

Abstract
KCNB1, a voltage-gated potassium (K+) channel that conducts a major delayed rectifier current in the brain, pancreas and cardiovascular system is a key player in apoptotic programs associated with oxidative stress. As a result, this protein represents a bona fide drug target for limiting the toxic effects of oxygen radicals. Until recently the consensus view was that reactive oxygen species trigger a pro-apoptotic surge in KCNB1 current via phosphorylation and SNARE-dependent incorporation of KCNB1 channels into the plasma membrane. However, new evidence shows that KCNB1 can be modified by oxidants and that oxidized KCNB1 channels can directly activate pro-apoptotic signaling pathways. Hence, a more articulated picture of the pro-apoptotic role of KCNB1 is emerging in which the protein induces cell’s death through distinct molecular mechanisms and activation of multiple pathways. In this review article we discuss the diverse functional, toxic and protective roles that KCNB1 channels play in the major organs where they are expressed.
Keywords: Apoptosis, Kv2.1, Aging, Reactive oxygen species, Alzheimer’s disease
Core tip: KCNB1 is a K+ channel that plays a key role in the brain, pancreas and cardiovascular system. KCNB1 is unique in that it induces apoptosis in association with oxidative stress. In this review article we discuss the diverse roles of this channel in the organs where it is expressed including recent advances in the molecular mechanisms through which KCNB1 causes cytotoxicity.
KCNB1 IS A PROMINENT MEMBER OF THE SHAB-RELATED FAMILY OF VOLTAGE-GATED K+ CHANNELS
KCNB1 (HUGO nomenclature), formerly DRK1 and Kv2.1, is a Shab delayed rectifier voltage-gated K+ channel which was cloned by Frech et al[1] using size-fractionated mRNA extracted from rat brain. KCNB1 is expressed in the central nervous system, pancreas, pulmonary arteries, heart, auditory outer hair cells, stem cells and retina[2-21]. As in other voltage-gated K+ channels, KCNB1 spatial and temporal expressions are both developmentally regulated. For example, three distinct (4.4, 9.0, 11.5 kb) mRNA transcripts are expressed in the rat brain, with the 4.4 kb transcript being predominant in embryos and the 11.5 kb transcript being predominant in adults[15]. Accordingly, multiple KCNB1 isoforms are detected which differ in their developmental expression. Functional KCNB1 channels can result from the assembly of four identical pore-forming subunits along a symmetry axis[22]. However, this simple stoichiometry is not likely to be observed in nature. In order to serve the specific requisites of the tissues in which the channel is expressed, heterogeneity of KCNB1 current can be achieved by formation of heterometric complexes containing non-conducting, pore-forming subunits of the KCNG and KCNS families as well as by assembly with accessory subunits of the KCNAB and KCNE families[16,17,23-28]. KCNB1 exhibits an unusual large number of consensus sites for phosphorylation. Accordingly, the channel is a substrate for protein kinases of different families and is constitutively phosphorylated in native cells[29-32]. KCNB1 can also be SUMOylated and acetylated in nervous system and pancreas even though the physiological role of these regulations awaits elucidation[33-35]. Finally, mature KCNB1 channels are not glycosylated despite the presence of consensus sites in the N-terminus[36].
Because of the potential therapeutic implications, the pharmacology of KCNB1 to a variety of toxins and drugs has been extensively investigated. Thus, KCNB1 is blocked by tarantula toxins that belong to the same structural family of inhibitor cystine knot spider peptides reticulated by disulfide bridges. Hanatoxin from G. spatulata, was the first toxin to be shown to interact with KCNB1, followed in more recent years by heteroscordratoxin and stromatoxin 1 from H. maculata and S. calceata and jingzhaotoxin (JZTX-I, -III, and -V) and guangxitoxin (GxTx-1E), isolated from the venoms of the Chinese tarantulas C. jingzhao and P. guangxiensis[37-40]. All these structurally related toxins exhibit variable affinities for the channel in the nanomolar to micromolar range and act to alter its gating by interacting with the voltage sensor[41,42]. KCNB1 is susceptible to inhibition by a number of compounds of different classes including classic K+ channel blockers tetraethylammonium (TEA) and 4-aminopyridine and antipsychotic, anesthetic and antiarrhythmic compounds[43-53]. Of particular relevance to the topic of this review is the fact that acetylcholinesterase inhibitor Donepezil, a drug used in the treatment of Alzheimer’s disease and vascular dementia, protects neurons from apoptosis by inhibiting KCNB1[54]. The exact mechanism awaits elucidation but recent findings showing that KCNB1 is subject to direct oxidative modification may suggest that the protective effect of the drug may stem from its ability to prevent the oxidation of KCNB1[55].
In summary, toxins and synthetic drugs have significantly contributed to the effort of dissecting native KCNB1 currents in various tissues and probing channel’s structure and functional mechanisms of gating.
KCNB1 IS A CRITICAL MEDIATOR OF HIPPOCAMPAL AND CORTICAL EXCITABILITY
KCNB1 is broadly expressed in the brain and is a major contributor to the delayed rectifier somatodendritic K+ current in hippocampal and cortical neurons[14,56]. In electrically quiescent neurons, KCNB1 is mostly localized in microdomains in the membranes of dendrites and cell bodies where it is constitutively phosphorylated and poorly conducting[20,21,29-32,57-60]. Upon the onset of neuronal activity, a series of cellular events are initiated that lead to de-phosphorylation of the channel. This transition is associated with two major changes in channel’s status: (1) its threshold for voltage activation is lowered; and (2) KCNB1 is released from the microdomains and begins to diffuse in the membrane[30]. The net effect of these changes is that KCNB1 conducts a delayed rectifier K+ current that acts to slow down and/or terminate periods of high frequency firing. Activity-dependent phosphorylation/de-phosphorylation of KCNB1 is partly mediated by cyclin-dependent kinase 5 and the phosphatase calcineurin. The latter is activated by a calcium influx driven by the electrical activity of the neuron[29-32]. Using mass spectrometry, Trimmer and colleagues identified 16 phosphorylation sites in KCNB1, of which roughly half provided a substrate for calcineurin[32]. This indicates that modulation of KCNB1 by protein kinases is graded to reflect dynamic regulation of neuron firing properties. However, KCNB1 can also terminate periods of neuronal activity by being directly phosphorylated. For example, AMP-activated protein kinase (AMPK, which is activated by ATP depletion) can phosphorylate KCNB1 at residue S440 and induce hyperpolarizing shifts in the current-voltage relationship for activation, shifts that make the channel more conductive at negative voltages[61].
KCNB1 PROMOTES APOPTOSIS IN RESPONSE TO OXIDATIVE STRESS
KCNB1 is a specific mediator of apoptosis in a variety of neuronal cell types including hippocampal, cortical and granule neurons[62-65]. For example, a study investigating the molecular correlate of the apoptotic K+ current in hippocampal neurons found that among nine alpha and 3 beta Kv subunits screened, KCNB1 was the primary correlate[63]. Several groups have demonstrated that the key event triggering KCNB1-induced apoptosis is an increase in reactive oxygen species (ROS), either following acute oxidation, or as a consequence of cellular stresses such as serum deprivation and excitotoxicity[55,62-65]. It is currently accepted that dysregulated K+ homeostasis causes apoptosis by inducing mitochondrial swelling and depolarization, ROS generation, deficient energy production and cell volume decrease[66]. Accordingly, augmented insertion of KCNB1 channels into the plasma membrane is observed in neurons subjected to oxidative challenges[67]. The accompanying increase in KCNB1 current is thought to be a key step in the apoptotic program. The execution of the latter requires phosphorylation of KCNB1 by multiple types of protein kinases a fact that should not surprise considering the primary role that phosphorylation plays for the function of KCNB1. Zhou et al[68] investigated apoptosis induced by lack of serum in granular neurons and found that this was associated with upregulation of KCNB1 via the activation of a signaling pathway involving cAMP, protein kinase A and cAMP response element-binding protein (CREB). Aras et al[69] have identified several kinases including apoptosis signal-regulating kinase 1 (ASK1), p38 MAPK-dependent kinase, c-Src tyrosine kinase, and Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) that interact with KCNB1 in response to oxidative stress[70-73]. Their studies have provided a model that predicts that oxygen radicals induce simultaneous increases in cytosolic levels of Zn2+ and Ca2+. These increases activate the previously listed kinases and accelerate KCNB1 forward trafficking by modulating and facilitating its interaction with SNARE family protein syntaxin. This apoptotic program is tightly regulated: knock down of just a single phosphorylation site (S800 for p-38, Y124 for c-Src) is sufficient to suppress the pro-apoptotic influence of KCNB1[70]. However, Src tyrosine kinases and protein tyrosine phosphatase epsilon (PTP epsilon) also play a role in the physiological modulation of KCNB1. In the Schwann cells of mice, Src-mediated phosphorylation of Y124, (the same residue responsible for Zn2+/Ca2+ induced apoptosis), causes specific augmentation of KCNB1 current which appears to be critical for Schwann cell proliferation and myelination[74,75]. In fact, de-phosphorylation of KCNB1 at Y124 by PTP epsilon reduces KCNB1 activity and stops KCNB1-induced myelination[76,77]. Accordingly, mice lacking PTP epsilon exhibit hypomyelination of sciatic nerve axons at an early post-natal age, an effect due to constitutive activation of KCNB1 by Src tyrosine kinases[78]. Moreover, a number of K+ channels can cause apoptosis via dysregulated K+ homeostasis in a variety of cell types[66]. Therefore, increased K+ current may not be the key feature responsible for the specific ability of KCNB1 to promote apoptosis, but rather a consequence of it. Recent work from our laboratory may shed light on this issue. Cotella et al[55] showed that oxygen radicals directly modify KCNB1 channels, leading to the formation of oligomers held together by disulfide bridges[55]. A KCNB1 variant which does not form oligomers, obtained by mutating an N-terminal cysteine (C73A), fails to increase apoptosis in mammalian cells. Cotella et al[55] further showed that in inside-out patches, oxidants inhibit KCNB1 current. These findings imply that the formation of oligomers, rather than KCNB1 current, is the event that triggers an initial pro-apoptotic stimulus. To answer this question, Wu et al[79] have investigated the fate of the KCNB1 oligomers. They found that they accumulate in the plasma membrane as a result of defective internalization. Notably, accumulation is transient, and normal endocytosis/surface expression are mostly restored within one hour post-oxidation. The transient accumulation of KCNB1 oligomers is associated with activation of c-Src and JNK kinases coupled to a steady increase in the levels of free radicals. Thus, oligomer-induced activation of a “death pathway” appears to trigger the initial pro-apoptotic stimulus. As apoptosis progresses and ROS levels increase in the cell, the surge of KCNB1 current follows to further execute the apoptotic program (Figure 1).
Figure 1.
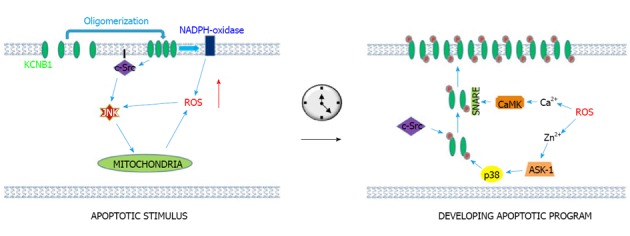
A two-step model for the pro-apoptotic actions of KCNB1. Upon exposure to oxidants, KCNB1 oligomers are formed. They accumulate in the plasma membrane thereby perturbing the organization of lipid rafts. This results in activation of an apoptotic stimulus mediated by c-Src and downstream, JNK kinases. As result of activation of c-Src and JNK kinases and in part of NADPH-oxidase (Xilong Wu, private communication) which is localized in the plasma membrane, ROS levels increase in the cell. ROS induce a raise in cytosolic Ca2+ and Zn2+ that initiate a phosphorylation-mediated surge of KCNB1 channels that further drives apoptosis. The signaling pathway activated by Zn2+ proceeds through activation of p38 by ASK-1 and independently, of c-Src tyrosine kinases (Zn2+ inhibits the activity of the tyrosine phosphatase PTP epsilon) which phosphorylate KCNB1 at S800 and Y124 thereby allowing interaction with SNARE family protein syntaxin. The Ca2+ signaling pathway results in activation of CaMKII kinase which in turn acts to modulate the interaction of KCNB1 with syntaxin. It is not known whether Src and p38 phosphorylation directly act to increase KCNB1 current. ROS: Reactive oxygen species.
INHIBITION OF KCNB1 MAY REPRESENT A VALID ANTI-APOPTOTIC STRATEGY
Pharmacological inhibition of KCNB1 current may represent a valid approach to preventing apoptosis. Accordingly, Peers and colleagues have shown that carbon monoxide (CO) can provide neuronal protection against an increase in KCNB1 current via regulating ROS and protein kinase G activity[80]. The same group has further proposed that the anti-apoptotic effect of CO may also be partially responsible for the etiology of cancer, as many oncogenic cells constitutively express heme oxygenase-1 (HO-1), which generates CO as a by product of its catalytic activity[81]. Chronic viruses, which establish a state of persistent infection by rendering infected cells resistant to apoptosis also appear to exploit inhibition of KCNB1 current. In human hepatocytes infected with hepatitis C virus (HCV), oxidative insults fail to initiate apoptosis because the HCV NS5A protein inhibits phosphorylation of KCNB1 by p38 MAPK and thus suppresses the current surge[82,83]. Furthermore, a neuronal NS5A isoform from HCV genotype 1b, NS5A1b, protects rat neurons against apoptosis by inhibiting KCNB1[73]. However, while NS5A acts on tyrosine kinase phosphorylation at residue Y124, NS5A1b inhibits p38-MAPK at residue S800 suggesting that the actions of these viral proteins are genotype-selective probably reflecting the characteristic of these viruses to target specific tissues.
VASOCONSTRICTION OF SMALL PULMONARY ARTERIES MAY PROCEED THROUGH DIRECT INHIBITION OF KCNB1 CURRENT BY ROS
Hypoxic pulmonary vasoconstriction is a physiological response to alveolar hypoxia, in which blood flow is redirected to better ventilated lobes via constriction of small pulmonary arteries. The mechanical force leading to vasoconstriction is exerted by pulmonary arteries smooth muscle cells (PASMCs). Hypoxia initially promotes PASMCs depolarization via inhibition of an oxygen-sensitive K+ current. This leads to the activation of L-type Ca2+ channels, which elevate cytosolic calcium thereby triggering PASMCs contraction. Biochemical, pharmacological, electrophysiological and genetic evidence designates KCNB1 - alone or mixed with KCNS3 silent subunits-as one of the major molecular correlates of the oxygen-sensitive K+ current in PASMCs[16-18,84,85]. Studies using the human ductus arteriosus as model system have provided a detailed picture of the cellular and molecular events leading to vasoconstriction during hypoxia[86-88]. Changes in O2 levels are translated to the mitochondrial electron transport chain (KCNB1 is insensitive to O2[89]) which responds by speeding the synthesis of diffusible hydrogen peroxide (H2O2). H2O2 causes smooth muscle cell depolarization, via inhibition of K+ current which further results in influx of calcium through L-type channels. The molecular details of the mechanism that links H2O2 to K+ current inhibition were not known previously but the fact that KCNB1 can be directly oxidized by H2O2 and most importantly, that its current is suppressed by oxidants may now provide a natural explanation for this mechanism of inhibition. It is worth noticing that chronic hypoxia is characterized by depolarized resting potential and elevated cytosolic Ca2+. Chronic depolarization is achieved by downregulation of KCNB1 protein[90-93] through mechanisms not completely understood, even though studies have implicated 15-lipoxygenase catalysis of arachidonic acid and hypoxia-inducible factor 1 in the mechanism[94,95]. Thus, different regulations of KCNB1 appear to mediate acute versus chronic conditions of hypoxia.
CONCLUSION
KCNB1 is a channel with a double-hedged sword nature: it is essential to the physiology of multiple organs, including the brain, pancreas and cardiovascular system and further acts as a mediator of apoptosis in response to oxidative stresses[2-21]. Dysregulated K+ homeostasis is a well established mechanism through which K+ channels contribute to an apoptotic program with a great deal of evidence implicating that KCNB1 do indeed work in this mechanism[55,62-65,67]. However recent findings have unveiled new ways through which KCNB1 mediates cell death: by giving rise to cytotoxic protein aggregates that result from direct oxidation of the protein[79]. The accumulation of these KCNB1 oligomers in the plasma membrane is transient but sufficient to trigger a pro-apoptotic signal via activation of a c-Src/JNK kinases pathway. As the apoptotic program progresses, a surge of KCNB1 current follows to induce mitochondrial destabilization, ROS generation, deficient energy production and cell volume decrease. Hence, KCNB1 plays a double role as both initiator and later executor of the apoptotic program.
Aging pathologies pose new challenges to health care, because even as advances in medicine are increasing lifespan, health problems become more prevalent as people age. A recent survey done by Harvard University School of Public Health and the Alzheimer’s Europe Consortium suggests that senile dementia is the second leading health concern after cancer[96]. Aging is also the most important risk factor in neurodegenerative conditions such as Alzheimer’s disease, the third most costly disease in the United Sates[97]. It is projected that the number of Western elders suffering from dementia and related neurodegenerative disease will increase by 350% by the midcentury[98,99]. Therefore, because of the impact of increasing lifespan on global human health issues, it is important to elucidate the cellular and molecular processes involved in aging. Oxidative modifications of KCNB1 are pervasive in the aging nervous system[55]. Hence, KCNB1 oxidation has the potential to impact all those conditions characterized by an imbalance in the redox status of the cell, from normal senescence to neuropathies such as Alzheimer’s disease. Understanding how oxidation of KCNB1 influences the function of the brain during aging may provide the insight necessary to design better pharmacological strategies; these include targeting KCNB1 for the potential therapeutic use of antioxidants in neurological treatments or targeting other components of the signaling pathways activated by oxidation of KCNB1.
ACKNOWLEDGEMENTS
We thank Ms. Aileen Baffo for critical reading of the manuscript.
Footnotes
Supported by National Science Foundation Grant to Sesti F, No. 1026958
P- Reviewers: Echtay KS, Martin-Romero FG, Lei S, Utkin YN, Zhang WZ S- Editor: Zhai HH L- Editor: A
E- Editor: Lu YJ
References
- 1.Frech GC, VanDongen AM, Schuster G, Brown AM, Joho RH. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature. 1989;340:642–645. doi: 10.1038/340642a0. [DOI] [PubMed] [Google Scholar]
- 2.Dixon JE, McKinnon D. Quantitative analysis of potassium channel mRNA expression in atrial and ventricular muscle of rats. Circ Res. 1994;75:252–260. doi: 10.1161/01.res.75.2.252. [DOI] [PubMed] [Google Scholar]
- 3.Barry DM, Trimmer JS, Merlie JP, Nerbonne JM. Differential expression of voltage-gated K+ channel subunits in adult rat heart. Relation to functional K+ channels? Circ Res. 1995;77:361–369. doi: 10.1161/01.res.77.2.361. [DOI] [PubMed] [Google Scholar]
- 4.Kyle B, Bradley E, Ohya S, Sergeant GP, McHale NG, Thornbury KD, Hollywood MA. Contribution of Kv2.1 channels to the delayed rectifier current in freshly dispersed smooth muscle cells from rabbit urethra. Am J Physiol Cell Physiol. 2011;301:C1186–C1200. doi: 10.1152/ajpcell.00455.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X, Surguchev A, Bian S, Navaratnam D, Santos-Sacchi J. Extracellular chloride regulation of Kv2.1, contributor to the major outward Kv current in mammalian outer hair cells. Am J Physiol Cell Physiol. 2012;302:C296–C306. doi: 10.1152/ajpcell.00177.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.You MH, Song MS, Lee SK, Ryu PD, Lee SY, Kim DY. Voltage-gated K+ channels in adipogenic differentiation of bone marrow-derived human mesenchymal stem cells. Acta Pharmacol Sin. 2013;34:129–136. doi: 10.1038/aps.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yazulla S, Studholme KM. Differential distribution of Shaker-like and Shab-like K+-channel subunits in goldfish retina and retinal bipolar cells. J Comp Neurol. 1998;396:131–140. doi: 10.1002/(sici)1096-9861(19980622)396:1<131::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 8.Tamarina NA, Kuznetsov A, Fridlyand LE, Philipson LH. Delayed-rectifier (KV2.1) regulation of pancreatic beta-cell calcium responses to glucose: inhibitor specificity and modeling. Am J Physiol Endocrinol Metab. 2005;289:E578–E585. doi: 10.1152/ajpendo.00054.2005. [DOI] [PubMed] [Google Scholar]
- 9.Yan L, Figueroa DJ, Austin CP, Liu Y, Bugianesi RM, Slaughter RS, Kaczorowski GJ, Kohler MG. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes. 2004;53:597–607. doi: 10.2337/diabetes.53.3.597. [DOI] [PubMed] [Google Scholar]
- 10.MacDonald PE, Ha XF, Wang J, Smukler SR, Sun AM, Gaisano HY, Salapatek AM, Backx PH, Wheeler MB. Members of the Kv1 and Kv2 voltage-dependent K(+) channel families regulate insulin secretion. Mol Endocrinol. 2001;15:1423–1435. doi: 10.1210/mend.15.8.0685. [DOI] [PubMed] [Google Scholar]
- 11.Su J, Yu H, Lenka N, Hescheler J, Ullrich S. The expression and regulation of depolarization-activated K+ channels in the insulin-secreting cell line INS-1. Pflugers Arch. 2001;442:49–56. doi: 10.1007/s004240000508. [DOI] [PubMed] [Google Scholar]
- 12.Roe MW, Worley JF, Mittal AA, Kuznetsov A, DasGupta S, Mertz RJ, Witherspoon SM, Blair N, Lancaster ME, McIntyre MS, et al. Expression and function of pancreatic beta-cell delayed rectifier K+ channels. Role in stimulus-secretion coupling. J Biol Chem. 1996;271:32241–32246. doi: 10.1074/jbc.271.50.32241. [DOI] [PubMed] [Google Scholar]
- 13.Du J, Tao-Cheng JH, Zerfas P, McBain CJ. The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience. 1998;84:37–48. doi: 10.1016/s0306-4522(97)00519-8. [DOI] [PubMed] [Google Scholar]
- 14.Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trimmer JS. Expression of Kv2.1 delayed rectifier K+ channel isoforms in the developing rat brain. FEBS Lett. 1993;324:205–210. doi: 10.1016/0014-5793(93)81394-f. [DOI] [PubMed] [Google Scholar]
- 16.Patel AJ, Lazdunski M, Honoré E. Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J. 1997;16:6615–6625. doi: 10.1093/emboj/16.22.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan XJ, Wang J, Juhaszova M, Golovina VA, Rubin LJ. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. Am J Physiol. 1998;274:L621–L635. doi: 10.1152/ajplung.1998.274.4.L621. [DOI] [PubMed] [Google Scholar]
- 18.Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest. 1998;101:2319–2330. doi: 10.1172/JCI333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Archer SL, Weir EK, Reeve HL, Michelakis E. Molecular identification of O2 sensors and O2-sensitive potassium channels in the pulmonary circulation. Adv Exp Med Biol. 2000;475:219–240. doi: 10.1007/0-306-46825-5_21. [DOI] [PubMed] [Google Scholar]
- 20.Trimmer JS. Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc Natl Acad Sci USA. 1991;88:10764–10768. doi: 10.1073/pnas.88.23.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang PM, Fotuhi M, Bredt DS, Cunningham AM, Snyder SH. Contrasting immunohistochemical localizations in rat brain of two novel K+ channels of the Shab subfamily. J Neurosci. 1993;13:1569–1576. doi: 10.1523/JNEUROSCI.13-04-01569.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- 23.Zhong XZ, Abd-Elrahman KS, Liao CH, El-Yazbi AF, Walsh EJ, Walsh MP, Cole WC. Stromatoxin-sensitive, heteromultimeric Kv2.1/Kv9.3 channels contribute to myogenic control of cerebral arterial diameter. J Physiol. 2010;588:4519–4537. doi: 10.1113/jphysiol.2010.196618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hristov KL, Chen M, Soder RP, Parajuli SP, Cheng Q, Kellett WF, Petkov GV. KV2.1 and electrically silent KV channel subunits control excitability and contractility of guinea pig detrusor smooth muscle. Am J Physiol Cell Physiol. 2012;302:C360–C372. doi: 10.1152/ajpcell.00303.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bocksteins E, Labro AJ, Snyders DJ, Mohapatra DP. The electrically silent Kv6.4 subunit confers hyperpolarized gating charge movement in Kv2.1/Kv6.4 heterotetrameric channels. PLoS One. 2012;7:e37143. doi: 10.1371/journal.pone.0037143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aimond F, Kwak SP, Rhodes KJ, Nerbonne JM. Accessory Kvbeta1 subunits differentially modulate the functional expression of voltage-gated K+ channels in mouse ventricular myocytes. Circ Res. 2005;96:451–458. doi: 10.1161/01.RES.0000156890.25876.63. [DOI] [PubMed] [Google Scholar]
- 27.McCrossan ZA, Lewis A, Panaghie G, Jordan PN, Christini DJ, Lerner DJ, Abbott GW. MinK-related peptide 2 modulates Kv2.1 and Kv3.1 potassium channels in mammalian brain. J Neurosci. 2003;23:8077–8091. doi: 10.1523/JNEUROSCI.23-22-08077.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bocksteins E, Labro AJ, Mayeur E, Bruyns T, Timmermans JP, Adriaensen D, Snyders DJ. Conserved negative charges in the N-terminal tetramerization domain mediate efficient assembly of Kv2.1 and Kv2.1/Kv6.4 channels. J Biol Chem. 2009;284:31625–31634. doi: 10.1074/jbc.M109.039479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerda O, Trimmer JS. Activity-dependent phosphorylation of neuronal Kv2.1 potassium channels by CDK5. J Biol Chem. 2011;286:28738–28748. doi: 10.1074/jbc.M111.251942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- 31.Misonou H, Mohapatra DP, Menegola M, Trimmer JS. Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci. 2005;25:11184–11193. doi: 10.1523/JNEUROSCI.3370-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- 33.Plant LD, Dowdell EJ, Dementieva IS, Marks JD, Goldstein SA. SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J Gen Physiol. 2011;137:441–454. doi: 10.1085/jgp.201110604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai XQ, Kolic J, Marchi P, Sipione S, Macdonald PE. SUMOylation regulates Kv2.1 and modulates pancreatic beta-cell excitability. J Cell Sci. 2009;122:775–779. doi: 10.1242/jcs.036632. [DOI] [PubMed] [Google Scholar]
- 35.Kim SJ, Widenmaier SB, Choi WS, Nian C, Ao Z, Warnock G, McIntosh CH. Pancreatic β-cell prosurvival effects of the incretin hormones involve post-translational modification of Kv2.1 delayed rectifier channels. Cell Death Differ. 2012;19:333–344. doi: 10.1038/cdd.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi G, Trimmer JS. Differential asparagine-linked glycosylation of voltage-gated K+ channels in mammalian brain and in transfected cells. J Membr Biol. 1999;168:265–273. doi: 10.1007/s002329900515. [DOI] [PubMed] [Google Scholar]
- 37.Yuan C, Yang S, Liao Z, Liang S. Effects and mechanism of Chinese tarantula toxins on the Kv2.1 potassium channels. Biochem Biophys Res Commun. 2007;352:799–804. doi: 10.1016/j.bbrc.2006.11.086. [DOI] [PubMed] [Google Scholar]
- 38.Swartz KJ, MacKinnon R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- 39.Escoubas P, Diochot S, Célérier ML, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol Pharmacol. 2002;62:48–57. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]
- 40.Herrington J, Zhou YP, Bugianesi RM, Dulski PM, Feng Y, Warren VA, Smith MM, Kohler MG, Garsky VM, Sanchez M, et al. Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes. 2006;55:1034–1042. doi: 10.2337/diabetes.55.04.06.db05-0788. [DOI] [PubMed] [Google Scholar]
- 41.Chen R, Robinson A, Chung SH. Binding of hanatoxin to the voltage sensor of Kv2.1. Toxins (Basel) 2012;4:1552–1564. doi: 10.3390/toxins4121552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee S, Milescu M, Jung HH, Lee JY, Bae CH, Lee CW, Kim HH, Swartz KJ, Kim JI. Solution structure of GxTX-1E, a high-affinity tarantula toxin interacting with voltage sensors in Kv2.1 potassium channels. Biochemistry. 2010;49:5134–5142. doi: 10.1021/bi100246u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopatin AN, Nichols CG. Block of delayed rectifier (DRK1) K+ channels by internal 2,3-butanedione monoxime in Xenopus oocytes. Receptors Channels. 1993;1:279–286. [PubMed] [Google Scholar]
- 44.Taglialatela M, Vandongen AM, Drewe JA, Joho RH, Brown AM, Kirsch GE. Patterns of internal and external tetraethylammonium block in four homologous K+ channels. Mol Pharmacol. 1991;40:299–307. [PubMed] [Google Scholar]
- 45.Wible B, Murawsky MK, Crumb WJ, Rampe D. Stable expression and characterization of the human brain potassium channel Kv2.1: blockade by antipsychotic agents. Brain Res. 1997;761:42–50. doi: 10.1016/s0006-8993(97)00315-6. [DOI] [PubMed] [Google Scholar]
- 46.Zhang ZH, Lee YT, Rhodes K, Wang K, Argentieri TM, Wang Q. Inhibitory effects of pimozide on cloned and native voltage-gated potassium channels. Brain Res Mol Brain Res. 2003;115:29–38. doi: 10.1016/s0169-328x(03)00175-x. [DOI] [PubMed] [Google Scholar]
- 47.Kulkarni RS, Zorn LJ, Anantharam V, Bayley H, Treistman SN. Inhibitory effects of ketamine and halothane on recombinant potassium channels from mammalian brain. Anesthesiology. 1996;84:900–909. doi: 10.1097/00000542-199604000-00018. [DOI] [PubMed] [Google Scholar]
- 48.Madeja M, Steffen W, Mesic I, Garic B, Zhorov BS. Overlapping binding sites of structurally different antiarrhythmics flecainide and propafenone in the subunit interface of potassium channel Kv2.1. J Biol Chem. 2010;285:33898–33905. doi: 10.1074/jbc.M110.159897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frolov RV, Bondarenko VE, Singh S. Mechanisms of Kv2.1 channel inhibition by celecoxib--modification of gating and channel block. Br J Pharmacol. 2010;159:405–418. doi: 10.1111/j.1476-5381.2009.00539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirsch GE, Drewe JA. Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J Gen Physiol. 1993;102:797–816. doi: 10.1085/jgp.102.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gordon E, Cohen JL, Engel R, Abbott GW. 1,4-Diazabicyclo[2.2.2]octane derivatives: a novel class of voltage-gated potassium channel blockers. Mol Pharmacol. 2006;69:718–726. doi: 10.1124/mol.105.018663. [DOI] [PubMed] [Google Scholar]
- 52.Salvador-Recatala V, Kim Y, Zaks-Makhina E, Levitan ES. Voltage-gated k+ channel block by catechol derivatives: defining nonselective and selective pharmacophores. J Pharmacol Exp Ther. 2006;319:758–764. doi: 10.1124/jpet.106.107607. [DOI] [PubMed] [Google Scholar]
- 53.Yoshikawa H, Ma Z, Björklund A, Grill V. Short-term intermittent exposure to diazoxide improves functional performance of beta-cells in a high-glucose environment. Am J Physiol Endocrinol Metab. 2004;287:E1202–E1208. doi: 10.1152/ajpendo.00255.2004. [DOI] [PubMed] [Google Scholar]
- 54.Yuan H, Wang WP, Feng N, Wang L, Wang XL. Donepezil attenuated oxygen-glucose deprivation insult by blocking Kv2.1 potassium channels. Eur J Pharmacol. 2011;657:76–83. doi: 10.1016/j.ejphar.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 55.Cotella D, Hernandez-Enriquez B, Wu X, Li R, Pan Z, Leveille J, Link CD, Oddo S, Sesti F. Toxic role of K+ channel oxidation in mammalian brain. J Neurosci. 2012;32:4133–4144. doi: 10.1523/JNEUROSCI.6153-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du J, Haak LL, Phillips-Tansey E, Russell JT, McBain CJ. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol. 2000;522 Pt 1:19–31. doi: 10.1111/j.1469-7793.2000.t01-2-00019.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scannevin RH, Murakoshi H, Rhodes KJ, Trimmer JS. Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K+ channel. J Cell Biol. 1996;135:1619–1632. doi: 10.1083/jcb.135.6.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fox PD, Loftus RJ, Tamkun MM. Regulation of Kv2.1 K(+) conductance by cell surface channel density. J Neurosci. 2013;33:1259–1270. doi: 10.1523/JNEUROSCI.3008-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deutsch E, Weigel AV, Akin EJ, Fox P, Hansen G, Haberkorn CJ, Loftus R, Krapf D, Tamkun MM. Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol Biol Cell. 2012;23:2917–2929. doi: 10.1091/mbc.E12-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Connell KM, Rolig AS, Whitesell JD, Tamkun MM. Kv2.1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J Neurosci. 2006;26:9609–9618. doi: 10.1523/JNEUROSCI.1825-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ikematsu N, Dallas ML, Ross FA, Lewis RW, Rafferty JN, David JA, Suman R, Peers C, Hardie DG, Evans AM. Phosphorylation of the voltage-gated potassium channel Kv2.1 by AMP-activated protein kinase regulates membrane excitability. Proc Natl Acad Sci USA. 2011;108:18132–18137. doi: 10.1073/pnas.1106201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yao H, Zhou K, Yan D, Li M, Wang Y. The Kv2.1 channels mediate neuronal apoptosis induced by excitotoxicity. J Neurochem. 2009;108:909–919. doi: 10.1111/j.1471-4159.2008.05834.x. [DOI] [PubMed] [Google Scholar]
- 63.Shen QJ, Zhao YM, Cao DX, Wang XL. Contribution of Kv channel subunits to glutamate-induced apoptosis in cultured rat hippocampal neurons. J Neurosci Res. 2009;87:3153–3160. doi: 10.1002/jnr.22136. [DOI] [PubMed] [Google Scholar]
- 64.Jiao S, Liu Z, Ren WH, Ding Y, Zhang YQ, Zhang ZH, Mei YA. cAMP/protein kinase A signalling pathway protects against neuronal apoptosis and is associated with modulation of Kv2.1 in cerebellar granule cells. J Neurochem. 2007;100:979–991. doi: 10.1111/j.1471-4159.2006.04261.x. [DOI] [PubMed] [Google Scholar]
- 65.Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu SP. Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol. 2003;70:363–386. doi: 10.1016/s0301-0082(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 67.Pal SK, Takimoto K, Aizenman E, Levitan ES. Apoptotic surface delivery of K+ channels. Cell Death Differ. 2006;13:661–667. doi: 10.1038/sj.cdd.4401792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou MH, Yang G, Jiao S, Hu CL, Mei YA. Cholesterol enhances neuron susceptibility to apoptotic stimuli via cAMP/PKA/CREB-dependent up-regulation of Kv2.1. J Neurochem. 2012;120:502–514. doi: 10.1111/j.1471-4159.2011.07593.x. [DOI] [PubMed] [Google Scholar]
- 69.Aras MA, Aizenman E. Obligatory role of ASK1 in the apoptotic surge of K+ currents. Neurosci Lett. 2005;387:136–140. doi: 10.1016/j.neulet.2005.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Redman PT, Hartnett KA, Aras MA, Levitan ES, Aizenman E. Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J Physiol. 2009;587:4393–4404. doi: 10.1113/jphysiol.2009.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Redman PT, He K, Hartnett KA, Jefferson BS, Hu L, Rosenberg PA, Levitan ES, Aizenman E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci USA. 2007;104:3568–3573. doi: 10.1073/pnas.0610159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McCord MC, Aizenman E. Convergent Ca2+ and Zn2+ signaling regulates apoptotic Kv2.1 K+ currents. Proc Natl Acad Sci USA. 2013;110:13988–13993. doi: 10.1073/pnas.1306238110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Norris CA, He K, Springer MG, Hartnett KA, Horn JP, Aizenman E. Regulation of neuronal proapoptotic potassium currents by the hepatitis C virus nonstructural protein 5A. J Neurosci. 2012;32:8865–8870. doi: 10.1523/JNEUROSCI.0937-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sobko A, Peretz A, Attali B. Constitutive activation of delayed-rectifier potassium channels by a src family tyrosine kinase in Schwann cells. EMBO J. 1998;17:4723–4734. doi: 10.1093/emboj/17.16.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peretz A, Sobko A, Attali B. Tyrosine kinases modulate K+ channel gating in mouse Schwann cells. J Physiol. 1999;519 Pt 2:373–384. doi: 10.1111/j.1469-7793.1999.0373m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tiran Z, Peretz A, Attali B, Elson A. Phosphorylation-dependent regulation of Kv2.1 Channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase epsilon. J Biol Chem. 2003;278:17509–17514. doi: 10.1074/jbc.M212766200. [DOI] [PubMed] [Google Scholar]
- 77.Tiran Z, Peretz A, Sines T, Shinder V, Sap J, Attali B, Elson A. Tyrosine phosphatases epsilon and alpha perform specific and overlapping functions in regulation of voltage-gated potassium channels in Schwann cells. Mol Biol Cell. 2006;17:4330–4342. doi: 10.1091/mbc.E06-02-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peretz A, Gil-Henn H, Sobko A, Shinder V, Attali B, Elson A. Hypomyelination and increased activity of voltage-gated K(+) channels in mice lacking protein tyrosine phosphatase epsilon. EMBO J. 2000;19:4036–4045. doi: 10.1093/emboj/19.15.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu X, Hernandez-Enriquez B, Banas M, Xu R, Sesti F. Molecular mechanisms underlying the apoptotic effect of KCNB1 K+ channel oxidation. J Biol Chem. 2013;288:4128–4134. doi: 10.1074/jbc.M112.440933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dallas ML, Boyle JP, Milligan CJ, Sayer R, Kerrigan TL, McKinstry C, Lu P, Mankouri J, Harris M, Scragg JL, et al. Carbon monoxide protects against oxidant-induced apoptosis via inhibition of Kv2.1. FASEB J. 2011;25:1519–1530. doi: 10.1096/fj.10-173450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Al-Owais MM, Scragg JL, Dallas ML, Boycott HE, Warburton P, Chakrabarty A, Boyle JP, Peers C. Carbon monoxide mediates the anti-apoptotic effects of heme oxygenase-1 in medulloblastoma DAOY cells via K+ channel inhibition. J Biol Chem. 2012;287:24754–24764. doi: 10.1074/jbc.M112.357012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mankouri J, Dallas ML, Hughes ME, Griffin SD, Macdonald A, Peers C, Harris M. Suppression of a pro-apoptotic K+ channel as a mechanism for hepatitis C virus persistence. Proc Natl Acad Sci USA. 2009;106:15903–15908. doi: 10.1073/pnas.0906798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amako Y, Igloi Z, Mankouri J, Kazlauskas A, Saksela K, Dallas M, Peers C, Harris M. Hepatitis C virus NS5A inhibits mixed lineage kinase 3 to block apoptosis. J Biol Chem. 2013;288:24753–24763. doi: 10.1074/jbc.M113.491985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hulme JT, Coppock EA, Felipe A, Martens JR, Tamkun MM. Oxygen sensitivity of cloned voltage-gated K(+) channels expressed in the pulmonary vasculature. Circ Res. 1999;85:489–497. doi: 10.1161/01.res.85.6.489. [DOI] [PubMed] [Google Scholar]
- 85.McDaniel SS, Platoshyn O, Yu Y, Sweeney M, Miriel VA, Golovina VA, Krick S, Lapp BR, Wang JY, Yuan JX. Anorexic effect of K+ channel blockade in mesenteric arterial smooth muscle and intestinal epithelial cells. J Appl Physiol (1985) 2001;91:2322–2333. doi: 10.1152/jappl.2001.91.5.2322. [DOI] [PubMed] [Google Scholar]
- 86.Michelakis ED, Rebeyka I, Wu X, Nsair A, Thébaud B, Hashimoto K, Dyck JR, Haromy A, Harry G, Barr A, et al. O2 sensing in the human ductus arteriosus: regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ Res. 2002;91:478–486. doi: 10.1161/01.res.0000035057.63303.d1. [DOI] [PubMed] [Google Scholar]
- 87.Thébaud B, Michelakis ED, Wu XC, Moudgil R, Kuzyk M, Dyck JR, Harry G, Hashimoto K, Haromy A, Rebeyka I, et al. Oxygen-sensitive Kv channel gene transfer confers oxygen responsiveness to preterm rabbit and remodeled human ductus arteriosus: implications for infants with patent ductus arteriosus. Circulation. 2004;110:1372–1379. doi: 10.1161/01.CIR.0000141292.28616.65. [DOI] [PubMed] [Google Scholar]
- 88.Archer SL, Wu XC, Thébaud B, Moudgil R, Hashimoto K, Michelakis ED. O2 sensing in the human ductus arteriosus: redox-sensitive K+ channels are regulated by mitochondria-derived hydrogen peroxide. Biol Chem. 2004;385:205–216. doi: 10.1515/BC.2004.014. [DOI] [PubMed] [Google Scholar]
- 89.Conforti L, Takimoto K, Petrovic M, Pongs O, Millhorn D. The pore region of the Kv1.2alpha subunit is an important component of recombinant Kv1.2 channel oxygen sensitivity. Biochem Biophys Res Commun. 2003;306:450–456. doi: 10.1016/s0006-291x(03)00989-6. [DOI] [PubMed] [Google Scholar]
- 90.Michelakis ED, Dyck JR, McMurtry MS, Wang S, Wu XC, Moudgil R, Hashimoto K, Puttagunta L, Archer SL. Gene transfer and metabolic modulators as new therapies for pulmonary hypertension. Increasing expression and activity of potassium channels in rat and human models. Adv Exp Med Biol. 2001;502:401–418. doi: 10.1007/978-1-4757-3401-0_26. [DOI] [PubMed] [Google Scholar]
- 91.Hong Z, Weir EK, Nelson DP, Olschewski A. Subacute hypoxia decreases voltage-activated potassium channel expression and function in pulmonary artery myocytes. Am J Respir Cell Mol Biol. 2004;31:337–343. doi: 10.1165/rcmb.2003-0386OC. [DOI] [PubMed] [Google Scholar]
- 92.Wang J, Weigand L, Wang W, Sylvester JT, Shimoda LA. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1049–L1058. doi: 10.1152/ajplung.00379.2004. [DOI] [PubMed] [Google Scholar]
- 93.Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ, Yuan JX. Chronic hypoxia decreases K(V) channel expression and function in pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol. 2001;280:L801–L812. doi: 10.1152/ajplung.2001.280.4.L801. [DOI] [PubMed] [Google Scholar]
- 94.Dong Q, Zhao N, Xia CK, Du LL, Fu XX, Du YM. Hypoxia induces voltage-gated K+ (Kv) channel expression in pulmonary arterial smooth muscle cells through hypoxia-inducible factor-1 (HIF-1) Bosn J Basic Med Sci. 2012;12:158–163. doi: 10.17305/bjbms.2012.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guo L, Qiu Z, Zhang L, Chen S, Zhu D. Hypoxia suppresses Kv 2.1 channel expression through endogenous 15-hydroxyeicosatetraenoic acid in rat pulmonary artery. J Physiol Sci. 2010;60:373–381. doi: 10.1007/s12576-010-0105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Swaninathan N. How to save your brain. Psych Today. 2012;45:74–79. [Google Scholar]
- 97.Alzheimer’s Drug Discovery Foundation. Annual Report, 2005. Available from: http: //www.alzdiscovery.org/assets/content/publications/ADDF_AR_3-22B.pdf.
- 98.Alzheimer's Association. 2010 Alzheimer’s disease facts and figures. Alzheimers Dement. 2010;6:158–194. doi: 10.1016/j.jalz.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 99.Alzheimer’s Association. Diagniostic Center for Alzheimer’s Disease. Available from: http: //www.alz.org/alzheimers_disease_diagnosis.asp.