

Abstract
Signal transducer and activator of transcription 3 (STAT3), a member of the STAT family, is a key regulator of many physiological and pathological processes. Significant progress has been made in understanding the transcriptional control, posttranslational modification, cellular localization and functional regulation of STAT3. STAT3 can translocate into the nucleus and bind to specific promoter sequences, thereby exerting transcriptional regulation. Recent studies have shown that STAT3 can also translocate into mitochondria, participating in aerobic respiration and apoptosis. In addition, STAT3 plays an important role in inflammation and tumorigenesis by regulating cell proliferation, differentiation and metabolism. Conditional knockout mouse models make it possible to study the physiological function of STAT3 in specific tissues and organs. This review summarizes the latest advances in the understanding of the expression, regulation and function of STAT3 in physiological and tumorigenic processes.
Keywords: Signal transducer and activator of transcription 3, Phosphorylation, Acetylation, Signal pathway, Tumor
Core tip: The differential subcellular localization of signal transducer and activator of transcription 3 makes it play distinct functions in transcriptional regulation, cell proliferation and cellular respiration, thus contributing to development, reproduction and tumorigenesis in physiological and pathological conditions.
INTRODUCTION
Signal transducer and activator of transcription factors (STATs) are a family of transcription factors that regulate cell growth, survival, differentiation, and motility. Structural studies identified that STAT proteins consist of an N-terminal domain, a coiled-coil domain, a DNA-binding domain, a Src homology 2 (SH2) domain and a transactivation domain, of which the DNA-binding domain is required for the recognition of specific binding sequences. Until now, seven members of the STAT family have been identified and characterized, including STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6. Despite the difference from canonical oncogenes, STAT3 has been recognized as a critical regulator in tumor cells since its identification[1]. STAT3 is over-expressed or activated by various carcinogenic agents, and can induce cell proliferation, differentiation and anti-apoptosis by activating the target genes, including STAT3, c-Myc and p53[2]. STAT3 exists in two main isoforms, full-length STAT3α and truncated STAT3β generated by alternative splicing. Under normal circumstances, STAT3α is the main isoform expressed in cells. STAT3β can competitively bind to the promoter of STAT3α target genes and inhibit the transactivation function of STAT3α. Additionally, STAT3β has its own specific target genes that differ from those of STAT3α[3].
STAT3 protein exists in a latent or inactive form in the cytoplasm. STAT3 can be activated by receptor-associated kinases and phosphorylated at various phosphorylation sites, particularly at Tyr-705 and Ser-727. Previous studies suggested that only phosphorylated STAT3 (p-STAT3) can translocate into the nucleus. However, recent data indicated that the nuclear translocation and transcriptional activity are partially independent of phosphorylation pathways[4]. Furthermore, STAT3 may translocate into mitochondria to control cell metabolism independent of its transcriptional regulatory activity[5]. Here we review the emerging biochemical and biological data on STAT3 and discuss its comprehensive roles in animal development and etiopathology of various diseases.
TRANSCRIPTIONAL REGULATION OF STAT3
STAT3 protein is expressed at a basal level in cells but rapidly increases once activated by specific cytokines. STAT3 is a critical factor in interleukin-6 (IL-6) induced gene regulation. STAT3 can be phosphorylated by IL-6 signal pathway, whereas IL-6 can also activate STAT3 at the transcriptional level. The level of STAT3 mRNA increases 1 h after IL-6 treatment and reaches to the maximum value at 3 h. There is an IL-6 response element (IL-6RE) in the promoter of STAT3 which contains a low affinity STAT3-binding element and a cAMP-responsive element (CRE). STAT3 executes its regulation in cooperation with this CRE-binding protein through self-activation[6].
In diabetic mice, estrogen administration can increase the level of STAT3 mRNA. There is a binding site of estrogen receptor α (ERα) in STAT3 promoter. Estrogen treatment induces the accumulation of ERα on STAT3 promoter and regulates the expression of STAT3[7]. STAT3 overexpression in tumor cells is related to the cytoplasmic/nuclear accumulation of β-catenin and the activation of β-catenin/T-cell factors (TCF) pathway. β-catenin is a key mediator in cell adhesion and signal transduction. Overexpression of β-catenin enhances both STAT3 mRNA and protein levels. There is a functional TCF binding element in STAT3 promoter, indicating that β-catenin/TCF may participate in the regulation of STAT3 expression[8].
The suppressors of cytokine signaling (SOCS) family consists of eight members, including SOCS1 to SOCS7 and cytokine-inducible SH2 domain proteins (CIS)[9]. SOCS proteins exist at low levels in resting cells and dramatically increase after STAT activation. SOCS proteins serve as classic negative regulatory factors of STAT activation[10]. Among them, SOCS3, a target gene of STAT3, contributes to negative feedback regulation of the JAK/STAT3 signal pathway, and inhibits the self-activation of STAT3[11]. Bone marrow SOCS3 deficient mice exhibit overexpression of STAT3 and continuous activation of the JAK/STAT3 signal pathway, suggesting that STAT3 expression is negatively regulated by SOCS3[12].
POST-TRANSCRIPTIONAL REGULATION OF STAT3 EXPRESSION
Human STAT3 gene is located on the long (q) arm of chromosome 17 at position 21.31. The encoding product of the STAT3 gene is an 89 kDa protein[13]. Further study identified a cDNA clone encoding a variant of STAT3 (named STAT3β), which is different from classic STAT3 (named STAT3α). Compared to STAT3α, STAT3β is the truncated form and lacks the internal domain of 50 base pairs located near the C-terminus (Figure 1). The encoding product of STAT3β is an 80 kDa protein. Under normal conditions, STAT3β exists in various cells, such as monocytes, lymphocytes and neutrophil granulocytes. In COS cells, STAT3β is phosphorylated at tyrosine sites by IL-5R treatment and binds to the palindromic IL-6/interferon-γ response element (pIRE) located in the promoter of intercellular adhesion molecule-1 (ICAM-1). However, this phosphorylated STAT3β exhibits a negative transcriptional regulation through inhibiting the transactivation potential of STAT3α, suggesting that STAT3β may be a dominant-negative regulator of transcription and promotes apoptosis[14].
Figure 1.
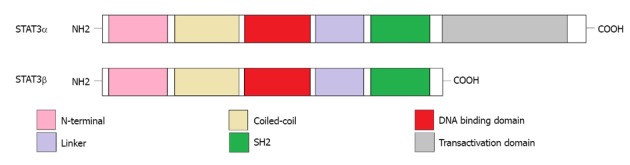
Domain structure of signal transducer and activator of transcription (3α and 3β). The signal transducer and activator of transcription 3α (STAT3α) protein is composed of N-terminal, coiled-coil, DNA binding, linker, SH2, and transactivation domains. However, the transactivation domain is absent in the alternative splicing variant, STAT3β.
Depending on context, truncated STAT3β can be phosphorylated at tyrosine 705 and bind to DNA sequence that is equal to that bound by STAT3α with negative transcriptional regulation. Overexpression of STAT3β can induce apoptosis and inhibit tumor growth[15,16]. However, alternative splicing regulation by antisense oligodeoxynucleotides targeting STAT3 can specifically shift the expression from STAT3α to STAT3β. High expression of endogenous STAT3β promotes cell apoptosis and leads to cell cycle arrest. This apoptosis-promoting effect of STAT3β is independent on the inhibition of STAT3α target genes. Several genes that differ from classic STAT3α target genes are specifically decreased by STAT3β knockdown, including lens epithelium-derived growth factor, p300/CBP-associated factor, Cyclin C, peroxisomal biogenesis factor 1 and STAT1β[3], indicating that STAT3β may promote cell apoptosis through regulating its own specific target genes in addition to negative transcriptional regulation of STAT3α.
POST-TRANSLATIONAL MODIFICATION OF STAT3
STAT3 phosphorylation
STAT3 protein exists in the cytoplasm as an inactive form until phosphorylation by receptor-associated kinases. Activated JAK kinases phosphorylate STAT3 through binding of the SH2 domain to a phosphorylated tyrosine residue, by which the C-terminus of p-STAT3 triggers its release from receptor, and form a homo- or hetero-dimerization of p-STAT3. Dimerized STAT3 translocates to the nucleus and binds to the promoters bearing cognate DNA-binding sequences[17]. STAT3 can be also phosphorylated by other tyrosine kinases, such as the Src family. However, such Src-induced STAT3 phosphorylation does not always result in STAT3 activation[18]. Tyrosine phosphorylation is necessary for STAT3 activity. In addition, serine phosphorylation at residue 727 of STAT3 also leads to the up-regulation of the transcriptional activity. STAT3 phosphorylation at Ser-727 is mediated by MAPK, P38 and c-Jun N-terminal kinase (JNK) pathways, and involved in transcriptional regulation of the target genes of STAT3[19]. Ser-727 mutant STAT3 knock-in mice display impaired development and survival process[20]. Recently, several articles reported that un-phosphorylated STAT3 can interact with nuclear factor-κB (NF-κB). Un-phosphorylated STAT3 (U-STAT3)/NF-κB complex translocates into the nucleus and activates the expression of NF-κB target genes[21].
STAT3 acetylation
Protein acetylation is a crucial post-translational modification of gene expression and involved in extensive physiological and pathological processes[22]. Investigation on protein acetylation is focused on the alteration of chromatin structure and activation of transcription factors. The inhibition of histone deacetylases (HDACs) can induce the acetylation of STAT3 at Lys-685, and acetylated STAT3 (Ac-STAT3) regulates the function of dendritic cells through activating the transcription of indoleamine 2,3-dioxygenase[23].
The significant increase in STAT3 acetylation at Lys-685 is detected in tumor tissues. CD44, a transmembrane glycoprotein, has been recognized as a marker for tumor cells. Activated CD44 can bind STAT3 and p300 in the nucleus and acetylate STAT3 at Lys-685. CD44/Ac-STAT3 complex activates cyclinD1 expression by binding to its promoter, leading to cell proliferation[24]. Additionally, Ac-STAT3 may be the major determinant for promoter methylation of tumor suppressor genes. DNA methyltransferase 1 (DNMT1) is primarily involved in the maintenance of methylation. Ac-STAT3/DNMT1 complex can induce gene silencing through binding to target genes, leading to increased CpG island methylation. STAT3 mutant at Lys685 exhibits impaired STAT3 acetylation and tumor growth. Acetylation inhibitors and HDAC activators can inhibit STAT3 acetylation with demethylation and reactivation of several tumor-suppressor genes, including cyclin-dependent kinase inhibitor 2A (CDKN2A), deleted in lung and esophageal cancer 1 (DLEC1) and STAT1. In triple-negative breast cancer cells and melanoma, Ac-STAT3 is related to the methylation of the ERα gene. Therefore, inhibition of Ac-STAT3 is favorable for hormone therapy through reactivating ERα expression[25].
Other post-translational modification of STAT3
Except for phosphorylation and acetylation, STAT1 and STAT3 are also subjected to SUMOylation through binding to small ubiquitin-like modifier (SUMO). STAT3 SUMOylation suppresses the transcriptional activity of STAT3 by affecting STAT3 phosphorylation and dimerization[26].
STAT3 LOCALIZATION AND FUNCTION
Nucleo-cytoplasmic shuttling of p-STAT3
Since protein synthesis and modification are processed in the cytoplasm, most transcription factors need to pass through the nuclear pore complex and enter into the nucleus to exert their transcriptional activity. In general, proteins that have a molecular weight greater than 50 kDa require specific structural domain named nuclear localization sequence (NLS) and nuclear export sequence (NES). Both NLS- and NES-containing proteins can recognize and combine with specific soluble carriers to mediate the nucleo-cytoplasmic trafficking[27]. Most NLS can recognize importin α and co-regulate the shuttling of proteins through interacting with importin β1[28].
The transcriptional regulatory activity of STAT3 is dependent on nuclear translocation. The distinction between STAT3 and other STAT members is that activated STAT3 can shuttle between the cytoplasm and nucleus, and accumulate in the nucleus to play the role in transcriptional activation. In the canonical nuclear translocation, p-STAT3 is released from the receptor, forms a homo- or hetero-dimer, and translocates into the nucleus. Importin α3 can specifically recognize the coiled-coil domain and mediate the nucleo-cytoplasmic shuttling of STAT3 protein[29].
Nucleo-cytoplasmic shuttling of U-STAT3
Previous studies showed that STAT3 protein acquires its DNA binding activity only in a phosphorylated form. However, recent studies indicated that the transcriptional activation of STAT3 in the nucleus is also independent of phosphorylation[21]. Both phosphorylated and unphosphorylated STAT3 proteins exist in the nucleus and regulate different target genes. Data from fluorescently-labeled STAT3 mutants in STAT3 deficient cells show that U-STAT3 can shuttle constitutively between the cytoplasm and nucleus under the condition of NLS and NES mutation, indicating that the nuclear accumulation of U-STAT3 is independent of the binding of NLS or NES and importins. Both native gel electrophoresis and dual-focus fluorescence correlation spectroscopy identify that the N-terminal domain is essential for dimer formation and nuclear accumulation of U-STAT3. The monomeric N-terminal deletion mutant can be phosphorylated and dimerized in response to IL-6 treatment without nuclear accumulation. Therefore, the N-terminal domain has an important role in nucleo-cytoplasmic trafficking of U-STAT3[30].
STAT3 in mitochondria
Except for the classic transcriptional regulation during cell proliferation and differentiation through nuclear translocation, STAT3 translocation in different organelles may regulate cell metabolism and be involved in a broad range of biological functions independent of transcriptional activity. For instance, phosphorylated STAT3 at Serine 727 (P-Ser-STAT3) is localized to the mitochondria of hepatocytes and myocardial cells. STAT3 deficient cells exhibit a low activity of complex I and II[31], suggesting that STAT3 regulates mitochondrial respiration via electron transport chain. Data from co-immunoprecipitation indicate that the translocation of STAT3 to mitochondria is mediated by the presequence receptor Tom20[32]. However, the mechanism that STAT3 alters mitochondrial respiration is controversial. There is an unfavorable ratio of complexes I/II and STAT3 in cardiac tissue, which implied the existence of an additional mechanism of STAT3 regulation of ATP production in vivo[33]. The sirtuin 1 (SIRT1), a NAD-dependent deacetylase, is located in the nucleus and known as a key factor regulating and controlling the mitochondrial bioenergetics by means of activating gene expression through deacetylating some important signal molecules, such as STAT3. In Sirt1-null cells, there is a significantly higher serine-phosphorylated STAT3 level in mitochondria with an increase in the mitochondrial bioenergetics and ATP formation[34].
In eukaryotes, the primary function of mitochondria is aerobic respiration and energy production, in which the reactive oxygen species (ROS) is the inevitable by-products. During the process of ischemia-reperfusion injury in the myocardium, the opening of mitochondrial permeability transition pore (MPTP) is a major response to cardiomyocyte death, while the ROS from respiratory chain is the primary endogenous reason for MPTP opening. Mitochondria play a major role in cardio-protection, most likely by preventing MPTP opening, while mitochondrial STAT3 has an impact on inhibiting MPTP opening and cardio-protection. In calcium-induced MPTP opening model, STAT3-KO mitochondria tolerate less induction of MPTP opening. The function of STAT3 in MPTP stability may be carried out through binding to cyclophilin D[32]. Another study found that GRIM-19-induced mitochondrial STAT3 location may involve in TNF-mediated necroptosis[35].
It is identified that cancer cells have the feature of metabolic turnover in aerobic glycolysis - the Warburg effect[36], in which STAT3 acts as a central mediator of cell metabolism through both HIF-1α-dependent and -independent mechanisms. Oncogenic signals activate STAT3 phosphorylation and induce STAT3 translocation into the nucleus where it regulates HIF-1α expression. Mitochondrial STAT3 displays Serine 727 phosphorylation, while tyrosine phosphorylation or DNA binding activity is not detected, unlike canonical transcriptional activation. p-Ser-STAT3 located in mitochondria shows many metabolic functions and induces malignant transformation mediated by oncogenic Ras[37]. Fibroblast growth factor receptor 4-R388 (FGFR4-R388), a known single nucleotide polymorphism which promotes breast cancer cell motility and invasiveness, can promote mitochondrial cytochrome c activity and induce pituitary tumor cell growth through STAT3 serine phosphorylation. Therefore, serine phosphorylation of STAT3 and mitochondrial translocation may contribute to tumor cell transformation and tumorigenesis[38].
FUNCTION OF STAT3 IN PATHOPHYSIOLOGY AND DEVELOPMENT
STAT3 in stem cells
Mouse embryonic stem cells (ES cells) are pluripotent cells derived from the inner cell mass of blastocysts. The self-renewal and pluripotency of ES cells depend on leukemia inhibitory factor (LIF) and bone morphogenetic protein 2 (BMP2) during in vitro culture[39]. Based on chromatin immunoprecipitation-deep sequencing (ChIP-seq), 13 specific transcriptional factors (Nanog, Oct4, STAT3, Smad1, Sox2, Zfx, c-Myc, n-Myc, Klf4, Esrrb, Tcfcp2l1, E2f1, and CTCF) and 2 transcription regulators (p300 and Suz12) are identified in the regulatory network of ES cells, and these factors are involved in LIF and BMP signaling pathways, and play important roles in self-renewal, reprogramming and pluripotency of ES cells[40].
LIF activates STAT3 through the Janus kinase (JAK) signal pathway. p-STAT3 is functionally associated with the transcriptional regulation of target genes for the self-renewal of ES cells, including Kruppel-like factors (Klf4 and Klf5)[41]. Furthermore, persistently activated STAT3 can maintain the self-renewal process without LIF[42]. Transcriptional factors Nanog and STAT3 are the molecular markers of ES cells. Nanog and STAT3 co-regulate the transcriptional activation of STAT3 target genes through binding to their promoters, such as α2M and Nanog promoters. This activation is abrogated by eliminating LIF, indicating that the function of Nanog and STAT3 is dependent on the LIF signal pathway[43]. Overexpression of STAT3 target genes, such as Klf4 and Klf5[41], has been shown to promote self-renewal of ES cells, while knockdown of these genes has no impact on the self-renewal in the presence of LIF or STAT3[44]. Gastrulation brain homeobox 2 (Gbx2), a LIF/STAT3 target gene, can facilitate the pluripotency of ES cells when over-expressed without LIF and STAT3[45]. These results illustrated that LIF/STAT3 may act upstream to trigger the maintenance of ES cells through activating a range of downstream target genes.
STAT3 in proliferation and apoptosis
P-STAT3 can activate proliferation-related genes to promote cell proliferation. Moreover, U-STAT3 can bind to the promoters of pro-apoptotic genes and inhibit their expression in tumor cells, but not in normal cells. Inhibitors of STAT3 phosphorylation or dominant-negative STAT3 mutants facilitate the expression of pro-apoptosis factors, suggesting that STAT3 plays a dominant role in regulating cell proliferation and anti-apoptosis[46]. STAT3 knockout mice exhibit complete embryonic lethality. STAT3 deficient embryos show a rapid degeneration on day 7 of pregnancy, highlighting the important role of STAT3 in embryo development[47]. Conditional ablation of STAT3 in myocardial cells leads to higher susceptibility to drug-induced heart failure[48]. In addition, ischemic preconditioning can induce the phosphorylation of STAT3 at Tyr-705 and Ser-727 in myocardial cells. However, the expression of cardio-protective factor (COX-2 and HO-1) and anti-apoptotic proteins [Mcl-1, Bcl-x (L) and c-FLIP (S)] is elevated in normal cells 24 h later, but not in STAT3 deficient cells[49]. These results illustrated the function of STAT3 in anti-inflammation and anti-apoptosis.
Mammary gland involution initiates at the ending of lactation, involving extensive apoptosis of the secretory alveolar epithelium and inflammatory response. Although STAT3 is expressed in the mammary gland throughout the whole reproductive cycle, it is only activated by LIF on the day of delivery and at 6-12 h after weaning[50]. STAT3 has an important role in mammary gland involution. Conditional ablation of STAT3 in mammary cells causes delayed involution of the mammary gland[51]. STAT3 is involved in the apoptotic process of mammary epithelial cells and tissue remodeling through inducing the expression of pro-apoptotic factors and regulating the balance of matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP)[52]. Mammary STAT3 deficient mice have impaired accumulation of inflammatory factors, macrophages and mastocytes in the mammary gland[53]. In addition, p-STAT3 in mammary epithelial cells is also involved in lysosomal-mediated cell death pathway through up-regulating the expression of lysosomal proteases cathepsin B and L[54]. Therefore, STAT3 expression in the mammary gland may participate in apoptosis under physiological conditions.
STAT3 in tumorigenesis and cancer-related inflammation
As a key transcriptional factor, p-STAT3 can translocate into the nucleus and bind to specific DNA sequences to activate the expression of target genes, including c-Myc and FGFR2, consequently regulating the proliferation, differentiation and anti-apoptosis of tumor cells[55,56]. Furthermore, acetylated STAT3 can induce the down-regulation of tumor suppressor genes through promoter methylation and facilitate tumorigenesis. MicroRNAs are short non-coding RNAs (ncRNAs) mediating post-transcriptional down-regulation of target genes and functioning in cell proliferation and apoptosis. MicroRNA-21 (miR-21) is an oncogene that contributes to anti-apoptosis in most tumor cells. There are two strictly conserved STAT3 binding sites in the enhancer of miR-21. MiR-21 induction by IL-6 is STAT3-dependent. ChIP results also confirm the accumulation of STAT3 in the upstream enhancer of miR-21[57], indicating that IL-6/STAT3 pathway contributes to miR-21 induction.
Chronic infection and inflammation contribute to about 15% of human cancers. The inflammatory response can induce necrotic cell death accompanied with activation of numerous cytokines, growth factors and chemokines which facilitate cell proliferation and survival[58]. The STAT3 signal pathway is the major intrinsic pathway for inflammation in tumor cells. STAT3 activates many inflammatory-related genes including BCL-XL, intercellular adhesion molecule 1 and vascular endothelial growth factor, and is involved in the maintenance of inflammatory environment[59]. NF-κB has the ability to induce the expression of inflammatory mediators, and is the major pathway functioning in inflammation-induced carcinogenesis and anti-tumor immunity. The signaling pathways of STATs, especially STAT3, are closely related with NF-κB signaling[60]. The inflammatory factor IL-6, the target gene of NF-κB, is the important STAT3 activator. In tumor cells, STAT3 directly interacts with NF-κB, translocates into the nucleus and contributes to the constitutive NF-κB activation in cancer. In addition, STAT3 binding to NF-κB also regulates numerous oncogenic and inflammatory genes[61].
Targeting the STAT3 pathway should be a promising and novel form of treatment for human cancers. Blocking STAT3 by siRNAs, antisense oligonucleotides, dominant-negative mutants, and specific inhibitors of STAT3 in combination with chemotherapeutics can synergistically inhibit the growth, invasion and metastasis of carcinoma cells[62-64]. Therefore, inhibiting STAT3 signals are a promising therapeutic target for most types of human cancers with constitutively activated STAT3.
STAT3 in reproduction
In mammals, the uterus is receptive to blastocyst during a restricted time termed as “implantation window”. LIF is expressed at a high level during implantation window in humans and mice. LIF deficient mice display embryo implantation failure[65]. In mouse uterus, STAT3 protein is expressed and phosphorylated in the luminal epithelium on day 4 of pregnancy. LIF treatment induces the STAT3 phosphorylation in mouse uterine luminal epithelium isolated from day 4 of pregnancy but not for days 3 and 5[66]. LIF antagonist (LA, truncated LIF protein) injection led to the failure of mouse embryo implantation through inhibiting STAT3 phosphorylation[67]. In humans, LIF and STAT3 are expressed in decidual tissues during early pregnancy. LIF can activate STAT3 phosphorylation in both non-decidualized and decidualized human endometrial stromal cells in vitro[68], indicating that LIF/STAT3 signaling is involved in human embryo implantation and decidualization.
To investigate the function of STAT3 during embryo implantation, a cell-permeable STAT3 peptide inhibitor is injected into mouse uterine lumen before implantation, which significantly reduces embryo implantation by 70%. STAT3 phosphorylation in uterine luminal epithelium activated by LIF and some LIF targeted genes, such as Irg1, is significantly inhibited by STAT3 inhibitors both in vivo and in vitro[69]. Meanwhile, the injection of STAT3 decoy into uterine lumen during implantation also causes implantation failure[70]. Co-immunoprecipitation assay showed that STAT3 can bind to progesterone receptor A (PR-A) and co-regulate the embryo implantation and decidualization in mice. Conditional ablation of STAT3 only in PR-positive cells (PRcre/+Stat3f/f; Stat3d/d) is used to investigate the role of STAT3 in reproduction. Conditional ablation of STAT3 in the uterus (Stat3d/d) results in embryo implantation failure. Furthermore, Stat3d/d mice are also defective in hormonally induced decidual reaction[71], suggesting that the interaction between STAT3 and PR is essential for successful implantation.
CONCLUSION
STAT3 is a key transcription factor and regulates a multitude of genes important for proliferation, differentiation, apoptosis, inflammation and tumorigenesis. STAT3 expression and activity are regulated through alternative splicing, post-translational modification and subcellular localization. STAT3β, the new isoform of STAT3, participates in apoptosis and plays a role distinct from STAT3α. Despite the different mechanism, STAT3 activation through phosphorylation or acetylation can facilitate tumorigenesis synergistically. STAT3 shuttles among the cytoplasm, nucleus, mitochondria and some other possible organelles, and exerts its diverse functions in transcriptional regulation, cellular respiration, proliferation and apoptosis. A variety of animal models reveal that STAT3 is essential for embryo development, pluripotency maintenance of stem cells, embryo implantation and decidualization. Increasing evidence confirms that STAT3 is a key modulator of cancer and inflammation (Figure 2). Hence, further clarification of the biological function of STAT3 will validate its promising application prospect for gene therapy in multi-directions.
Figure 2.
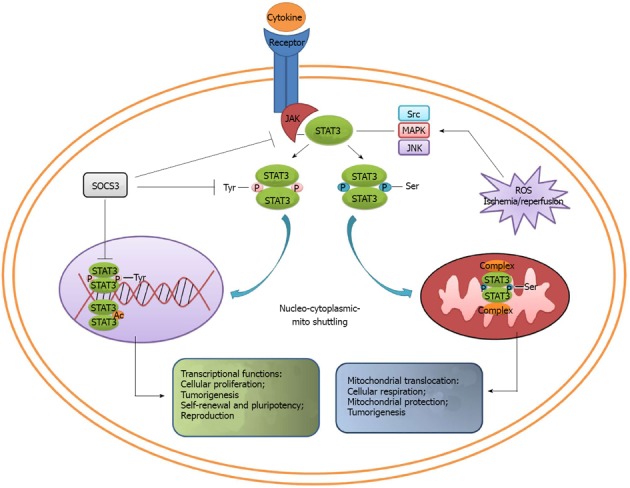
Converging roles of signal transducer and activator of transcription 3. Different signals can selectively trigger signal transducer and activator of transcription 3 phosphorylation. Tyr-phosphorylated STAT3 translocates into the nucleus and regulates gene expression, thus playing an important role in cell proliferation, tumorigenesis, self-renewal and pluripotency. On the other hand, Ser-phosphorylated STAT3 translocates into mitochondria, binds with the complexes in respiratory chain, and ultimately maintains the cellular respiration and mitochondrial protection. MAPK: Mitogen-activated protein kinase; ROS: Reactive oxygen species; JNK: c-Jun N-terminal kinase; JAK: Janus kinase.
Footnotes
Supported by National Natural Science Foundation of China, No. 30930013
P- Reviewers: Aggarwal A, JuergensKU S- Editor: Zhai HH L- Editor: Wang TQ E- Editor: Lu YJ
References
- 1.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15:79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kathiria AS, Neumann WL, Rhees J, Hotchkiss E, Cheng Y, Genta RM, Meltzer SJ, Souza RF, Theiss AL. Prohibitin attenuates colitis-associated tumorigenesis in mice by modulating p53 and STAT3 apoptotic responses. Cancer Res. 2012;72:5778–5789. doi: 10.1158/0008-5472.CAN-12-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zammarchi F, de Stanchina E, Bournazou E, Supakorndej T, Martires K, Riedel E, Corben AD, Bromberg JF, Cartegni L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc Natl Acad Sci USA. 2011;108:17779–17784. doi: 10.1073/pnas.1108482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, Tarasov SG, Zhang X, Korostyshevskiy VR, Cheema A, Zhang L, et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J Biol Chem. 2012;287:14192–14200. doi: 10.1074/jbc.M111.323899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw PE. Could STAT3 provide a link between respiration and cell cycle progression? Cell Cycle. 2010;9:4294–4296. doi: 10.4161/cc.9.21.13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J Biol Chem. 1998;273:6132–6138. doi: 10.1074/jbc.273.11.6132. [DOI] [PubMed] [Google Scholar]
- 7.Gao H, Bryzgalova G, Hedman E, Khan A, Efendic S, Gustafsson JA, Dahlman-Wright K. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol. 2006;20:1287–1299. doi: 10.1210/me.2006-0012. [DOI] [PubMed] [Google Scholar]
- 8.Yan S, Zhou C, Zhang W, Zhang G, Zhao X, Yang S, Wang Y, Lu N, Zhu H, Xu N. beta-Catenin/TCF pathway upregulates STAT3 expression in human esophageal squamous cell carcinoma. Cancer Lett. 2008;271:85–97. doi: 10.1016/j.canlet.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 9.Haan S, Wüller S, Kaczor J, Rolvering C, Nöcker T, Behrmann I, Haan C. SOCS-mediated downregulation of mutant Jak2 (V617F, T875N and K539L) counteracts cytokine-independent signaling. Oncogene. 2009;28:3069–3080. doi: 10.1038/onc.2009.155. [DOI] [PubMed] [Google Scholar]
- 10.Leroith D, Nissley P. Knock your SOCS off! J Clin Invest. 2005;115:233–236. doi: 10.1172/JCI24228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji Y, Wang Z, Li Z, Li K, Le X, Zhang T. Angiotensin II induces angiogenic factors production partly via AT1/JAK2/STAT3/SOCS3 signaling pathway in MHCC97H cells. Cell Physiol Biochem. 2012;29:863–874. doi: 10.1159/000171034. [DOI] [PubMed] [Google Scholar]
- 12.Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox TH, Park K, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci USA. 2012;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 14.Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JA, Lammers JW, Koenderman L, de Groot RP. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J Biol Chem. 1996;271:13221–13227. doi: 10.1074/jbc.271.22.13221. [DOI] [PubMed] [Google Scholar]
- 15.Huang Y, Qiu J, Dong S, Redell MS, Poli V, Mancini MA, Tweardy DJ. Stat3 isoforms, alpha and beta, demonstrate distinct intracellular dynamics with prolonged nuclear retention of Stat3beta mapping to its unique C-terminal end. J Biol Chem. 2007;282:34958–34967. doi: 10.1074/jbc.M704548200. [DOI] [PubMed] [Google Scholar]
- 16.Ng IH, Ng DC, Jans DA, Bogoyevitch MA. Selective STAT3-α or -β expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem J. 2012;447:125–136. doi: 10.1042/BJ20120941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L, Nam S, Tian Y, Yang F, Wu J, Wang Y, Scuto A, Polychronopoulos P, Magiatis P, Skaltsounis L, et al. 6-Bromoindirubin-3’-oxime inhibits JAK/STAT3 signaling and induces apoptosis of human melanoma cells. Cancer Res. 2011;71:3972–3979. doi: 10.1158/0008-5472.CAN-10-3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michels S, Trautmann M, Sievers E, Kindler D, Huss S, Renner M, Friedrichs N, Kirfel J, Steiner S, Endl E, et al. SRC signaling is crucial in the growth of synovial sarcoma cells. Cancer Res. 2013;73:2518–2528. doi: 10.1158/0008-5472.CAN-12-3023. [DOI] [PubMed] [Google Scholar]
- 19.Tworkoski K, Singhal G, Szpakowski S, Zito CI, Bacchiocchi A, Muthusamy V, Bosenberg M, Krauthammer M, Halaban R, Stern DF. Phosphoproteomic screen identifies potential therapeutic targets in melanoma. Mol Cancer Res. 2011;9:801–812. doi: 10.1158/1541-7786.MCR-10-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen Y, Schlessinger K, Zhu X, Meffre E, Quimby F, Levy DE, Darnell JE. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol. 2004;24:407–419. doi: 10.1128/MCB.24.1.407-419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–451. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 22.Hohl M, Wagner M, Reil JC, Müller SA, Tauchnitz M, Zimmer AM, Lehmann LH, Thiel G, Böhm M, Backs J, et al. HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest. 2013;123:1359–1370. doi: 10.1172/JCI61084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Y, Chin YE, Weisiger E, Malter C, Tawara I, Toubai T, Gatza E, Mascagni P, Dinarello CA, Reddy P. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J Immunol. 2009;182:5899–5903. doi: 10.4049/jimmunol.0804388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JL, Wang MJ, Chen JY. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J Cell Biol. 2009;185:949–957. doi: 10.1083/jcb.200812060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, Hoon DS, Forman SJ, Jove R, Riggs AD, et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci USA. 2012;109:7765–7769. doi: 10.1073/pnas.1205132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Droescher M, Begitt A, Marg A, Zacharias M, Vinkemeier U. Cytokine-induced paracrystals prolong the activity of signal transducers and activators of transcription (STAT) and provide a model for the regulation of protein solubility by small ubiquitin-like modifier (SUMO) J Biol Chem. 2011;286:18731–18746. doi: 10.1074/jbc.M111.235978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Q, Lu Z, Liu Q, Guo L, Ren H, Fu J, Jiang Q, Clarke PR, Zhang C. Chromatin-bound NLS proteins recruit membrane vesicles and nucleoporins for nuclear envelope assembly via importin-α/β. Cell Res. 2012;22:1562–1575. doi: 10.1038/cr.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi K, Morita T. Differences in the nuclear export mechanism between myocardin and myocardin-related transcription factor A. J Biol Chem. 2013;288:5743–5755. doi: 10.1074/jbc.M112.408120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci USA. 2005;102:8150–8155. doi: 10.1073/pnas.0501643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogt M, Domoszlai T, Kleshchanok D, Lehmann S, Schmitt A, Poli V, Richtering W, Müller-Newen G. The role of the N-terminal domain in dimerization and nucleocytoplasmic shuttling of latent STAT3. J Cell Sci. 2011;124:900–909. doi: 10.1242/jcs.072520. [DOI] [PubMed] [Google Scholar]
- 31.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105:771–785. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillips D, Reilley MJ, Aponte AM, Wang G, Boja E, Gucek M, Balaban RS. Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein-protein interactions. J Biol Chem. 2010;285:23532–23536. doi: 10.1074/jbc.C110.152652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernier M, Paul RK, Martin-Montalvo A, Scheibye-Knudsen M, Song S, He HJ, Armour SM, Hubbard BP, Bohr VA, Wang L, et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J Biol Chem. 2011;286:19270–19279. doi: 10.1074/jbc.M110.200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shulga N, Pastorino JG. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J Cell Sci. 2012;125:2995–3003. doi: 10.1242/jcs.103093. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Haigis MC, Deng CX, Finley LW, Kim HS, Gius D. SIRT3 is a mitochondrial tumor suppressor: a scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res. 2012;72:2468–2472. doi: 10.1158/0008-5472.CAN-11-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tateno T, Asa SL, Zheng L, Mayr T, Ullrich A, Ezzat S. The FGFR4-G388R polymorphism promotes mitochondrial STAT3 serine phosphorylation to facilitate pituitary growth hormone cell tumorigenesis. PLoS Genet. 2011;7:e1002400. doi: 10.1371/journal.pgen.1002400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 41.Bourillot PY, Aksoy I, Schreiber V, Wianny F, Schulz H, Hummel O, Hubner N, Savatier P. Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem Cells. 2009;27:1760–1771. doi: 10.1002/stem.110. [DOI] [PubMed] [Google Scholar]
- 42.Matsuda T, Nakamura T, Nakao K, Arai T, Katsuki M, Heike T, Yokota T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999;18:4261–4269. doi: 10.1093/emboj/18.15.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torres J, Watt FM. Nanog maintains pluripotency of mouse embryonic stem cells by inhibiting NFkappaB and cooperating with Stat3. Nat Cell Biol. 2008;10:194–201. doi: 10.1038/ncb1680. [DOI] [PubMed] [Google Scholar]
- 44.Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, Ng HH. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. 2008;10:353–360. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 45.Tai CI, Ying QL. Gbx2, a LIF/Stat3 target, promotes reprogramming to and retention of the pluripotent ground state. J Cell Sci. 2013;126:1093–1098. doi: 10.1242/jcs.118273. [DOI] [PubMed] [Google Scholar]
- 46.Timofeeva OA, Tarasova NI, Zhang X, Chasovskikh S, Cheema AK, Wang H, Brown ML, Dritschilo A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc Natl Acad Sci USA. 2013;110:1267–1272. doi: 10.1073/pnas.1211805110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindler CW. Series introduction. JAK-STAT signaling in human disease. J Clin Invest. 2002;109:1133–1137. doi: 10.1172/JCI15644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA. 2003;100:12929–12934. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolli R, Stein AB, Guo Y, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu Y, Xuan YT. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol. 2011;50:589–597. doi: 10.1016/j.yjmcc.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Kritikou EA, Sharkey A, Abell K, Came PJ, Anderson E, Clarkson RW, Watson CJ. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development. 2003;130:3459–3468. doi: 10.1242/dev.00578. [DOI] [PubMed] [Google Scholar]
- 51.Humphreys RC, Bierie B, Zhao L, Raz R, Levy D, Hennighausen L. Deletion of Stat3 blocks mammary gland involution and extends functional competence of the secretory epithelium in the absence of lactogenic stimuli. Endocrinology. 2002;143:3641–3650. doi: 10.1210/en.2002-220224. [DOI] [PubMed] [Google Scholar]
- 52.Baxter FO, Neoh K, Tevendale MC. The beginning of the end: death signaling in early involution. J Mammary Gland Biol Neoplasia. 2007;12:3–13. doi: 10.1007/s10911-007-9033-9. [DOI] [PubMed] [Google Scholar]
- 53.Hughes K, Wickenden JA, Allen JE, Watson CJ. Conditional deletion of Stat3 in mammary epithelium impairs the acute phase response and modulates immune cell numbers during post-lactational regression. J Pathol. 2012;227:106–117. doi: 10.1002/path.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW, Watson CJ. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011;13:303–309. doi: 10.1038/ncb2171. [DOI] [PubMed] [Google Scholar]
- 55.Ochi A, Graffeo CS, Zambirinis CP, Rehman A, Hackman M, Fallon N, Barilla RM, Henning JR, Jamal M, Rao R, et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J Clin Invest. 2012;122:4118–4129. doi: 10.1172/JCI63606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei W, Liu W, Cassol CA, Zheng W, Asa SL, Ezzat S. The breast cancer susceptibility gene product fibroblast growth factor receptor 2 serves as a scaffold for regulation of NF-κB signaling. Mol Cell Biol. 2012;32:4662–4673. doi: 10.1128/MCB.00935-12. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Löffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermüller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–1333. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 58.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee H, Deng J, Xin H, Liu Y, Pardoll D, Yu H. A requirement of STAT3 DNA binding precludes Th-1 immunostimulatory gene expression by NF-κB in tumors. Cancer Res. 2011;71:3772–3780. doi: 10.1158/0008-5472.CAN-10-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He G, Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma J, Wang S, Zhao M, Deng XS, Lee CK, Yu XD, Liu B. Therapeutic potential of cladribine in combination with STAT3 inhibitor against multiple myeloma. BMC Cancer. 2011;11:255. doi: 10.1186/1471-2407-11-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Souissi I, Najjar I, Ah-Koon L, Schischmanoff PO, Lesage D, Le Coquil S, Roger C, Dusanter-Fourt I, Varin-Blank N, Cao A, et al. A STAT3-decoy oligonucleotide induces cell death in a human colorectal carcinoma cell line by blocking nuclear transfer of STAT3 and STAT3-bound NF-κB. BMC Cell Biol. 2011;12:14. doi: 10.1186/1471-2121-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, Lee H, Scuto A, Liu Y, Yang C, et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27:925–932. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang S, Lin H, Kong S, Wang S, Wang H, Wang H, Armant DR. Physiological and molecular determinants of embryo implantation. Mol Aspects Med. 2013;34:939–980. doi: 10.1016/j.mam.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng JG, Chen JR, Hernandez L, Alvord WG, Stewart CL. Dual control of LIF expression and LIF receptor function regulate Stat3 activation at the onset of uterine receptivity and embryo implantation. Proc Natl Acad Sci USA. 2001;98:8680–8685. doi: 10.1073/pnas.151180898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.White CA, Zhang JG, Salamonsen LA, Baca M, Fairlie WD, Metcalf D, Nicola NA, Robb L, Dimitriadis E. Blocking LIF action in the uterus by using a PEGylated antagonist prevents implantation: a nonhormonal contraceptive strategy. Proc Natl Acad Sci USA. 2007;104:19357–19362. doi: 10.1073/pnas.0710110104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shuya LL, Menkhorst EM, Yap J, Li P, Lane N, Dimitriadis E. Leukemia inhibitory factor enhances endometrial stromal cell decidualization in humans and mice. PLoS One. 2011;6:e25288. doi: 10.1371/journal.pone.0025288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Catalano RD, Johnson MH, Campbell EA, Charnock-Jones DS, Smith SK, Sharkey AM. Inhibition of Stat3 activation in the endometrium prevents implantation: a nonsteroidal approach to contraception. Proc Natl Acad Sci USA. 2005;102:8585–8590. doi: 10.1073/pnas.0502343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakamura H, Kimura T, Koyama S, Ogita K, Tsutsui T, Shimoya K, Taniguchi T, Koyama M, Kaneda Y, Murata Y. Mouse model of human infertility: transient and local inhibition of endometrial STAT-3 activation results in implantation failure. FEBS Lett. 2006;580:2717–2722. doi: 10.1016/j.febslet.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 71.Lee JH, Kim TH, Oh SJ, Yoo JY, Akira S, Ku BJ, Lydon JP, Jeong JW. Signal transducer and activator of transcription-3 (Stat3) plays a critical role in implantation via progesterone receptor in uterus. FASEB J. 2013;27:2553–2563. doi: 10.1096/fj.12-225664. [DOI] [PMC free article] [PubMed] [Google Scholar]