecipitation of sFlt1 in cells derived from placental villous explants normalized their angiogenic responses[129].
In addition, the VEGF-signaling changes in the TGF-β signal transduction pathway promotes the development of pre-eclampsia. Placentas of pre-eclamptic women show increased levels of membrane-bound Eng and sol-Eng[14]. Hypoxia and oxidative stress seem to be important triggers for the release of sol-Eng, as shown in a study where oxysterol activation promoted MMP-14-mediated cleavage of sol-Eng in cells of trophoblast origin[132]. sol-Eng antagonizes TGF-β1 induced vasodilatation, leading to vascular hypertension[133-135]. The increase of systemic sol-Eng in pregnant women is a factor that prequels the onset of pre-eclampsia[106,136,137]. Modulating the TGF-β pathway, endoglin can, alone or together with sFlt1, induce pre-eclampsia symptoms in pregnant rats[14].
The pathogenesis of pre-eclampsia is defined by the imbalance of the anti-angiogenic factors, sFlt1 and sol-Eng, and the proangiogenic factors, placental growth factor, TGF-β and VEGF[138]. Current treatment concepts therefore include the use of antibodies and small molecules to sequester or limit synthesis of anti-angiogenic molecules. Improvement in blood pressure and renal function could be achieved after administration of exogenous VEGF in a preclinical model of pre-eclampsia, modulating the balance of angio- and anti-angiogenic factors[139]. Recently, a study using a dextran sulfate column to remove sFlt1 from the maternal circulation by extracorporeal apheresis showed a potential therapeutic approach for the treatment of pre-eclampsia[140]. Other studies using induction of hemoxygenase-1 with cobalt protoporphyrin in pre-eclamptic rats[141] and prevention of the release of sol-Eng by direct inhibition of MMP-14 showed promising results[142]. As mentioned before, any therapeutic approach must be safe for mother and fetus and should be evaluated by controlled studies. Currently these problems still limit any effective therapy.
HELLP Syndrome
The HELLP syndrome is a complex of maternal symptoms in pregnancy, including hemolysis, elevated liver enzymes and low platelet count. HELLP syndrome occurs in 0.2%-0.8% of pregnancies and is a serious threat for mother and child. 70%-80% of women expressing HELLP symptoms also suffer from pre-eclampsia[143]. As in pre-eclampsia, a previous HELLP pregnancy increases the risk of HELLP as well as pre-eclampsia in subsequent pregnancies, suggesting related pathogenetics. Anti-angiogenic factors play an important role in both symptom complexes. In comparison to pre-eclampsia, maternal blood levels of anti-angiogenic sFlt1 are similar, but HELLP shows higher sol-Eng levels[144]. The pathogenesis of symptoms defining HELLP is driven by those angiopathogenic mechanisms. Activated vascular endothelium leads to an inflammatory response, including coagulation and complement activation, increased white blood count and elevated levels of inflammatory cytokines such as TNF-α and von Willebrand factor, leading to clinical symptoms of disseminated coagulation in microvessels[144,145]. Activation of these inflammatory signaling cascades leads to hemolysis in response to microangiopathy, reduced liver blood flow with elevated liver enzymes and low platelet counts due to consumption of platelets by microvessel thrombosis (= HELLP).
Cystic fibrosis associated liver disease
Cystic fibrosis (CF, mucoviscidosis) is an autosomal recessive genetic disorder affecting lungs, pancreas, liver and intestine. A mutation in the gene for the protein cystic fibrosis transmembrane conductance regulator (CFTR) causes an abnormal transport of chloride and sodium across an epithelium, resulting in viscous secretions[146]. The most severe symptoms affect the lungs, often causing lung transplantation or death in those patients. Gastrointestinal symptoms due to thick mucus are common[147] and cystic fibrosis associated liver disease (CFLD) is often (30%) diagnosed, accounting for 2.5% of overall mortality, representing the third most common cause of death in these patients[148].
Rath et al[28] showed in a recent study that patients suffering of CFLD show elevated serum levels of TIMP-4 and endoglin. Expression levels correlate with hepatic staging, therefore allowing, together with transient elastography, to increase the sensitivity for the non-invasive diagnosis of CFLD in patients suffering from CF. High endoglin levels showed a significant association with the severity of liver injury, suggesting an active role for endoglin in the pathology of liver fibrosis.
Endoglin in liver fibrosis and HCC
Liver fibrosis and cirrhosis is the outcome of most types of chronic liver injury. The excessive accumulation of extracellular matrix (ECM) proteins promotes hepatic scarring and eventually leads to organ failure[149]. In the pathogenesis of liver fibrosis, TGF-β is the most potent fibrogenic cytokine. It induces fibrosis through multiple mechanisms, including direct activation of HSC, stimulation of ECM production, as well as prompting the synthesis of tissue inhibitors of matrixmetalloproteases (TIMPs) and thereby inhibiting ECM degradation[150]. Knock-out mice with deletions in components of the TGF-β signaling cascade (TGF-β1, SMAD3 and MMP13) develop less severe fibrosis[151]. TGF-β ligands and receptors form a complex signaling network, which can be modulated by endoglin and betaglycan (TGF-β type III receptor). By inhibiting ALK5-Smad2/3 and promoting ALK1-Smad1/5 signaling, endoglin can shift TGF-β downstream signals to pro-fibrogenic effects[40]. Presently, there is not much knowledge how the expression of the different endoglin isoforms and sol-Eng is regulated in diverse liver cell subpopulations but it was reported that the concentration of sol-Eng increases during hepatic fibrogenesis (see below). In previous studies, we could show that endoglin expression is increased in activated HSC in vitro and in murine models of liver injury (carbon tetrachloride application and bile duct ligation) in vivo[41]. HSC are the major source for ECM production in liver fibrosis. Endoglin overexpression leads to enhanced TGF-β-driven Smad1/5 phosphorylation and α-smooth muscle actin expression without affecting Smad2/3 signaling in these cells. By shifting TGF-β signaling from ALK5-Smad2/3 to ALK1-Smad1/5 pathway, endoglin exceeds a central role in TGF-β signal modulation and the development of liver fibrosis.
HCC develops most often (80%) in cirrhotic livers. Angiogenesis and irregular capillary distribution are a key feature for malignant lesions[152]. Blood vessels are needed to supply nutrients and oxygen to the growing tumors. Most malignant tumors as well as HCCs have developed efficient strategies to promote fast vessel growth. Angiogenesis is a highly regulated, complex process modulated by many intersecting pathways, including vascular endothelial growth factor (VEGF), TGF-β and endoglin[26], angiopoietins[153], Notch[154] and integrins[155]. Usually, pro-angiogenic and anti-angiogenic factors are tightly balanced. In contrast to physiological angiogenesis (i.e., in wound healing), tumor angiogenesis is not controlled by normal physiological inhibition, resulting in an imbalance of pro-angiogenic and anti-angiogenic factors. By modulating TGF-β signaling, endoglin plays a crucial role in angiogenesis and tumor growth and could be linked to HCC[108], as well as esophageal cancer[156], breast carcinoma[157], colorectal cancer[158] and tumor angiogenesis[44].
EXPRESSION OF ENDOGLIN IN ISOLATED LIVER CELLS AND LIVER TISSUE
Endoglin expression has been studied in many different tissues and diseases. It is highly expressed on proliferating vascular endothelial cells[159,160]. However, Meurer et al[40,41] showed that endoglin is expressed on HSC and activated MFB as well. By molecular cloning of endoglin cDNA, surface labeling, immunoprecipitation and immunocytochemistry experiments, it could be shown that endoglin plays a significant role in liver injury and fibrosis development[40,41]. Endoglin expression is differentially regulated at the plasma membrane of HSCs and in activated myofibroblasts (MFB)[40,41]. Endoglin expression is increased in transdifferentiating HSC and in two models of liver fibrosis but not in hepatocytes. Furthermore, endoglin is expressed in cultured portal fibroblasts, representing another important fibrogenic cell type in biliary types of liver disease. Transient overexpression of endoglin leads to significantly increased TGF-β1-driven Smad1/5 phosphorylation and α-smooth muscle actin expression, while Smad2 phosphorylation is not changed[40]. These results are in line with a study by Lebrin et al[88] which showed endoglin promoting TGF-β1/ALK1-Smad1/5 signaling in endothelial cells.
To further investigate the influence of endoglin on TGF-β signal transduction, we recently established and characterized a new mouse HSC line expressing collagen 1(I) promoter/enhancer driven green fluorescent protein (GFP). These cells, originating from quiescent HSC, show an activated MFB phenotype in culture and express low endogenous endoglin concentrations. By selective overexpression of endoglin in these cells, stimulation with TGF-β and PDGF, and specific inhibition of endoglin/ALK signaling with antagonists, the differential effect of endoglin on downstream Smad-signaling could be shown[77].
Because of the complexity of endoglin and TGF-β signaling pathways, it is important to investigate the modulation of TGF-β signal transduction in cells of different origin. For example, Velasco et al[87] showed the differential effects of endoglin isoforms in L6E9 myoblasts[85]. Because these cells have no endogenous endoglin expression, this cell line is an ideal tool to selectively express specific isoforms of endoglin and show a different and sometimes opposing effect of L- and S-Eng isoforms on downstream regulation of TGF-β-induced responses. While endoglin expression is well investigated in vascular endothelial cells, HHT and tumor angiogenesis, the role of endoglin in liver disease is poorly understood. Liver cell lines overexpressing endoglin or single members of the TGF-β pathway, as well as cells with low endogenous endoglin expression and specifically induced endoglin expression are needed to further dissect the functional roles of endoglin in liver injury and fibrosis.
ANIMAL MODELS IN UNDERSTANDING ENDOGLIN FUNCTION
Endoglin deficiency in humans has a strong phenotype and is responsible for many diseases, such as HHT, pre-eclampsia liver fibrosis and cancer. To study its impact on the pathogenesis of those diseases, murine endoglin knockout models were needed. Because a complete homozygous endoglin knockout is embryonically lethal, several alternative strategies were established. Endoglin plays an important role in angiogenesis; a complete endoglin deficiency has fatal consequences in the development of heart and major vessels. To study the role of endoglin in vivo and its impact on HHT-1, Arthur et al[161] established a mouse carrying a targeted nonsense mutation (deletion of exons 9-11) in the endoglin gene. These mice already showed that endoglin expression is critical for early vascular development. Embryos with two mutated endoglin genes die at day 10 - 10.5 post coitum (dpc) due to cardiac malformations and a failure to form mature blood vessels in the yolk sac. Homozygous endoglin knockout embryos generated by a deletion of 609 bp including exon 1 show a similar phenotype as mice lacking TGF-β1 and the TGF-β receptor II, suggesting that endoglin plays a crucial role in TGF-β signaling in early vascular development[162,163]. Li and co-workers reported that mice lacking functionally active endoglin by replacing the first two exons die from defective vascular development but do not show defective vasculogenesis, which is observed in mice lacking TGF-β1[163]. Loss of endoglin caused poor vascular smooth muscle cell (vSMC) development and arrested endothelial remodelling. Therefore, endoglin is required for the differential growth and sprouting of endothelial tubes and recruitment and differentiation of mesenchymal cells into vSMC and pericytes[164]. Both studies show slight differences in vascular embryonic development. Eng deficient mice generated by Li et al[163] die at day 11.5 dpc. While Arthur et al[161] used embryonic stem cells of 129/Ola origin, Li et al[163] generated endoglin knockout mice by targeting embryonic stem cells from 129/SVJ background. Those different approaches already suggest a strong impact of genetic background on murine models of Eng deficiency.
To overcome the problems of embryonic lethality and to study the effect of endoglin in disease, several groups have used alternative approaches to generate endoglin deficient mice. Allinson et al[164] for example generated a mouse in which the endoglin gene is flanked by loxP sites at exons 5 and 6. These mice show a normal phenotype comparable to wild type littermates. Using the Cre-loxP genetic recombination system and an appropriate Cre expressing mouse line, specific endoglin knockout mice can be created. To generate a null allele of the endoglin gene, the floxed construct was designed to allow a conditional deletion of exons 5 and 6, which would also lead to frameshift mutation in exon 7 before reaching a stop codon, resulting in a functional inactive endoglin[164].
Using this approach, two mouse models were generated expressing Cre in smooth muscle (SM22αcre) and endothelial cells (Tie2cre) to evaluate the role of endoglin in vascular smooth muscle and endothelial cells during angiogenesis[165]. In this study, endoglin null embryos show ectopic arterial expression of the venous specific marker COUPTFII (chicken ovalbumin upstream promoter transcription factor II). Normal expression of COUPTFII was restored after endoglin re-expression in endothelial cells. COUPTFII plays an important role in vascular development, including heart, blood vessels and smooth muscle cell differentiation. Endoglin induces changes in COUPTFII expression patterns and therefore can influence vSMC recruitment and differentiation in angiogenesis.
Other groups used heterozygous endoglin knockout mice to investigate the function of endoglin and avoid embryonic lethality. Bourdeau et al[166] developed a mouse model with a single copy of the endoglin gene and another mouse line with a homozygous deletion of the endoglin gene. As already observed by Arthur et al[161], mice lacking any functional endoglin die at day 10.0-10.5 dpc due to defects in vessel and heart development. Embryos show a normal angiogenesis and vessel formation until hemorrhage occurs in the yolk sac around 9.0-10.5 dpc. Heart development stopped at day 9.0 and the atrioventricular canal endocardium did not undergo mesenchymal transformation and cushion-tissue formation. Similar to the study published by Arthur et al[161], Bourdeau et al[166] used 129/Ola origin on C57BL/6 background. The heterozygous mouse displays a multiorgan vascular phenotype similar to the human HHT, which is often caused by endoglin haploinsufficiency. To evaluate the impact of the genetic background on endoglin deficiency, different Eng/null mouse strains were generated. The 129/Ola strain developed HHT symptoms at an earlier age and with greater severity than C57BL/6 mice. The F2 strain intercrosses between both strains showed an intermediate phenotype. As in humans, Eng deficiency shows variable penetrance. Of 171 mice observed in this study over a 12 mo period, 50 developed clinical signs of HHT. Disease prevalence was high in the 129/Ola strain (72%), intermediate in the intercrosses (36%), and low in C57BL/6 backcrosses (7%)[166].
Using the heterozygous Eng null mouse generated by Bordeau et al[166], another study showed that endoglin is required for paracrine TGF-β signaling between endothelial cells and adjacent smooth muscle cells to promote smooth muscle cell differentiation[167].
In primary cultures of endothelial cells generated from mice carrying only one functional Eng allele, a significantly reduced migration and proliferation along with increased collagen production, vascular endothelial growth factor (VEGF) secretion and decreased NO synthase expression was observed[168]. This again highlights the important role of endoglin in vascular pathology.
As outlined above, endoglin modulates both the ALK1 and ALK5 pathways. Park et al[169] generated an ALK1 conditional knockout mouse line. The specific deletion of ALK1 in vascular endothelial cells by an endothelial specific Cre was lethal through massive hemorrhage in the lungs. ALK1 deficient mice showed heavy pulmonary vascular malformations mimicking all pathological features of HHT-2, such as dilation of vessel lumen, thinning of vascular walls, loss of capillaries, development of excessive tortuous vessels, and AVM[169].
Dolinsec et al[170] used another approach to investigate endoglin deficiency in murine models without affecting embryonical vascular development[170]. By applying siRNA against endoglin to human and murine endothelial cells (HMEC-1, 2H11) in vitro and in TS/A mammary adenocarcinoma growing in BALB/c mice, they evaluated the therapeutic potential of siRNA in cancer treatment. In vitro, the transfection resulted in reduced levels of endoglin mRNA and protein, leading to a 60% decrease of endothelial cell proliferation. In vivo silencing of endoglin expression showed lower endoglin mRNA levels and a decreased number of tumor blood vessels resulting in significantly reduced TS/A tumor growth. The study demonstrated that siRNA molecules against endoglin have a good anti-angiogenic therapeutic potential[171].
The endoglin gene gives rise to two different isoforms resulting from differential splicing, i.e., S- and L-Eng (for details see above). Pérez-Gómez et al[43] investigated the role of S-Eng in vivo using a mouse with ICAM-2 driven overexpression of human S-Eng on the vascular endothelium. Interestingly, breeding these mice to endoglin deficient mice did not rescue the embryogenic lethal phenotype. Furthermore, this study investigates the impact of S-Eng on carcinogenesis. Therefore, Lewis lung carcinoma cells were transplanted into mice expressing S-Eng. Carcinoma cells in these mice showed reduced tumor growth and less neovascularization. Additionally, benign papilloma formation was reduced significantly in respective S-Eng positive mice. These results show that S-Eng has anti-angiogenic properties in cancer development, showing new potential approaches for tumor therapy[43].
DIAGNOSTIC VALUE OF ENDOGLIN IN LIVER-ASSOCIATED DISEASES
Genetic testing
HHT is phenotypically heterogeneous both between affected families and amongst members of the same family in regard to penetrance and age of disease onset. There are hundreds of different mutations in the human ENG gene known that affect proper gene function. Although HHT is most common in Caucasians, disease causing mutations with ethnic-related differences also occur in Asians, Africans and Middle Eastern[171]. The overall incidence of HHT in North America is more frequent than initially estimated and ranges between 1:5000 and 1:10000[172], while the frequency in Europe varies between 1:2500 to 1:40000[173-175]. In a cohort of the northern part of Japan, the prevalence of HHT in the population was estimated to be 1:8000[176], demonstrating that HHT is more common among Asians than often assumed.
HHT is a dominantly inherited autosomal disorder and genetic testing of individuals with a known family history is generally performed for disease confirmation (Figure 7). In addition, pre-symptomatic screening of relatives of patients with a positive molecular diagnosis and in patients with suggestive (but not confirmatory) clinical features of HHT is well established[177].
Figure 7.
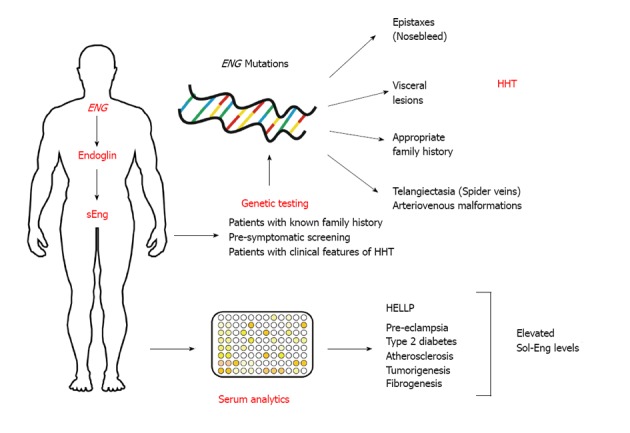
Endoglin in diagnostics. Several distinct mutations in the endoglin gene (ENG) give rise to hereditary hemorrhagic telangiectasia (HHT) that is mainly characterized by epistaxes (nosebleed), various visceral lesions, telangiectasia (spider veins) and arteriovenous malformations. Patients often show an appropriate family history. The clinical diagnosis “HHT” is made if three of the four classical signs (i.e., epistaxes, visceral lesions, telangiectasia and family history) occur. Elevated levels of soluble endoglin have been reported in patients suffering from hemolysis, elevated liver enzymes and low platelets syndrome (HELLP), pre-eclampsia, type 2 diabetes, atherosclerosis, tumorgenesis in several organs, and fibrogenesis.
At the molecular level, there is a large spectrum of different gene mutations that influence the expression, integrity and stability of the endoglin protein. Missense (nonsynonymous) mutations introducing different aa, nonsense mutations introducing premature stop codons, splice-site mutations that affect consensus splice donor sites and provoke exon skipping, frame shift and in frame deletions resulting in proteins with markedly different sizes, and several intronic mutations are rather common and show an ethnic and regional distribution[7-10,178-180]. However, the penetrance of the different mutations and gene variations are rather different and subtle genotype-phenotype correlations in HHT-1 have been reported, revealing that truncating mutations in ENG are associated with more affected organs and more severe hemorrhage than ENG missense mutations[13]. Pulse-chase experimentation and overexpression studies have further shown that several endoglin gene mutations form proteins that are only barely detectable, do not form heterodimers with normal endoglin, and are further unable to interfere with endoglin trafficking to the cell surface and remain intracellular as a precursor form[12,181]. On the contrary, another study that investigated six different missense and two truncation mutations have shown that not all mutants are unable to dimerize with normal endoglin, suggesting that haploinsufficiency and dominant-negative protein interactions both can cause HHT-1[12,182]. No homozygotes that carry two abnormal copies of the ENG gene have been reported so far, suggesting that this constellation is not compatible with life[183]. Likewise, mice lacking both copies of the ENG gene die at gestational day 10.0-10.5 due to defects in vessel and heart development[161].
However, there are four other genetic types of HHT identified that are not associated with alterations in the ENG gene. It is essential to know that there are likely to be differences in the normal requirements for the individual disease-causing genes in different vascular beds and cell types that, when affected by mutation, result in somewhat diverse clinical features and symptoms[183]. The onset of epistaxis for example was found to have an earlier onset in patients with HHT-1 than those with HHT-2 and AVM of the brain and lungs were more common in respective patients, while hepatic and spinal AVM were noticed at a lower frequency in patients with HHT-2[13,178,184,185]. Based on all these findings, several guidelines were proposed in which the ENG gene should be first targeted for mutational screening when large visceral AVM in the lungs in patients younger than 45 years occur[185]. However, based on the fact that all 15 exons and their non-coding introns can be easily sequenced, it is self-evident that these molecular diagnostic tests have refined and supplemented the criteria that were first proposed for clinical diagnosis of HHT[114].
Serum measurements
Based on the finding that the serum or plasma concentration of sol-Eng is increased dramatically in several disease conditions, its predictive value for the outcome of various diseases is presently intensively discussed and a large variety of commercially available ELISA test systems that allow reliable and accurate detection of endoglin in biological fluids have been established by many companies. It was shown that serum sol-Eng that plays a major role as an anti-angiogenic factor increases two- and three-fold in preterm and term pregnancy compared to non-pregnant controls and further dramatically increases two to three months before the onset of pre-eclampsia and in patients with HELLP syndrome, suggesting that sol-Eng alone or in combination with other variables is usable as a biomarker with a high predictive value in pregnancy complications[14,106,186,187]. Other studies demonstrated that plasma sol-Eng levels are significant higher in patients with diabetes than in healthy control subjects and that the duration of diabetes is an independent predictor of plasma sol-Eng increase[17]. The measurement of sol-Eng also has predictive value for the progression of the atherosclerotic process and correlates well with the expression of eNOS in endothelium, repair of the vessel wall, plaque neoangiogenesis, production of collagen and stabilization of atherosclerotic lesions[19]. As an indicator of endothelial dysfunction, the measurement of sol-Eng was proposed to monitor the therapy efficacy during extracorporeal LDL-cholesterol elimination therapy for familial hypercholesterolemia[18]. Since endoglin expression was shown to be extremely relevant for cancer formation[159], it is not surprising that sol-Eng is a potential angiogenic marker to indicate and predict diseases associated with metastases[32,188-190]. Patients suffering from Alzheimer’s disease were also found to have elevated levels of sol-Eng combined with decreased levels of TGF-β, possibly indicating impairment of cerebral circulation that is associated with this neurodegenerative process[24]. Of course, the wide expression pattern of endoglin that encompasses endothelial cells, subsets of bone marrow cells, activated macrophages, fibroblasts, chondrocytes, smooth muscle cells and pro-fibrogenic cells (e.g., HSC) as well as its linkage with the TGF-β signaling pathways has further offered several new avenues in which sol-Eng measurements might be beneficial. In regards to liver, it is well established that intrahepatic and circulating levels of endoglin are elevated in patients suffering from chronic hepatitis C infection, liver cirrhosis and carcinoma. In addition, there is a correlation of histological and serum markers of hepatic fibrosis and endoglin is abundantly expressed in hepatic sinusoidal endothelium of non-tumor tissues with cirrhosis[108,191,192]. Increased endoglin expression was recently also documented by proteomic profiling in patients suffering from cystic fibrosis associated liver disease[28]. Likewise, high circulating endoglin concentrations are correlated with a poor outcome for biliary atresia that represents a chronic progressive disorder of the extrahepatic and intrahepatic biliary system[27]. Therefore, there is no doubt that these measurements enrich the panel of available diagnostic options to identify proliferative disorders, including organ diseases that are associated with fibrogenesis.
CONCLUSION
Endoglin is found on many cell surfaces and plays a crucial role in TGF-β signaling. It forms homodimers and consists of a large extracellular domain, a hydrophobic transmembrane domain and a short cytoplasmic tail. This receptor binds to a large variety of extra- and intracellular binding partners and modulates numerous cellular properties, including morphology, migration, endocytic vesicular transport, microtubular structures and functionality of focal adhesion proteins. Several hundred independent ENG gene mutations result in HHT that is associated with various vascular lesions, mainly on the face, lips, hands and gastrointestinal mucosa. Recent work has demonstrated that endoglin expression is also altered during ongoing hepatic fibrogenesis. The unravelling of the underlying pathways that are associated with alterations in endoglin expression will be of fundamental interest, not only for establishment of potential new therapeutic options for HHT treatment, but might allow re-establishing the activities of Smad2/3 and Smad1/5/8 that are both part of TGF-β homeostasis and pathologically altered in ongoing and established organ fibrosis.
Footnotes
Supported by Deutsche Forschungsgemeinschaft SFB/TRR57, P13 and P26; A grant from the Interdisciplinary Centre for Clinical Research within the faculty of Medicine at the RWTH Aachen University IZKF Aachen, Project E6-11, to Weiskirchen R
P- Reviewers: Antonio L, Fabre JM, Gao BL S- Editor: Qi Y L- Editor: Roemmele A E- Editor: Lu YJ
References
- 1.Gougos A, Letarte M. Identification of a human endothelial cell antigen with monoclonal antibody 44G4 produced against a pre-B leukemic cell line. J Immunol. 1988;141:1925–1933. [PubMed] [Google Scholar]
- 2.Gougos A, Letarte M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem. 1990;265:8361–8364. [PubMed] [Google Scholar]
- 3.Bellón T, Corbí A, Lastres P, Calés C, Cebrián M, Vera S, Cheifetz S, Massague J, Letarte M, Bernabéu C. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur J Immunol. 1993;23:2340–2345. doi: 10.1002/eji.1830230943. [DOI] [PubMed] [Google Scholar]
- 4.Fernández-Ruiz E, St-Jacques S, Bellón T, Letarte M, Bernabéu C. Assignment of the human endoglin gene (END) to 9q34--& gt; qter. Cytogenet Cell Genet. 1993;64:204–207. doi: 10.1159/000133576. [DOI] [PubMed] [Google Scholar]
- 5.Pilz A, Woodward K, Povey S, Abbott C. Comparative mapping of 50 human chromosome 9 loci in the laboratory mouse. Genomics. 1995;25:139–149. doi: 10.1016/0888-7543(95)80119-7. [DOI] [PubMed] [Google Scholar]
- 6.Qureshi ST, Gros P, Letarte M, Malo D. The murine endoglin gene (Eng) maps to chromosome 2. Genomics. 1995;26:165–166. doi: 10.1016/0888-7543(95)80099-8. [DOI] [PubMed] [Google Scholar]
- 7.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 8.Lesca G, Plauchu H, Coulet F, Lefebvre S, Plessis G, Odent S, Rivière S, Leheup B, Goizet C, Carette MF, et al. Molecular screening of ALK1/ACVRL1 and ENG genes in hereditary hemorrhagic telangiectasia in France. Hum Mutat. 2004;23:289–299. doi: 10.1002/humu.20017. [DOI] [PubMed] [Google Scholar]
- 9.Gallione CJ, Scheessele EA, Reinhardt D, Duits AJ, Berg JN, Westermann CJ, Marchuk DA. Two common endoglin mutations in families with hereditary hemorrhagic telangiectasia in the Netherlands Antilles: evidence for a founder effect. Hum. Genet. 2000;107:40–44. doi: 10.1007/s004390000326. [DOI] [PubMed] [Google Scholar]
- 10.Cymerman U, Vera S, Karabegovic A, Abdalla S, Letarte M. Characterization of 17 novel endoglin mutations associated with hereditary hemorrhagic telangiectasia. Hum Mutat. 2003;21:482–492. doi: 10.1002/humu.10203. [DOI] [PubMed] [Google Scholar]
- 11.Pece N, Vera S, Cymerman U, White RI, Wrana JL, Letarte M. Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest. 1997;100:2568–2579. doi: 10.1172/JCI119800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pece-Barbara N, Cymerman U, Vera S, Marchuk DA, Letarte M. Expression analysis of four endoglin missense mutations suggests that haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Hum Mol Genet. 1999;8:2171–2181. doi: 10.1093/hmg/8.12.2171. [DOI] [PubMed] [Google Scholar]
- 13.Bayrak-Toydemir P, McDonald J, Markewitz B, Lewin S, Miller F, Chou LS, Gedge F, Tang W, Coon H, Mao R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A. 2006;140:463–470. doi: 10.1002/ajmg.a.31101. [DOI] [PubMed] [Google Scholar]
- 14.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 15.Luft FC. Soluble endoglin (sEng) joins the soluble fms-like tyrosine kinase (sFlt) receptor as a pre-eclampsia molecule. Nephrol Dial Transplant. 2006;21:3052–3054. doi: 10.1093/ndt/gfl439. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Hao Q, Kim H, Su H, Letarte M, Karumanchi SA, Lawton MT, Barbaro NM, Yang GY, Young WL. Soluble endoglin modulates aberrant cerebral vascular remodeling. Ann Neurol. 2009;66:19–27. doi: 10.1002/ana.21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Motawi TK, Rizk SM, Ibrahim IA, El-Emady YF. Alterations in circulating angiogenic and anti-angiogenic factors in type 2 diabetic patients with neuropathy. Cell Biochem Funct. 2013;32:155–163. doi: 10.1002/cbf.2987. [DOI] [PubMed] [Google Scholar]
- 18.Blaha M, Cermanova M, Blaha V, Jarolim P, Andrys C, Blazek M, Maly J, Smolej L, Zajic J, Masin V, et al. Elevated serum soluble endoglin (sCD105) decreased during extracorporeal elimination therapy for familial hypercholesterolemia. Atherosclerosis. 2008;197:264–270. doi: 10.1016/j.atherosclerosis.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 19.Nachtigal P, Zemankova Vecerova L, Rathouska J, Strasky Z. The role of endoglin in atherosclerosis. Atherosclerosis. 2012;224:4–11. doi: 10.1016/j.atherosclerosis.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Bassyouni IH, El-Shazly R, Azkalany GS, Zakaria A, Bassyouni RH. Clinical significance of soluble-endoglin levels in systemic lupus erythematosus: possible association with anti-phospholipid syndrome. Lupus. 2012;21:1565–1570. doi: 10.1177/0961203312460115. [DOI] [PubMed] [Google Scholar]
- 21.Kopczyńska E, Dancewicz M, Kowalewski J, Makarewicz R, Kardymowicz H, Kaczmarczyk A, Tyrakowski T. Influence of surgical resection on plasma endoglin (CD105) level in non-small cell lung cancer patients. Exp Oncol. 2012;34:53–56. [PubMed] [Google Scholar]
- 22.Blázquez-Medela AM, García-Ortiz L, Gómez-Marcos MA, Recio-Rodríguez JI, Sánchez-Rodríguez A, López-Novoa JM, Martínez-Salgado C. Increased plasma soluble endoglin levels as an indicator of cardiovascular alterations in hypertensive and diabetic patients. BMC Med. 2010;8:86. doi: 10.1186/1741-7015-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wipff J, Avouac J, Borderie D, Zerkak D, Lemarechal H, Kahan A, Boileau C, Allanore Y. Disturbed angiogenesis in systemic sclerosis: high levels of soluble endoglin. Rheumatology (Oxford) 2008;47:972–975. doi: 10.1093/rheumatology/ken100. [DOI] [PubMed] [Google Scholar]
- 24.Juraskova B, Andrys C, Holmerova I, Solichova D, Hrnciarikova D, Vankova H, Vasatko T, Krejsek J. Transforming growth factor beta and soluble endoglin in the healthy senior and in Alzheimer’s disease patients. J Nutr Health Aging. 2010;14:758–761. doi: 10.1007/s12603-010-0325-1. [DOI] [PubMed] [Google Scholar]
- 25.Davidson B, Stavnes HT, Førsund M, Berner A, Staff AC. CD105 (Endoglin) expression in breast carcinoma effusions is a marker of poor survival. Breast. 2010;19:493–498. doi: 10.1016/j.breast.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 26.Bellone G, Gramigni C, Vizio B, Mauri FA, Prati A, Solerio D, Dughera L, Ruffini E, Gasparri G, Camandona M. Abnormal expression of Endoglin and its receptor complex (TGF-β1 and TGF-β receptor II) as early angiogenic switch indicator in premalignant lesions of the colon mucosa. Int J Oncol. 2010;37:1153–1165. doi: 10.3892/ijo_00000767. [DOI] [PubMed] [Google Scholar]
- 27.Preativatanyou K, Honsawek S, Chongsrisawat V, Vejchapipat P, Theamboonlers A, Poovorawan Y. Correlation of circulating endoglin with clinical outcome in biliary atresia. Eur J Pediatr Surg. 2010;20:237–241. doi: 10.1055/s-0030-1249695. [DOI] [PubMed] [Google Scholar]
- 28.Rath T, Hage L, Kügler M, Menendez Menendez K, Zachoval R, Naehrlich L, Schulz R, Roderfeld M, Roeb E. Serum proteome profiling identifies novel and powerful markers of cystic fibrosis liver disease. PLoS One. 2013;8:e58955. doi: 10.1371/journal.pone.0058955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaiworapongsa T, Romero R, Kusanovic JP, Savasan ZA, Kim SK, Mazaki-Tovi S, Vaisbuch E, Ogge G, Madan I, Dong Z, et al. Unexplained fetal death is associated with increased concentrations of anti-angiogenic factors in amniotic fluid. J Matern Fetal Neonatal Med. 2010;23:794–805. doi: 10.3109/14767050903443467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dietmann A, Helbok R, Lackner P, Fischer M, Reindl M, Lell B, Issifou S, Kremsner PG, Schmutzhard E. Endoglin in African children with Plasmodium falciparum malaria: a novel player in severe malaria pathogenesis? J Infect Dis. 2009;200:1842–1848. doi: 10.1086/648476. [DOI] [PubMed] [Google Scholar]
- 31.Fujita K, Ewing CM, Chan DY, Mangold LA, Partin AW, Isaacs WB, Pavlovich CP. Endoglin (CD105) as a urinary and serum marker of prostate cancer. Int J Cancer. 2009;124:664–669. doi: 10.1002/ijc.24007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuiper P, Hawinkels LJ, de Jonge-Muller ES, Biemond I, Lamers CB, Verspaget HW. Angiogenic markers endoglin and vascular endothelial growth factor in gastroenteropancreatic neuroendocrine tumors. World J Gastroenterol. 2011;17:219–225. doi: 10.3748/wjg.v17.i2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 34.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 35.Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42:577–582. doi: 10.1136/jmg.2004.028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayrak-Toydemir P, McDonald J, Akarsu N, Toydemir RM, Calderon F, Tuncali T, Tang W, Miller F, Mao R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140:2155–2162. doi: 10.1002/ajmg.a.31450. [DOI] [PubMed] [Google Scholar]
- 37.St-Jacques S, Cymerman U, Pece N, Letarte M. Molecular characterization and in situ localization of murine endoglin reveal that it is a transforming growth factor-beta binding protein of endothelial and stromal cells. Endocrinology. 1994;134:2645–2657. doi: 10.1210/endo.134.6.8194490. [DOI] [PubMed] [Google Scholar]
- 38.Roy-Chaudhury P, Simpson JG, Power DA. Endoglin, a transforming growth factor-beta-binding protein, is upregulated in chronic progressive renal disease. Exp Nephrol. 1997;5:55–60. [PubMed] [Google Scholar]
- 39.Leask A, Abraham DJ, Finlay DR, Holmes A, Pennington D, Shi-Wen X, Chen Y, Venstrom K, Dou X, Ponticos M, et al. Dysregulation of transforming growth factor beta signaling in scleroderma: overexpression of endoglin in cutaneous scleroderma fibroblasts. Arthritis Rheum. 2002;46:1857–1865. doi: 10.1002/art.10333. [DOI] [PubMed] [Google Scholar]
- 40.Meurer SK, Tihaa L, Borkham-Kamphorst E, Weiskirchen R. Expression and functional analysis of endoglin in isolated liver cells and its involvement in fibrogenic Smad signalling. Cell Signal. 2011;23:683–699. doi: 10.1016/j.cellsig.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Meurer SK, Tihaa L, Lahme B, Gressner AM, Weiskirchen R. Identification of endoglin in rat hepatic stellate cells: new insights into transforming growth factor β receptor signaling. J Biol Chem. 2005;280:3078–3087. doi: 10.1074/jbc.M405411200. [DOI] [PubMed] [Google Scholar]
- 42.Scherner O, Meurer SK, Tihaa L, Gressner AM, Weiskirchen R. Endoglin differentially modulates antagonistic transforming growth factor-beta1 and BMP-7 signaling. J Biol Chem. 2007;282:13934–13943. doi: 10.1074/jbc.M611062200. [DOI] [PubMed] [Google Scholar]
- 43.Pérez-Gómez E, Eleno N, López-Novoa JM, Ramirez JR, Velasco B, Letarte M, Bernabéu C, Quintanilla M. Characterization of murine S-endoglin isoform and its effects on tumor development. Oncogene. 2005;24:4450–4461. doi: 10.1038/sj.onc.1208644. [DOI] [PubMed] [Google Scholar]
- 44.Blanco FJ, Bernabeu C. Alternative splicing factor or splicing factor-2 plays a key role in intron retention of the endoglin gene during endothelial senescence. Aging Cell. 2011;10:896–907. doi: 10.1111/j.1474-9726.2011.00727.x. [DOI] [PubMed] [Google Scholar]
- 45.Blanco FJ, Grande MT, Langa C, Oujo B, Velasco S, Rodriguez-Barbero A, Perez-Gomez E, Quintanilla M, López-Novoa JM, Bernabeu C. S-endoglin expression is induced in senescent endothelial cells and contributes to vascular pathology. Circ Res. 2008;103:1383–1392. doi: 10.1161/CIRCRESAHA.108.176552. [DOI] [PubMed] [Google Scholar]
- 46.ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11:79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 47.Llorca O, Trujillo A, Blanco FJ, Bernabeu C. Structural model of human endoglin, a transmembrane receptor responsible for hereditary hemorrhagic telangiectasia. J Mol Biol. 2007;365:694–705. doi: 10.1016/j.jmb.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 48.Gougos A, Letarte M. Biochemical characterization of the 44G4 antigen from the HOON pre-B leukemic cell line. J Immunol. 1988;141:1934–1940. [PubMed] [Google Scholar]
- 49.Bork P, Sander C. A large domain common to sperm receptors (Zp2 and Zp3) and TGF-beta type III receptor. FEBS Lett. 1992;300:237–240. doi: 10.1016/0014-5793(92)80853-9. [DOI] [PubMed] [Google Scholar]
- 50.Jovine L, Darie CC, Litscher ES, Wassarman PM. Zona pellucida domain proteins. Annu Rev Biochem. 2005;74:83–114. doi: 10.1146/annurev.biochem.74.082803.133039. [DOI] [PubMed] [Google Scholar]
- 51.Letamendía A, Lastres P, Botella LM, Raab U, Langa C, Velasco B, Attisano L, Bernabeu C. Role of endoglin in cellular responses to transforming growth factor-beta. A comparative study with betaglycan. J Biol Chem. 1998;273:33011–33019. doi: 10.1074/jbc.273.49.33011. [DOI] [PubMed] [Google Scholar]
- 52.Bernabeu C, Lopez-Novoa JM, Quintanilla M. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim Biophys Acta. 2009;1792:954–973. doi: 10.1016/j.bbadis.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Ge AZ, Butcher EC. Cloning and expression of a cDNA encoding mouse endoglin, an endothelial cell TGF-beta ligand. Gene. 1994;138:201–206. doi: 10.1016/0378-1119(94)90808-7. [DOI] [PubMed] [Google Scholar]
- 54.Gregory A. Structural and Functional Characteristics of a Soluble Form of Endoglin in the Context of Preeclampsia. A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Immunology, University of Toronto, Toronto, Canada. 2011. Available from: https: //tspace.library.utoronto.ca/bitstream/1807/30615/1/Gregory_Allison_L_201111_MSc_thesis.pdf. [Google Scholar]
- 55.Van Le B, Franke D, Svergun DI, Han T, Hwang HY, Kim KK. Structural and functional characterization of soluble endoglin receptor. Biochem Biophys Res Commun. 2009;383:386–391. doi: 10.1016/j.bbrc.2009.02.162. [DOI] [PubMed] [Google Scholar]
- 56.Guerrero-Esteo M, Sanchez-Elsner T, Letamendia A, Bernabeu C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-beta receptors I and II. J Biol Chem. 2002;277:29197–29209. doi: 10.1074/jbc.M111991200. [DOI] [PubMed] [Google Scholar]
- 57.Raab U, Velasco B, Lastres P, Letamendía A, Calés C, Langa C, Tapia E, López-Bote JP, Páez E, Bernabéu C. Expression of normal and truncated forms of human endoglin. Biochem J. 1999;339(Pt 3):579–588. [PMC free article] [PubMed] [Google Scholar]
- 58.Li CG, Wilson PB, Bernabeu C, Raab U, Wang JM, Kumar S. Immunodetection and characterisation of soluble CD105-TGFbeta complexes. J Immunol Methods. 1998;218:85–93. doi: 10.1016/s0022-1759(98)00118-5. [DOI] [PubMed] [Google Scholar]
- 59.Hawinkels LJ, Kuiper P, Wiercinska E, Verspaget HW, Liu Z, Pardali E, Sier CF, ten Dijke P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010;70:4141–4150. doi: 10.1158/0008-5472.CAN-09-4466. [DOI] [PubMed] [Google Scholar]
- 60.Kumar S, Pan CC, Bloodworth JC, Nixon A, Theuer C, Hoyt DG, Lee NY. Antibody-directed coupling of endoglin and MMP-14 is a key mechanism for endoglin shedding and deregulation of TGF-β signaling. Oncogene. 2013:Sep 30; Epub ahead of print. doi: 10.1038/onc.2013.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Velasco-Loyden G, Arribas J, López-Casillas F. The shedding of betaglycan is regulated by pervanadate and mediated by membrane type matrix metalloprotease-1. J Biol Chem. 2004;279:7721–7733. doi: 10.1074/jbc.M306499200. [DOI] [PubMed] [Google Scholar]
- 62.Yamashita H, Ichijo H, Grimsby S, Morén A, ten Dijke P, Miyazono K. Endoglin forms a heteromeric complex with the signaling receptors for transforming growth factor-beta. J Biol Chem. 1994;269:1995–2001. [PubMed] [Google Scholar]
- 63.Sanz-Rodriguez F, Guerrero-Esteo M, Botella LM, Banville D, Vary CP, Bernabéu C. Endoglin regulates cytoskeletal organization through binding to ZRP-1, a member of the LIM family of proteins. J Biol Chem. 2004;279:32858–32868. doi: 10.1074/jbc.M400843200. [DOI] [PubMed] [Google Scholar]
- 64.Conley BA, Koleva R, Smith JD, Kacer D, Zhang D, Bernabéu C, Vary CP. Endoglin controls cell migration and composition of focal adhesions: function of the cytosolic domain. J Biol Chem. 2004;279:27440–27449. doi: 10.1074/jbc.M312561200. [DOI] [PubMed] [Google Scholar]
- 65.Meng Q, Lux A, Holloschi A, Li J, Hughes JM, Foerg T, McCarthy JE, Heagerty AM, Kioschis P, Hafner M, et al. Identification of Tctex2beta, a novel dynein light chain family member that interacts with different transforming growth factor-beta receptors. J Biol Chem. 2006;281:37069–37080. doi: 10.1074/jbc.M608614200. [DOI] [PubMed] [Google Scholar]
- 66.Lee NY, Blobe GC. The interaction of endoglin with beta-arrestin2 regulates transforming growth factor-beta-mediated ERK activation and migration in endothelial cells. J Biol Chem. 2007;282:21507–21517. doi: 10.1074/jbc.M700176200. [DOI] [PubMed] [Google Scholar]
- 67.Lee NY, Ray B, How T, Blobe GC. Endoglin promotes transforming growth factor beta-mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J Biol Chem. 2008;283:32527–32533. doi: 10.1074/jbc.M803059200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blanco FJ, Santibanez JF, Guerrero-Esteo M, Langa C, Vary CP, Bernabeu C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J Cell Physiol. 2005;204:574–584. doi: 10.1002/jcp.20311. [DOI] [PubMed] [Google Scholar]
- 69.Lastres P, Martín-Perez J, Langa C, Bernabéu C. Phosphorylation of the human-transforming-growth-factor-beta-binding protein endoglin. Biochem J. 1994;301(Pt 3):765–768. doi: 10.1042/bj3010765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koleva RI, Conley BA, Romero D, Riley KS, Marto JA, Lux A, Vary CP. Endoglin structure and function: Determinants of endoglin phosphorylation by transforming growth factor-beta receptors. J Biol Chem. 2006;281:25110–25123. doi: 10.1074/jbc.M601288200. [DOI] [PubMed] [Google Scholar]
- 71.Ray BN, Lee NY, How T, Blobe GC. ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis. 2010;31:435–441. doi: 10.1093/carcin/bgp327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tian H, Mythreye K, Golzio C, Katsanis N, Blobe GC. Endoglin mediates fibronectin/α5β1 integrin and TGF-β pathway crosstalk in endothelial cells. EMBO J. 2012;31:3885–3900. doi: 10.1038/emboj.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang X, Abraham S, McKenzie JA, Jeffs N, Swire M, Tripathi VB, Luhmann UF, Lange CA, Zhai Z, Arthur HM, et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature. 2013;499:306–311. doi: 10.1038/nature12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blobe GC, Liu X, Fang SJ, How T, Lodish HF. A novel mechanism for regulating transforming growth factor beta (TGF-beta) signaling. Functional modulation of type III TGF-beta receptor expression through interaction with the PDZ domain protein, GIPC. J Biol Chem. 2001;276:39608–39617. doi: 10.1074/jbc.M106831200. [DOI] [PubMed] [Google Scholar]
- 75.Chen W, Kirkbride KC, How T, Nelson CD, Mo J, Frederick JP, Wang XF, Lefkowitz RJ, Blobe GC. Beta-arrestin 2 mediates endocytosis of type III TGF-beta receptor and down-regulation of its signaling. Science. 2003;301:1394–1397. doi: 10.1126/science.1083195. [DOI] [PubMed] [Google Scholar]
- 76.Lee NY, Golzio C, Gatza CE, Sharma A, Katsanis N, Blobe GC. Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Mol Biol Cell. 2012;23:2412–2423. doi: 10.1091/mbc.E11-12-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136:3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 78.Wiercinska E, Wickert L, Denecke B, Said HM, Hamzavi J, Gressner AM, Thorikay M, ten Dijke P, Mertens PR, Breitkopf K, et al. Id1 is a critical mediator in TGF-beta-induced transdifferentiation of rat hepatic stellate cells. Hepatology. 2006;43:1032–1041. doi: 10.1002/hep.21135. [DOI] [PubMed] [Google Scholar]
- 79.Meurer SK, Alsamman M, Sahin H, Wasmuth HE, Kisseleva T, Brenner DA, Trautwein C, Weiskirchen R, Scholten D. Overexpression of endoglin modulates TGF-β1-signalling pathways in a novel immortalized mouse hepatic stellate cell line. PLoS One. 2013;8:e56116. doi: 10.1371/journal.pone.0056116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lastres P, Letamendía A, Zhang H, Rius C, Almendro N, Raab U, López LA, Langa C, Fabra A, Letarte M, et al. Endoglin modulates cellular responses to TGF-beta 1. J Cell Biol. 1996;133:1109–1121. doi: 10.1083/jcb.133.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Meeteren LA, ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012;347:177–186. doi: 10.1007/s00441-011-1222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lastres P, Bellon T, Cabañas C, Sanchez-Madrid F, Acevedo A, Gougos A, Letarte M, Bernabeu C. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur J Immunol. 1992;22:393–397. doi: 10.1002/eji.1830220216. [DOI] [PubMed] [Google Scholar]
- 83.Ríus C, Smith JD, Almendro N, Langa C, Botella LM, Marchuk DA, Vary CP, Bernabéu C. Cloning of the promoter region of human endoglin, the target gene for hereditary hemorrhagic telangiectasia type 1. Blood. 1998;92:4677–4690. [PubMed] [Google Scholar]
- 84.Botella LM, Sánchez-Elsner T, Rius C, Corbí A, Bernabéu C. Identification of a critical Sp1 site within the endoglin promoter and its involvement in the transforming growth factor-beta stimulation. J Biol Chem. 2001;276:34486–34494. doi: 10.1074/jbc.M011611200. [DOI] [PubMed] [Google Scholar]
- 85.Botella LM, Sánchez-Elsner T, Sanz-Rodriguez F, Kojima S, Shimada J, Guerrero-Esteo M, Cooreman MP, Ratziu V, Langa C, Vary CP, et al. Transcriptional activation of endoglin and transforming growth factor-beta signaling components by cooperative interaction between Sp1 and KLF6: their potential role in the response to vascular injury. Blood. 2002;100:4001–4010. doi: 10.1182/blood.V100.12.4001. [DOI] [PubMed] [Google Scholar]
- 86.Sánchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabéu C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J Biol Chem. 2002;277:43799–43808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- 87.Velasco S, Alvarez-Muñoz P, Pericacho M, Dijke PT, Bernabéu C, López-Novoa JM, Rodríguez-Barbero A. L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J Cell Sci. 2008;121:913–919. doi: 10.1242/jcs.023283. [DOI] [PubMed] [Google Scholar]
- 88.Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004;23:4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jerkic M, Rivas-Elena JV, Santibanez JF, Prieto M, Rodríguez-Barbero A, Perez-Barriocanal F, Pericacho M, Arévalo M, Vary CP, Letarte M, et al. Endoglin regulates cyclooxygenase-2 expression and activity. Circ Res. 2006;99:248–256. doi: 10.1161/01.RES.0000236755.98627.69. [DOI] [PubMed] [Google Scholar]
- 90.Pece-Barbara N, Vera S, Kathirkamathamby K, Liebner S, Di Guglielmo GM, Dejana E, Wrana JL, Letarte M. Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor beta1 with higher affinity receptors and an activated Alk1 pathway. J Biol Chem. 2005;280:27800–27808. doi: 10.1074/jbc.M503471200. [DOI] [PubMed] [Google Scholar]
- 91.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21:1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Santibanez JF, Letamendia A, Perez-Barriocanal F, Silvestri C, Saura M, Vary CP, Lopez-Novoa JM, Attisano L, Bernabeu C. Endoglin increases eNOS expression by modulating Smad2 protein levels and Smad2-dependent TGF-beta signaling. J Cell Physiol. 2007;210:456–468. doi: 10.1002/jcp.20878. [DOI] [PubMed] [Google Scholar]
- 93.Diez-Marques L, Ortega-Velazquez R, Langa C, Rodriguez-Barbero A, Lopez-Novoa JM, Lamas S, Bernabeu C. Expression of endoglin in human mesangial cells: modulation of extracellular matrix synthesis. Biochim Biophys Acta. 2002;1587:36–44. doi: 10.1016/s0925-4439(02)00051-0. [DOI] [PubMed] [Google Scholar]
- 94.Guo B, Slevin M, Li C, Parameshwar S, Liu D, Kumar P, Bernabeu C, Kumar S. CD105 inhibits transforming growth factor-beta-Smad3 signalling. Anticancer Res. 2004;24:1337–1345. [PubMed] [Google Scholar]
- 95.Obreo J, Díez-Marques L, Lamas S, Düwell A, Eleno N, Bernabéu C, Pandiella A, López-Novoa JM, Rodríguez-Barbero A. Endoglin expression regulates basal and TGF-beta1-induced extracellular matrix synthesis in cultured L6E9 myoblasts. Cell Physiol Biochem. 2004;14:301–310. doi: 10.1159/000080340. [DOI] [PubMed] [Google Scholar]
- 96.Rodríguez-Barbero A, Obreo J, Alvarez-Munoz P, Pandiella A, Bernabéu C, López-Novoa JM. Endoglin modulation of TGF-beta1-induced collagen synthesis is dependent on ERK1/2 MAPK activation. Cell Physiol Biochem. 2006;18:135–142. doi: 10.1159/000095181. [DOI] [PubMed] [Google Scholar]
- 97.Schmidt-Weber CB, Letarte M, Kunzmann S, Rückert B, Bernabéu C, Blaser K. TGF-{beta} signaling of human T cells is modulated by the ancillary TGF-{beta} receptor endoglin. Int Immunol. 2005;17:921–930. doi: 10.1093/intimm/dxh272. [DOI] [PubMed] [Google Scholar]
- 98.Pickles M, Leask A. Analysis of CCN2 promoter activity in PANC-1 cells: regulation by ras/MEK/ERK. J Cell Commun Signal. 2007;1:85–90. doi: 10.1007/s12079-007-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang X, Arnott JA, Rehman S, Delong WG, Sanjay A, Safadi FF, Popoff SN. Src is a major signaling component for CTGF induction by TGF-beta1 in osteoblasts. J Cell Physiol. 2010;224:691–701. doi: 10.1002/jcp.22173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Santibanez JF, Pérez-Gómez E, Fernandez-L A, Garrido-Martin EM, Carnero A, Malumbres M, Vary CP, Quintanilla M, Bernabéu C. The TGF-beta co-receptor endoglin modulates the expression and transforming potential of H-Ras. Carcinogenesis. 2010;31:2145–2154. doi: 10.1093/carcin/bgq199. [DOI] [PubMed] [Google Scholar]
- 101.Holmes AM, Ponticos M, Shi-Wen X, Denton CP, Abraham DJ. Elevated CCN2 expression in scleroderma: a putative role for the TGFβ accessory receptors TGFβRIII and endoglin. J Cell Commun Signal. 2011;5:173–177. doi: 10.1007/s12079-011-0140-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pannu J, Nakerakanti S, Smith E, ten Dijke P, Trojanowska M. Transforming growth factor-beta receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem. 2007;282:10405–10413. doi: 10.1074/jbc.M611742200. [DOI] [PubMed] [Google Scholar]
- 103.Morris E, Chrobak I, Bujor A, Hant F, Mummery C, Ten Dijke P, Trojanowska M. Endoglin promotes TGF-β/Smad1 signaling in scleroderma fibroblasts. J Cell Physiol. 2011;226:3340–3348. doi: 10.1002/jcp.22690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. doi: 10.1053/jhep.2001.25349. [DOI] [PubMed] [Google Scholar]
- 105.Tang H, Low B, Rutherford SA, Hao Q. Thrombin induces endocytosis of endoglin and type-II TGF-beta receptor and down-regulation of TGF-beta signaling in endothelial cells. Blood. 2005;105:1977–1985. doi: 10.1182/blood-2004-08-3308. [DOI] [PubMed] [Google Scholar]
- 106.Pérez-Gómez E, Del Castillo G, Juan Francisco S, López-Novoa JM, Bernabéu C, Quintanilla M. The role of the TGF-β coreceptor endoglin in cancer. ScientificWorldJournal. 2010;10:2367–2384. doi: 10.1100/tsw.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, Epstein FH, Romero R, Thadhani R, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992–1005. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 108.Yagmur E, Rizk M, Stanzel S, Hellerbrand C, Lammert F, Trautwein C, Wasmuth HE, Gressner AM. Elevation of endoglin (CD105) concentrations in serum of patients with liver cirrhosis and carcinoma. Eur J Gastroenterol Hepatol. 2007;19:755–761. doi: 10.1097/MEG.0b013e3282202bea. [DOI] [PubMed] [Google Scholar]
- 109.Cheifetz S, Bellón T, Calés C, Vera S, Bernabeu C, Massagué J, Letarte M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–19030. [PubMed] [Google Scholar]
- 110.Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999;274:584–594. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- 111.De Crescenzo G, Pham PL, Durocher Y, O’Connor-McCourt MD. Transforming growth factor-beta (TGF-beta) binding to the extracellular domain of the type II TGF-beta receptor: receptor capture on a biosensor surface using a new coiled-coil capture system demonstrates that avidity contributes significantly to high affinity binding. J Mol Biol. 2003;328:1173–1183. doi: 10.1016/s0022-2836(03)00360-7. [DOI] [PubMed] [Google Scholar]
- 112.Castonguay R, Werner ED, Matthews RG, Presman E, Mulivor AW, Solban N, Sako D, Pearsall RS, Underwood KW, Seehra J, et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J Biol Chem. 2011;286:30034–30046. doi: 10.1074/jbc.M111.260133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genet Med. 2011;13:607–616. doi: 10.1097/GIM.0b013e3182136d32. [DOI] [PubMed] [Google Scholar]
- 114.Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am J Med Genet. 2000;91:66–67. doi: 10.1002/(sici)1096-8628(20000306)91:1<66::aid-ajmg12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 115.Sadick H, Hage J, Goessler U, Stern-Straeter J, Riedel F, Hoermann K, Bugert P. Mutation analysis of “Endoglin” and “Activin receptor-like kinase” genes in German patients with hereditary hemorrhagic telangiectasia and the value of rapid genotyping using an allele-specific PCR-technique. BMC Med Genet. 2009;10:53. doi: 10.1186/1471-2350-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43:97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Srinivasan S, Hanes MA, Dickens T, Porteous ME, Oh SP, Hale LP, Marchuk DA. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet. 2003;12:473–482. doi: 10.1093/hmg/ddg050. [DOI] [PubMed] [Google Scholar]
- 118.Bourdeau A, Cymerman U, Paquet ME, Meschino W, McKinnon WC, Guttmacher AE, Becker L, Letarte M. Endoglin expression is reduced in normal vessels but still detectable in arteriovenous malformations of patients with hereditary hemorrhagic telangiectasia type 1. Am J Pathol. 2000;156:911–923. doi: 10.1016/S0002-9440(10)64960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bauditz J, Lochs H. Angiogenesis and vascular malformations: antiangiogenic drugs for treatment of gastrointestinal bleeding. World J Gastroenterol. 2007;13:5979–5984. doi: 10.3748/wjg.v13.45.5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bowcock SJ, Patrick HE. Lenalidomide to control gastrointestinal bleeding in hereditary haemorrhagic telangiectasia: potential implications for angiodysplasias? Br J Haematol. 2009;146:220–222. doi: 10.1111/j.1365-2141.2009.07730.x. [DOI] [PubMed] [Google Scholar]
- 121.Dupuis-Girod S, Ginon I, Saurin JC, Marion D, Guillot E, Decullier E, Roux A, Carette MF, Gilbert-Dussardier B, Hatron PY, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307:948–955. doi: 10.1001/jama.2012.250. [DOI] [PubMed] [Google Scholar]
- 122.Albiñana V, Recio-Poveda L, Zarrabeitia R, Bernabéu C, Botella LM. Propranolol as antiangiogenic candidate for the therapy of hereditary haemorrhagic telangiectasia. Thromb Haemost. 2012;108:41–53. doi: 10.1160/TH11-11-0809. [DOI] [PubMed] [Google Scholar]
- 123.Yaniv E, Preis M, Shevro J, Nageris B, Hadar T. Anti-estrogen therapy for hereditary hemorrhagic telangiectasia - a long-term clinical trial. Rhinology. 2011;49:214–216. doi: 10.4193/Rhino09.201. [DOI] [PubMed] [Google Scholar]
- 124.Albiñana V, Bernabeu-Herrero ME, Zarrabeitia R, Bernabéu C, Botella LM. Estrogen therapy for hereditary haemorrhagic telangiectasia (HHT): Effects of raloxifene, on Endoglin and ALK1 expression in endothelial cells. Thromb Haemost. 2010;103:525–534. doi: 10.1160/TH09-07-0425. [DOI] [PubMed] [Google Scholar]
- 125.Naljayan MV, Karumanchi SA. New developments in the pathogenesis of preeclampsia. Adv Chronic Kidney Dis. 2013;20:265–270. doi: 10.1053/j.ackd.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Karnad DR, Guntupalli KK. Neurologic disorders in pregnancy. Crit Care Med. 2005;33:S362–S371. doi: 10.1097/01.ccm.0000182790.35728.f7. [DOI] [PubMed] [Google Scholar]
- 127.Brown MC, Best KE, Pearce MS, Waugh J, Robson SC, Bell R. Cardiovascular disease risk in women with pre-eclampsia: systematic review and meta-analysis. Eur J Epidemiol. 2013;28:1–19. doi: 10.1007/s10654-013-9762-6. [DOI] [PubMed] [Google Scholar]
- 128.Zhou Y, Damsky CH, Fisher SJ. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J Clin Invest. 1997;99:2152–2164. doi: 10.1172/JCI119388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ahmad S, Ahmed A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ Res. 2004;95:884–891. doi: 10.1161/01.RES.0000147365.86159.f5. [DOI] [PubMed] [Google Scholar]
- 130.Staff AC, Braekke K, Harsem NK, Lyberg T, Holthe MR. Circulating concentrations of sFlt1 (soluble fms-like tyrosine kinase 1) in fetal and maternal serum during pre-eclampsia. Eur J Obstet Gynecol Reprod Biol. 2005;122:33–39. doi: 10.1016/j.ejogrb.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 131.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Valbuena-Diez AC, Blanco FJ, Oujo B, Langa C, Gonzalez-Nuñez M, Llano E, Pendas AM, Díaz M, Castrillo A, Lopez-Novoa JM, et al. Oxysterol-induced soluble endoglin release and its involvement in hypertension. Circulation. 2012;126:2612–2624. doi: 10.1161/CIRCULATIONAHA.112.101261. [DOI] [PubMed] [Google Scholar]
- 133.Jerkic M, Rivas-Elena JV, Prieto M, Carrón R, Sanz-Rodríguez F, Pérez-Barriocanal F, Rodríguez-Barbero A, Bernabéu C, López-Novoa JM. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004;18:609–611. doi: 10.1096/fj.03-0197fje. [DOI] [PubMed] [Google Scholar]
- 134.López-Novoa JM, Bernabeu C. The physiological role of endoglin in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2010;299:H959–H974. doi: 10.1152/ajpheart.01251.2009. [DOI] [PubMed] [Google Scholar]
- 135.Toporsian M, Gros R, Kabir MG, Vera S, Govindaraju K, Eidelman DH, Husain M, Letarte M. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res. 2005;96:684–692. doi: 10.1161/01.RES.0000159936.38601.22. [DOI] [PubMed] [Google Scholar]
- 136.Gu Y, Lewis DF, Wang Y. Placental productions and expressions of soluble endoglin, soluble fms-like tyrosine kinase receptor-1, and placental growth factor in normal and preeclamptic pregnancies. J Clin Endocrinol Metab. 2008;93:260–266. doi: 10.1210/jc.2007-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gilbert JS, Gilbert SA, Arany M, Granger JP. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension. 2009;53:399–403. doi: 10.1161/HYPERTENSIONAHA.108.123513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Powers RW, Jeyabalan A, Clifton RG, Van Dorsten P, Hauth JC, Klebanoff MA, Lindheimer MD, Sibai B, Landon M, Miodovnik M. Soluble fms-Like tyrosine kinase 1 (sFlt1), endoglin and placental growth factor (PlGF) in preeclampsia among high risk pregnancies. PLoS One. 2010;5:e13263. doi: 10.1371/journal.pone.0013263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li Z, Zhang Y, Ying Ma J, Kapoun AM, Shao Q, Kerr I, Lam A, O’Young G, Sannajust F, Stathis P, et al. Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension. 2007;50:686–692. doi: 10.1161/HYPERTENSIONAHA.107.092098. [DOI] [PubMed] [Google Scholar]
- 140.Thadhani R, Kisner T, Hagmann H, Bossung V, Noack S, Schaarschmidt W, Jank A, Kribs A, Cornely OA, Kreyssig C, et al. Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation. 2011;124:940–950. doi: 10.1161/CIRCULATIONAHA.111.034793. [DOI] [PubMed] [Google Scholar]
- 141.Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, Devey LR, Wigmore SJ, Abbas A, Hewett PW, et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007;115:1789–1797. doi: 10.1161/CIRCULATIONAHA.106.660134. [DOI] [PubMed] [Google Scholar]
- 142.Devy L, Huang L, Naa L, Yanamandra N, Pieters H, Frans N, Chang E, Tao Q, Vanhove M, Lejeune A, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69:1517–1526. doi: 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- 143.Abildgaard U, Heimdal K. Pathogenesis of the syndrome of hemolysis, elevated liver enzymes, and low platelet count (HELLP): a review. Eur J Obstet Gynecol Reprod Biol. 2013;166:117–123. doi: 10.1016/j.ejogrb.2012.09.026. [DOI] [PubMed] [Google Scholar]
- 144.Reimer T, Rohrmann H, Stubert J, Pecks U, Glocker MO, Richter DU, Gerber B. Angiogenic factors and acute-phase proteins in serum samples of preeclampsia and HELLP patients: a matched-pair analysis. J Matern Fetal Neonatal Med. 2013;26:263–269. doi: 10.3109/14767058.2012.733747. [DOI] [PubMed] [Google Scholar]
- 145.Indraccolo U, Gentile G, Manfreda VM, Pomili G. The development of disseminated intravascular coagulation in hemolysis, elevated liver enzymes, and low platelet count syndrome (HELLP) at very early gestational age. Minerva Ginecol. 2008;60:445–450. [PubMed] [Google Scholar]
- 146.Lubamba B, Dhooghe B, Noel S, Leal T. Cystic fibrosis: insight into CFTR pathophysiology and pharmacotherapy. Clin Biochem. 2012;45:1132–1144. doi: 10.1016/j.clinbiochem.2012.05.034. [DOI] [PubMed] [Google Scholar]
- 147.Gelfond D, Borowitz D. Gastrointestinal complications of cystic fibrosis. Clin Gastroenterol Hepatol. 2013;11:333–342; quiz e30-e31. doi: 10.1016/j.cgh.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 148.Leeuwen L, Fitzgerald DA, Gaskin KJ. Liver disease in cystic fibrosis. Paediatr Respir Rev. 2013:Jun 13; Epub ahead of print. doi: 10.1016/j.prrv.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 149.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Brenner DA. Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc. 2009;120:361–368. [PMC free article] [PubMed] [Google Scholar]
- 151.Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P, Rose-John S, zum Büschenfelde KH, Blessing M. TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol. 1999;276:G1059–G1068. doi: 10.1152/ajpgi.1999.276.4.G1059. [DOI] [PubMed] [Google Scholar]
- 152.Fakih M. The evolving role of VEGF-targeted therapies in the treatment of metastatic colorectal cancer. Expert Rev Anticancer Ther. 2013;13:427–438. doi: 10.1586/era.13.20. [DOI] [PubMed] [Google Scholar]
- 153.Fagiani E, Christofori G. Angiopoietins in angiogenesis. Cancer Lett. 2013;328:18–26. doi: 10.1016/j.canlet.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 154.Benedito R, Hellström M. Notch as a hub for signaling in angiogenesis. Exp Cell Res. 2013;319:1281–1288. doi: 10.1016/j.yexcr.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 155.Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. 2013;319:1271–1280. doi: 10.1016/j.yexcr.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 156.Bellone G, Solerio D, Chiusa L, Brondino G, Carbone A, Prati A, Scirelli T, Camandona M, Palestro G, Dei Poli M. Transforming growth factor-beta binding receptor endoglin (CD105) expression in esophageal cancer and in adjacent nontumorous esophagus as prognostic predictor of recurrence. Ann Surg Oncol. 2007;14:3232–3242. doi: 10.1245/s10434-007-9528-z. [DOI] [PubMed] [Google Scholar]
- 157.Kumar S, Ghellal A, Li C, Byrne G, Haboubi N, Wang JM, Bundred N. Breast carcinoma: vascular density determined using CD105 antibody correlates with tumor prognosis. Cancer Res. 1999;59:856–861. [PubMed] [Google Scholar]
- 158.Yu JX, Zhang XT, Liao YQ, Zhang QY, Chen H, Lin M, Kumar S. Relationship between expression of CD105 and growth factors in malignant tumors of gastrointestinal tract and its significance. World J Gastroenterol. 2003;9:2866–2869. doi: 10.3748/wjg.v9.i12.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Duff SE, Li C, Garland JM, Kumar S. CD105 is important for angiogenesis: evidence and potential applications. FASEB J. 2003;17:984–992. doi: 10.1096/fj.02-0634rev. [DOI] [PubMed] [Google Scholar]
- 160.Wikström P, Lissbrant IF, Stattin P, Egevad L, Bergh A. Endoglin (CD105) is expressed on immature blood vessels and is a marker for survival in prostate cancer. Prostate. 2002;51:268–275. doi: 10.1002/pros.10083. [DOI] [PubMed] [Google Scholar]
- 161.Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, Charlton R, Parums DV, Jowett T, Marchuk DA, et al. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol. 2000;217:42–53. doi: 10.1006/dbio.1999.9534. [DOI] [PubMed] [Google Scholar]
- 162.Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- 164.Allinson KR, Carvalho RL, van den Brink S, Mummery CL, Arthur HM. Generation of a floxed allele of the mouse Endoglin gene. Genesis. 2007;45:391–395. doi: 10.1002/dvg.20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mancini ML, Terzic A, Conley BA, Oxburgh LH, Nicola T, Vary CP. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev Dyn. 2009;238:2479–2493. doi: 10.1002/dvdy.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bourdeau A, Faughnan ME, McDonald ML, Paterson AD, Wanless IR, Letarte M. Potential role of modifier genes influencing transforming growth factor-beta1 levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am J Pathol. 2001;158:2011–2020. doi: 10.1016/s0002-9440(10)64673-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Carvalho RL, Jonker L, Goumans MJ, Larsson J, Bouwman P, Karlsson S, Dijke PT, Arthur HM, Mummery CL. Defective paracrine signalling by TGFbeta in yolk sac vasculature of endoglin mutant mice: a paradigm for hereditary haemorrhagic telangiectasia. Development. 2004;131:6237–6247. doi: 10.1242/dev.01529. [DOI] [PubMed] [Google Scholar]
- 168.Jerkic M, Rodríguez-Barbero A, Prieto M, Toporsian M, Pericacho M, Rivas-Elena JV, Obreo J, Wang A, Pérez-Barriocanal F, Arévalo M, et al. Reduced angiogenic responses in adult Endoglin heterozygous mice. Cardiovasc Res. 2006;69:845–854. doi: 10.1016/j.cardiores.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 169.Park SO, Lee YJ, Nguyen HL, Han C, Choi EJ, Oh SP. ALK1 signaling plays a pivotal role in regulation of genes involved in angiogenesis and vascular tone: implication on the pathogenetic mechanism for hereditary hemorrhagic telangiectasia 2 (HHT2) FASEB J. 2008;22:318. [Google Scholar]
- 170.Dolinsek T, Markelc B, Sersa G, Coer A, Stimac M, Lavrencak J, Brozic A, Kranjc S, Cemazar M. Multiple delivery of siRNA against endoglin into murine mammary adenocarcinoma prevents angiogenesis and delays tumor growth. PLoS One. 2013;8:e58723. doi: 10.1371/journal.pone.0058723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Krex D, Ziegler A, Schackert HK, Schackert G. Lack of association between endoglin intron 7 insertion polymorphism and intracranial aneurysms in a white population: evidence of racial/ethnic differences. Stroke. 2001;32:2689–2694. doi: 10.1161/hs1101.098660. [DOI] [PubMed] [Google Scholar]
- 172.Daina E, D’Ovidio F, Sabbà C. Introduction: hereditary hemorrhagic telangiectasia as a rare disease. Curr Pharm Des. 2006;12:1171–1172. doi: 10.2174/138161206776361264. [DOI] [PubMed] [Google Scholar]
- 173.Plauchu H, de Chadarévian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. 1989;32:291–297. doi: 10.1002/ajmg.1320320302. [DOI] [PubMed] [Google Scholar]
- 174.Porteous ME, Burn J, Proctor SJ. Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet. 1992;29:527–530. doi: 10.1136/jmg.29.8.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schulte C, Geisthoff U, Lux A, Kupka S, Zenner HP, Blin N, Pfister M. High frequency of ENG and ALK1/ACVRL1 mutations in German HHT patients. Hum Mutat. 2005;25:595. doi: 10.1002/humu.9345. [DOI] [PubMed] [Google Scholar]
- 176.Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M, Nozaki J, Inoue S, Koizumi A. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat. 2002;19:140–148. doi: 10.1002/humu.10026. [DOI] [PubMed] [Google Scholar]
- 177.Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med. 2004;6:175–191. doi: 10.1097/01.gim.0000132689.25644.7c. [DOI] [PubMed] [Google Scholar]
- 178.Shovlin CL, Hughes JM, Scott J, Seidman CE, Seidman JG. Characterization of endoglin and identification of novel mutations in hereditary hemorrhagic telangiectasia. Am J Hum Genet. 1997;61:68–79. doi: 10.1086/513906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Brusgaard K, Kjeldsen AD, Poulsen L, Moss H, Vase P, Rasmussen K, Kruse TA, Hørder M. Mutations in endoglin and in activin receptor-like kinase 1 among Danish patients with hereditary haemorrhagic telangiectasia. Clin Genet. 2004;66:556–561. doi: 10.1111/j.1399-0004.2004.00341.x. [DOI] [PubMed] [Google Scholar]
- 180.Wehner LE, Folz BJ, Argyriou L, Twelkemeyer S, Teske U, Geisthoff UW, Werner JA, Engel W, Nayernia K. Mutation analysis in hereditary haemorrhagic telangiectasia in Germany reveals 11 novel ENG and 12 novel ACVRL1/ALK1 mutations. Clin Genet. 2006;69:239–245. doi: 10.1111/j.1399-0004.2006.00574.x. [DOI] [PubMed] [Google Scholar]
- 181.Paquet ME, Pece-Barbara N, Vera S, Cymerman U, Karabegovic A, Shovlin C, Letarte M. Analysis of several endoglin mutants reveals no endogenous mature or secreted protein capable of interfering with normal endoglin function. Hum Mol Genet. 2001;10:1347–1357. doi: 10.1093/hmg/10.13.1347. [DOI] [PubMed] [Google Scholar]
- 182.Lux A, Gallione CJ, Marchuk DA. Expression analysis of endoglin missense and truncation mutations: insights into protein structure and disease mechanisms. Hum Mol Genet. 2000;9:745–755. doi: 10.1093/hmg/9.5.745. [DOI] [PubMed] [Google Scholar]
- 183.Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17:860–871. doi: 10.1038/ejhg.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Berg J, Porteous M, Reinhardt D, Gallione C, Holloway S, Umasunthar T, Lux A, McKinnon W, Marchuk D, Guttmacher A. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet. 2003;40:585–590. doi: 10.1136/jmg.40.8.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sabbà C, Pasculli G, Lenato GM, Suppressa P, Lastella P, Memeo M, Dicuonzo F, Guant G. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost. 2007;5:1149–1157. doi: 10.1111/j.1538-7836.2007.02531.x. [DOI] [PubMed] [Google Scholar]
- 186.Stepan H, Geipel A, Schwarz F, Krämer T, Wessel N, Faber R. Circulatory soluble endoglin and its predictive value for preeclampsia in second-trimester pregnancies with abnormal uterine perfusion. Am J Obstet Gynecol. 2008;198:175.e1–175.e6. doi: 10.1016/j.ajog.2007.08.052. [DOI] [PubMed] [Google Scholar]
- 187.Abdelaziz A, Maher MA, Sayyed TM, Bazeed MF, Mohamed NS. Early pregnancy screening for hypertensive disorders in women without a-priori high risk. Ultrasound Obstet Gynecol. 2012;40:398–405. doi: 10.1002/uog.11205. [DOI] [PubMed] [Google Scholar]
- 188.Li C, Guo B, Wilson PB, Stewart A, Byrne G, Bundred N, Kumar S. Plasma levels of soluble CD105 correlate with metastasis in patients with breast cancer. Int J Cancer. 2000;89:122–126. doi: 10.1002/(sici)1097-0215(20000320)89:2<122::aid-ijc4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 189.Takahashi N, Kawanishi-Tabata R, Haba A, Tabata M, Haruta Y, Tsai H, Seon BK. Association of serum endoglin with metastasis in patients with colorectal, breast, and other solid tumors, and suppressive effect of chemotherapy on the serum endoglin. Clin Cancer Res. 2001;7:524–532. [PubMed] [Google Scholar]
- 190.Li C, Gardy R, Seon BK, Duff SE, Abdalla S, Renehan A, O‘Dwyer ST, Haboubi N, Kumar S. Both high intratumoral microvessel density determined using CD105 antibody and elevated plasma levels of CD105 in colorectal cancer patients correlate with poor prognosis. Br J Cancer. 2003;88:1424–1431. doi: 10.1038/sj.bjc.6600874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Clemente M, Núñez O, Lorente R, Rincón D, Matilla A, Salcedo M, Catalina MV, Ripoll C, Iacono OL, Bañares R, et al. Increased intrahepatic and circulating levels of endoglin, a TGF-beta1 co-receptor, in patients with chronic hepatitis C virus infection: relationship to histological and serum markers of hepatic fibrosis. J Viral Hepat. 2006;13:625–632. doi: 10.1111/j.1365-2893.2006.00733.x. [DOI] [PubMed] [Google Scholar]
- 192.Yu D, Zhuang L, Sun X, Chen J, Yao Y, Meng K, Ding Y. Particular distribution and expression pattern of endoglin (CD105) in the liver of patients with hepatocellular carcinoma. BMC Cancer. 2007;7:122. doi: 10.1186/1471-2407-7-122. [DOI] [PMC free article] [PubMed] [Google Scholar]