

Abstract
Flaviviruses, ss(+) RNA viruses, include many of mankind’s most important pathogens. Their pathogenicity derives from their ability to infect many types of cells including neurons, to replicate, and eventually to kill the cells. Flaviviruses can activate tumor necrosis factor α and both intrinsic (Bax-mediated) and extrinsic pathways to apoptosis. Thus they can use many approaches for activating these pathways. Infection can lead to necrosis if viral load is extremely high or to other types of cell death if routes to apoptosis are blocked. Dengue and Japanese Encephalitis Virus can also activate autophagy. In this case the autophagy temporarily spares the infected cell, allowing a longer period of reproduction for the virus, and the autophagy further protects the cell against other stresses such as those caused by reactive oxygen species. Several of the viral proteins have been shown to induce apoptosis or autophagy on their own, independent of the presence of other viral proteins. Given the versatility of these viruses to adapt to and manipulate the metabolism, and thus to control the survival of, the infected cells, we need to understand much better how the specific viral proteins affect the pathways to apoptosis and autophagy. Only in this manner will we be able to minimize the pathology that they cause.
Keywords: Flavivirus, Dengue virus, West Nile virus, Japanese encephalitis virus, Programmed cell death, Apoptosis, Extrinsic pathway, Intrinsic pathway, Autophagy, Necrosis
Core tip: The pathogenicity of Flaviviruses derives from their ability to infect many types of cells. They can activate both intrinsic and extrinsic pathways of apoptosis, by many means. Dengue and Japanese encephalitis virus can also activate autophagy, whereby autophagy temporarily spares the infected cell, allowing longer reproduction of virus and protecting the cell against other stresses. Given the versatility of these viruses, we need to understand much better how the specific viral proteins affect the pathways to apoptosis and autophagy. Only in this manner will we be able to minimize the pathology that they cause.
INTRODUCTION
The aim of a virus is to infect and propagate and in doing so, affect the cell survival pathways. A wide range of viruses from different families (Poxviridae, Adenoviridae, Retroviridae, Picornoviridae, Flaviviridae, Orthomyxoviridae) have life cycles that intertwine with critical pathways involved in cell death and survival[1]. In this review we focus our attention on Flavivirus (Flaviviridae).
Flaviviridae, a family of small and enveloped ss(+)-RNA virus, consists some of the worst pathogens known to mankind and mammals. The family is grouped into three genera, namely, Flavivirus, Hepacivirus and Pestivirus with each genus harboring potent killers, viz., dengue (DEN), hepatitis C (HCV) and classical swine fever virus (CSFV), respectively[2]. The largest and clinically the most relevant of three, Flavivirus contains almost 70 members, most of them transmitted to humans by mosquitos or ticks. Among the mosquito-borne are the most virulent viruses like dengue (DEN)[3,4], West Nile (WNV)[5], Japanese encephalitis (JEV) and Yellow fever (YFV)[6].
Although a few reviews address the role of cell death pathways during viral infection in general[1,7,8], there are none solely addressing Flavivirus. Here we summarize the most recent findings on survival and cell death pathways triggered by key members of Flavivirus. We focus on flaviviruses widely studied in relation to cell death - dengue, West Nile and Japanese encephalitis virus. We conclude that the viruses affect different parts of the apoptotic pathways in different cell types, and that dengue and JEV especially can protect cells by activating autophagy. Anti-viral therapeutics will have to address these issues.
CELL DEATH AND ITS PATHWAYS
The ascendance of programmed cell death (PCD) as a theme of modern biology has followed an exciting trail from the mid-19th century until the present[9]. The idea of a cell programming its death had few takers during the early half of 20th century, though evidence was gathering since 1842, when Carl Vogt observed loss of notochord in amphibian metamorphosis[10]. Since then, evidence of programmed cell death has surfaced in various organisms as diverse as Dictyostelium[11], insects[12], and chicken[13]. Recognition of apoptosis as the primary form of programmed cell death, in the early 1970’s[14] as well as recognition that apoptosis is conserved from C. elegans to humans)[15,16] has fueled interest among biologists. Moreover, association of apoptosis and other forms of cell death, notably the lysosomal (autophagic) cell death, with AIDS[17], cancer[18,19], Alzheimer’s[20], and viral infection[1] has catapulted cell death to the forefront of biomedical research.
The importance of cell death was not fully appreciated until the late 1960’s. This delay was partly due to the difficulty in documenting dying cells, as compared to dividing ones, as it was possible to monitor and finally trace a cell’s duplication into daughter cells. While cells that have undergone mitosis can be traced considerably thereafter, an apoptotic cell in an organism is visible only up to 20 min after death[12].
Programmed cell death contributes to the sculpting of digits (prenatal disappearance of interdigital epidermis), removal of unnecessary tissues (involution of mammary glands during post-lactation) or irrelevant (wolffian/mullerian ducts after sex determination) organs, elimination of toxic and harmful cells (self-reactive thymocytes, UV-irradiated cells), and winnowing to only a properly integrated cell population (as in the case of differentiated neurons)[21,22]. A cell may trigger its own death (intrinsic/cell autonomous) or it may be brought upon by signals from the microenvironment (extrinsic). Deregulation of the cell death machinery can inflict upon the organism severe consequences like anomalous or stalled development, tumor formation, autoimmune disorder or neurological disorders (Huntington, Parkinson). In contrast, the vestiges of dead cells in some plants may serve important functions[22,23].
Most biologists make a clear distinction between “programmed” physiological (beneficial) and ‘‘accidental’’ (hazardous) cell death. The former denotes death of cells essential for physiological events (development, organogenesis, homeostasis, and defense) whereas the latter may be used for loss of cells during tissue damage. Apart from this functional distinction, cell death can also be classified based on morphology (apoptosis, autophagy, necrosis, and cornification) and enzyme involvement (proteases like calpains, caspases, and endonucleases). The Nomenclature Committee on Cell Death (NCCD) encourages researchers to clearly distinguish between ‘‘dying cells’’ and ‘‘dead cells’’, and by using the latter term, they should denote cells that have gone past the threshold ‘‘point-of-no-return’’ into a state of irreversibility. The NCCD has also revised the defining hallmarks for a dead cell: dissolution of the plasma membrane and complete fragmentation and engulfment by phagocytosis, since the traditional parameters like activation of caspases, mitochondrial trans membrane permeabilization and flipping of phosphatidylserine (PS) have been associated with non-lethal events[24].
APOPTOSIS
The most studied form of programmed cell death (PCD), apoptosis (Greek: falling of leaves), was first reported by Walter Flemming[10]. Kerr et al[14] characterized apoptosis (later described by Majno and Joris as PCD type I) and described it as a general process mistakenly previously identified as an arcane form of death called “shrinkage necrosis”. While undergoing apoptosis, the cell separates from its neighboring cells, shrinks, undergoes chromatin condensation and DNA fragmentation, and is finally engulfed by a phagocyte (macrophage).
Apoptosis follows two distinct pathways, the extrinsic (death receptor) and intrinsic (mitochondrial) pathway[25]. The extrinsic branch of PCD is activated by external death signals. The cytotoxic effect is mediated by the binding of ligands [tumor necrosis factor-α (TNF-α), FasL, TRAIL] to the death receptors (TNF RI, Fas/CD95, DR3, TRAIL R1/DR4, or TRAIL R2/DR5) on the cell surface[26-28]. This binding leads to the trimerization of the membrane receptor, followed by the downstream activation of the DISC protein complex. The multi-protein complex initiates cleavage and activation of caspase-8, which in turn cleaves downstream zymogens (caspase-7, 10) and this sets forth a chain of reactions finally leading to activation of caspase-3 and cell death[25,28]. The caspase proteins (Cysteine-dependent Aspartate-directed Proteases = C-A-S-PASES) are central to the entire apoptotic machinery within the cell. They are also integral to the intrinsic pathway, are synthesized as inactive zymogens that are activated by cleavage.
Intrinsic apoptosis is activated proximately by damage to mitochondria, which releases cytochrome C and apoptosis-activating factor from mitochondria. These latter, together with pro-caspase-9, bind together into an apoptosome, in which caspase-9 is activated. By means of this complex, caspase-3 is activated and, as in extrinsically-activated apoptosis, caspases 3 and 7 destroy the substructure of the cell.
Like caspases, Bcl-2 family members are also essential for carrying out intrinsic apoptosis. Based on domain structure and function, the members are grouped into anti-apoptotic guardians (Bcl-2, Bcl-xL, MCL-1), pro-apoptotic effectors (Bax, Bak) and sensors (Bad/Bim/Bid/Noxa)[29-31]. The intrinsic pathway is initiated by intracellular stress signals like ER stress, oxidative stress, DNA damage, growth factor withdrawals, and loss of contact with the extracellular matrix. Once the decision to die is made, the effectors are set free from their negative interaction with guardians by the sensors. They insert into and disintegrate the mitochondrial membrane, a phenomenon known as the mitochondrial outer membrane permeabilization (MOMP). This releases pro-apoptotic factors (cytochrome C, Smac/Diablo, HTRA2/Omi, apoptosis-inducing factor, and endonuclease G) into the cytoplasm. Cytochrome C interacts with the APAF-1, recruiting pro-caspase-9 (zymogen) to form the apoptosome, where the latter is cleaved and activated. This event triggers cleavage and activation of downstream caspases (2, 3, 7, 8) and accomplishes the death of cell[32]. Certain cell death regulators like inhibitor of apoptosis (IAP) can bind and suppress the apoptotic function of caspases[33].
AUTOPHAGY
Autophagy or PCD type II, literally meaning ‘‘self-eating’’, is a highly conserved catabolic process that is thought to precede apoptosis in evolution[34]. It is a surveillance process that is involved in the recycling of basic biomolecules. It oversees the entire cell homeostasis, packaging degraded/misfolded proteins or organelles in specialized bilayer membranes (autophagosomes) which fuse with the lysosome for digestion. This process is induced under conditions of high stress like starvation, growth factor withdrawal, viral invasion and ER stress. Deregulation of the autophagy pathway has been observed in pathogenic conditions like cancer or Parkinson’s[35].
The induction of autophagy involves a set of multiprotein complexes, some of which have ubiquitin-like properties. mTORC1, a versatile signaling complex, strictly inhibits induction of autophagy by imposing an inhibitory phosphorylation on Unc-51-like kinase (ULK1). Under stress conditions, this block is removed by several factors, such as PTEN, AMPK, and TSC2. Activation of ULK1, which forms a complex with ATG13/FIP200/ATG101, leads to the nucleation of the pre-autophagosomal structure (PAS). This involves the phosphatidylinositol-3-kinase class III (PI3K III)-Vps34-Beclin 1 (ATG6) complex[36,37]. The subsequent elongation of the autophagosome is dependent on two ubiquitin-like conjugation systems. E1-like enzyme autophagy related gene 7 (ATG7) and E2-like enzymes ATG3, ATG10 are involved in the conjugation of ATG12-ATG5 and LC3 (ATG8)-phosphatidylethanolamine (PE). ATG12-ATG5 acts like an E3-like protein for the LC3-PE conjugation system, and then forms a complex with ATG16. These coordinated and combined steps accomplish the formation of a mature autophagosome which then fuses with a lysosome through a canonical endocytic pathway[25,38-40].
NECROSIS
Some forms of necrosis are programmed and controlled through a specific set of signal transduction pathways and degradative mechanisms. Cell death by specific necrosis can also contribute to embryonic development and adult tissue homeostasis[41]. Necrosis can be triggered by the same death signals that induce apoptosis[42]. The difference between apoptosis and specific necrosis lies in the rapid cytoplasmic swelling and release of extracellular components, seen in specific necrosis, which is often due to extreme physiochemical stress, osmotic shock, mechanical stress and high concentration of hydrogen peroxide[43]. When a cell is under such conditions, which can be produced by physiological or developmental situations, cell death occurs accidentally and uncontrolled. Necrosis signaling complex forms by interaction of receptor interacting protein 1 (RIPK1) with the receptor interacting protein 3 (RIPK3). This signaling complex forms by introducing death receptors either by inhibiting caspases or genotoxic stress[43]. In this type of cell death, unlike apoptosis, death is accidental and not programmed. Necrosis does not depend on caspase activation. In a study done by Nikoletopoulou et al[42], two different cell lines were treated with a tumor necrosis factor-α. In one cell line, apoptosis was triggered, whereas in another cell line it induced necrosis. In addition, necrosis can be in the form of regulated and programmed form of cell death. This phenomenon is referred to as necroptosis. Various death receptors associated with apoptosis, such as FAS, TNFR2, TRAILR1 and TRAILR2, have been shown to induce necroptosis in different cell types. Furthermore, necroptosis can be instigated by the members of the pathogen recognition receptor that are responsible for sensing pathogen-associated molecular patterns.
FLAVIVIRUS-STRUCTURE, INFECTIVITY, REPLICATION AND CELL SURVIVAL
Flaviviridae is a medically important family of animal virus, with members responsible for serious pathological conditions in human and other important mammals. This group IV family (positive sense RNA) consists of three genera: Flavivirus, Hepacivirus and Pestivirus. The largest of them, Flavivirus (with approximately 70 members), includes some of the deadliest arthropod-transmitted virus. They are icosahedral, enveloped (+)-ssRNA virus measuring approximately 500Å in diameter. The typical Flavivirus (Latin flavus - yellow, indicating Yellow Fever) virion is composed of the genetic material surrounded by the capsid protein and 180 copies of two glycoproteins. The average genome size of the Flavivirus is 11kb, coding for a single polyprotein. The amino terminal accounts for the structural proteins: capsid (C), membrane precursor (prM) and envelope (E), and the remaining genome gives rise to the non-structural proteins (NS1, 2A, 2B, 3, 4A, 4B, and 5) which form the viral replication complex (RC)[2,44].
Infection starts as virions bind to the cell membrane through receptor-mediated endocytosis, aided by primary receptors (DC-SIGN, Grp78/BiP, CD-14 associated molecules) and low-affinity co-receptors (heparin, glycosaminoglycan). Acidification of the vesicle triggers disassembly of virus, releasing the genetic material into the cytoplasm. The resultant polyprotein undergoes co- and post-translational processing by viral and host proteases to give rise to the individual proteins. The structural proteins then assemble on the ER surface along with the RNA which is replicated on intracellular membranes. The assembly of virus in the ER lumen is followed by the movement of these immature viral particles through the trans-Golgi network. These are cleaved by the host protease furin to form mature virions, and are subsequently released by exocytosis[45-51].
Dengue virus
Among the members of Flavivirus family, Dengue is transmitted to human (in urban areas) and primates (in forests) by the urban-adapted mosquito strain Aedes aegypti (primary vector) and the emerging Aedes albopictus[52]. Dengue has been declared endemic in approximately 100 countries with 40% of the global population susceptible to infection. Dengue infection has doubled over the last two decades, and current annual figures have risen to 50-100 million humans affected[53].
Dengue has a genome of 10.7 kb positive sense single strand RNA that contains a type I cap at its 5’ terminus[54]. The enveloped icosahedral virion measures 50 nm in diameter. The RNA is translated by the host cell machinery into a 3391-amino acid polyprotein that undergoes co- and post-translational processing by viral (NS2B-3) and cellular proteases[55-57]. The first quarter of the viral genome from the 5’ end codes for the structural proteins C (capsid), prM (membrane), and E (lipopolysaccharide envelope), thus leaving the rest to code for eight non-structural proteins (NS1, 2A, 2B, 3, 4A, 2K peptide, 4B, 5) which are expressed only inside the host cell[58].
Dengue from different regions of the globe show four antigenically distinct serotypes (DENV 1-4), each having multiple phenotypes[59]. The distribution of these serotypes has spread alarmingly throughout the globe since 1970, when only South Asia had all four[60]. This spread has added to the complexity of dengue-induced pathogenesis since very little cross-immunity has been recorded between these serotypes, leading to multiple sequential infections and overwhelmed immune response[61]. Outcomes of dengue infection may lead to diverse pathogenic conditions, ranging from the mild-flu like febrile syndrome (dengue fever) to the very serious conditions resulting from infection with a second serotype, the lethal hemorrhagic condition dengue hemorrhagic fever (DHF) or the dengue shock syndrome (DSS)[62]. Dengue fever, the most important arboviral disease in humans, features rapid onset of fever, accompanied by headache, retro-orbital pain, myalgia, gastrointestinal irritation[63,64]. DHF, which claims more lives (5% mortality) than any other hemorrhagic fever, is characterized by bleeding, thrombocytopenia, increased vascular permeability beyond the usual dengue fever symptoms[65]. An equally lethal condition DSS is also characterized by vascular leakage, which is more pronounced in young children, and very low blood pressure[66]. Autopsies conducted on patients (predominantly children) dying from DSS have revealed a broad range of dengue susceptible tissue as shown by virus infecting skin, liver, spleen, lymph node, kidney, bone marrow, lung, thymus and brain[67-70].
Cell death and survival after infection with dengue
Dengue has been shown to derive pathogenic effect from apoptotic cell death in several types of mammalian cells. The role of apoptosis in dengue infection has been seriously studied since the mid-1990s, along with the identification of Bcl-2 superfamily members. Dengue-induced apoptosis has been observed in cells from the nervous system (human and mice neuroblastoma, murine cortical and hippocampal neurons, human cerebral cells); liver (human hepatoma); immune system(human peripheral blood mononuclear cells like CD8+-T lymphocytes, monocyte-derived macrophages, human mast cells like KU812, HMC-1, and primary murine macrophages); vascular system (human umbilical cord vein endothelial cells/EA.hy296, human microvascular endothelial cells, pulmonary microvascular endothelial cells/MECs) and, digestive tract (intestinal cells); and kidney cells (human embryonic kidney HEK 293, green monkey kidney Vero). Of the four antigenically distinct serotypes infection with variants of dengue 1 (human isolates of dengue type 1 virus FGA/89 and BR/90, neurovirulent variant FGA/NA d1d), 2 (strain NGC, 16681) and 3 (DENV3/5532) lead to cell death and apoptosis within 25-36 h post infection.
Apoptosis is triggered by live virus or dengue proteins through components of both extrinsic and intrinsic apoptotic signaling (Figure 1). Death ligands and receptors participate in dengue-induced apoptosis. Increased levels of pro-apoptotic proinflammatory cytokines (TNF-α and interleukin-10) and Apo2L/TRAIL are observed after infection, which the virus possibly induces in a TNF-α-fashion[71]. Profiling of genes reveal the activation of death receptors FAS/CD 95, TNFR superfamily member 9/CD 137, TNFRI/TNF-α (caspase-independent) and IL-1β/ NFκB (caspase-dependent) pathways[72,73].
Figure 1.
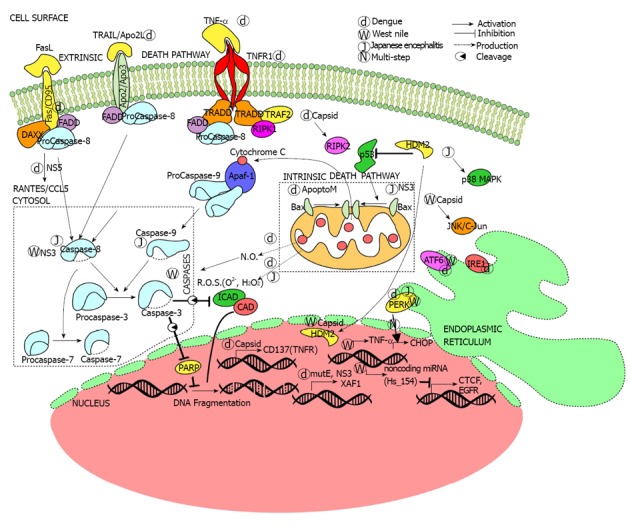
Flaviviruses target cell death and survival pathways. Extrinsic and intrinsic cell death pathways are activated during viral infection (d, w, and j are for dengue, west nile and japanese encephalitis live viruses respectively) or expression of specific viral proteins (d, w and j with viral protein). Expression of multiple genes including non-coding microRNAs (miRNA) also induced during flavivirus infections. FLaviviruses also activate ER stress signaling and increase metabolism related products (ROS and NO). TNF: Tumor necrosis factor; EGFR: Epidermal growth factor receptor.
Viral protein NS5 interacts with death protein 6 (Daxx), which among other functions interacts with death receptor FAS, to activate RANTES (CCL5), a cytokine closely associated with DHF[74,75]. Moreover, transfection with wild type capsid protein increased the expression of CD137, a member of the TNFR family. Receptor-interacting serine/threonine protein kinase 2 (RIPK2), a master regulator of stress pathways[76], is also necessary for capsid-induced apoptosis[76]. In addition to capsid protein modulation of death receptor expression, infection with live dengue virus leads to differential expression of several interferon-inducible genes, the most important being XAF1. XAF1 upregulates caspase 3 36 h after infection and mediates apoptosis[77]. The activation of caspases leads to the characteristic nuclear fragmentation and cytoplasmic blebbing of apoptosis.
Mitochondria-mediated or intrinsic apoptosis signaling also occurs after dengue infection. The reactive oxygen species (ROS) O2 and H2O2, which are predominantly produced in the mitochondria, increase during infection. Toxic levels of ROS can activate calpains and lead to apoptosis. Secondary messenger oxides like nitric oxide (NO) also mediate in dengue-triggered apoptosis in a caspase dependent manner[78]. Other dengue structural proteins are also involved in apoptosis. Intracellular production of the M protein from all dengue strains activated the intrinsic pathway apoptosis in mouse neuroblastoma (Neuro2a) and human hepatoma (HepG2) cells. ApoptoM, a nine-residue sequence (M-32 to -40) from the M ectodomain (M-1 to -40), is instrumental in the cytopathic effect of the flavivirus[79].
The activation of apoptosis at different levels of the extrinsic and intrinsic pathways by several variants of dengue virus implies an important role in the life cycle of the virus. As infected cells undergo apoptosis by multiple means the extrinsic and intrinsic apoptotic pathways converge at the activation of phosphatidylserine (PS) for phagocytic clearance during secondary dengue infection[80].
Apoptosis, supposedly an innate immune response, is often manipulated by the viruses like dengue to act against the immune system itself, as shown by the more numerous apoptotic peripheral blood mononuclear cells (PBMC) in dengue infected children. The proportion of apoptosis and its mediators (CD95) in the circulating PBMCs was much higher in individuals progressing towards hemorrhage (DHF) than those developing febrile symptoms (Dengue Fever), indicating a higher viral load in the former. A fact that most of the apoptotic PBMCs were CD8+-T lymphocytes bears testimony to the deranged immune machinery in infected individuals. The immune response to increased dengue-induced apoptosis does not curb virus proliferation. Apoptosis, in the context of dengue infection, fails to arrest viral reproduction and even correlates with increased virus production[72,73].
Unlike lytic viruses that indiscriminately trigger cell death, pro-apoptotic variants of dengue can lose their pathogenic ability in certain cells. For example the neurovirulent variant FGA/NA d1d, developed from the apoptosis inducing dengue 1 human isolate FGA/89, kills neuroblastoma but not hepatoma cells[81]. Apoptosis seen during infection of human umbilical cord vein endothelial cells (ECV304) and Swiss Webster primary macrophages by Dengue-2 virus strain 16681 is lost in MDCK, HeLa, HEK 293T, Vero and Swiss Webster primary mouse embryo fibroblasts (MEF) even after 144 h (6 d) post infection[82,83].
The differences in dengue outbreaks are partly explained by differences in cell killing by clinical isolates of virus from a fatal case (Paraguay 2007; DENV3/5532) had higher replication rate in monocyte-derived human dendritic cells (mdDCs) than isolates of virus from a non-fatal breakout (Brazil 2002; DENV3/290). The former also induced more proinflammatory cytokines associated with apoptosis[71]. Moreover, differences in cell toxicity among dengue variants have been attributed to mutations in the E and NS3[81]. Although adequate to explain certain differences in cell killing these mechanisms fail to explain the attenuated pathogenicity of immune/endothelial toxic dengue against other cells even in the presence of apoptotic agents like staurosporine, cycloheximide, camptothecin and influenza virus[83].
Involvement of autophagy in dengue infection is a relatively new finding, shown first in 2008. DENV2 caused ATG5-dependent autophagy in hepatic (Huh7) and fibroblast (MEF) cells. The virus’ ability to induce autophagy correlated positively with viral replication without a direct role in infectivity, as its downregulation did not increase amounts of intracellular virus[84,85]. Denv2-mediated autophagy protects from toxic stimuli canine kidney epithelial (MDCK) and mouse embryo fibroblast (MEF) cells but not murine macrophages, where infection leads to apoptotic cell death. Expression of dengue NS4A protein, like infection with live virus, induces PI3K-mediated autophagy and protects these cells against death from toxins[83]. Specific inhibitors of autophagy like spautin-1 have revealed the role autophagy plays in maturation of dengue virion. Blocking autophagy in Huh7.a.1, BHK21 cell lines and AG129 mice resulted in a heat-sensitive and non-infectious dengue virion[86].
West Nile virus
West Nile virus (WNV), first encountered in the New World in New York City (1999), has been the cause of three major arboviral neuroinvasive outbreaks in the United States[87]. It belongs to the same Flavivirus serocomplex as the Japanese encephalitis virus (JEV) and St. Louis encephalitis virus 15, following a bird-mosquito-bird transmission cycle. In the United States, Culex pipiens serves as the major arthropod vector. The human is a ‘‘dead-end host’’ for WNV due to low levels of serum viremia[88]. WNV consists of five phylogenetic lineages, of which 1, 2 have been associated with significant outbreaks. The primary targets are keratinocytes and dendritic cells, which upon infection migrate to visceral organs and the central nervous system. The neurovirulence of WNV is dependent on varying factors-its ability to cross the endothelium of blood-brain barrier (helped by cytokine mediated increased vascular permeability), import of infected macrophages into the CNS (Trojan horse mechanism) and viral retrograde transport from peripheral neurons to CNS[89-91]. Like dengue, outcome of infection varies from mild fever (WNV fever), accompanied by headache and diarrhea, to neurological symptoms (WNV neuroinvasive disease). While only 1% of infected individuals develop the latter, mild fever can be seen in 25%. However, neuroinvasive infections have a 10% fatality, which makes it extremely lethal. The serious pathological conditions (meningitis, encephalitis, acute flaccid paralysis) are also accompanied by chills, rash and visual disturbance. The severity is higher in elder patients, as is evident from the higher death rate (17%) in individuals aged at least 70 from those (0.8%) in their mid-40s[88,92,93]. Complete recovery following acute infection is extremely rare, and fatigue, cognitive difficulties, depression and muscle aches have been reported even after a year[94-97]. Diagnosis is dependent on detection of IgM levels in the cerebrospinal fluid by MAC-ELISA, although false positive results have been reported during infection with related Flavivirus[98,99]. To date, treatment has been supportive, relying on vector control, and no vaccine is licensed for human use. Human being the ‘‘dead-end host’’, future vaccinations will not prevent spreading of the virus in nature either[100-102]. It is extremely important that molecular mechanisms adopted by the virus, like manipulation of the cell survival pathway, be studied. This would help in developing an effective antiviral therapy.
Cell death and survival after infection with WNV
The relationship between WNV infectivity and cell survival pathways has been studied for more than a decade. WNV-mediated cell death and cytotoxicity depend on the severity of the initial infection. Vero cells infected with many virus particles (multiplicity of infection, moi > 10) showed signs of necrosis (leakage of HMGB1 and high LDH activity) within 8 h of infection. In contrast, cells infected with a lower load (moi < 10) showed signs of apoptotic cell death at a later stage (32 hpi)[103]. Very similar to dengue, WNV induces apoptotic cell death in several cell types, such as, immune cells (human leukemic -K562), neuronal cells (mouse neuroblastoma - Neuro 2a, brain tumors), epithelial cells (Vero, A549), fibroblasts (MEF, BHK21), and embryonic cells (HEK293T)[104-106].
The upstream events leading to apoptotic death in WNV infected cells include endoplasmic reticulum (ER) stress pathways. Infection of human neuroblastoma (SK-N-MC) cells and primary rat hippocampal neurons led to activation of two branches of ER stress-mediated unfolded protein response (UPR). ATF6 and PERK pathways were induced during infection, resulting in CHOP activation and downstream apoptosis[107]. A different effect on the UPR pathways has been observed. The West Nile virus Kunjin strain (WNVKUN) shuts off PERK pathway and interferon-mediated STAT phosphorylation in wild type MEFs. However, it activates the remaining two UPR (ATF6, IRE1) pathways. Studies with ATF6-/-, IRE1-/- MEFs point to the synergetic role these pathways play in WNVKUN pathogenesis. They contribute to increased cell viability and viral load, by restricting apoptotic cell death[108].
WNV can regulate both extrinsic and intrinsic pathways to launch pathogenesis (Figure 1). The virus induces Bax-dependent intracellular apoptosis in human leukemic (K562) and mouse neuroblastoma (Neuro 2a) cells. Strains that did not possess the ability to induce apoptosis, due to UV-inactivation, could not establish infectivity in cells[104]. WNV encephalitis in CNS-derived mouse neurons was highly dependent on the activation of caspase-3, and infection in the permissive T98G (brain-derived tumor) cells involved both extrinsic and intrinsic apoptotic pathways[105,106]. Tetracyclines are well established antiviral compounds, and minocycline strongly inhibited WNV infection in three CNS-derived human cell types (HBN, HRPE, and T98G).The antibiotic blocked viral replication, apoptosis and the viral activation of JNK/c-jun pathway, establishing a link among them[109]. Kobayashi et al[110] proposed that the presence of ubiquitinated proteins had functional implication in apoptosis of WNV-infected mouse neuroblastoma (Neuro-2a) cells. Migration of CD8+- T lymphocytes to drained lymph nodes (dLNs) was hindered in the CNS of Cd22-/- mice, which had a higher viral load than the wild type. This finding suggests a role for the B-cell marker, also an important component in cell survival, in modulating cellular immunity during infection[111].
Apoptosis often restricts viral replication and infection. Shrestha et al[112] showed the beneficial role of TNF-α related apoptosis inducing ligand (TRAIL), produced by CD8+- T cells, in limiting WNV infection in mouse central nervous system. CD8+- T cells in TRAIL-/- mice encountered difficulty in clearing the viral particles from the neurons. Zhang et al[113] demonstrated, using mouse neuron as an infection model, rise in the levels of TNF-α during infection. The rise served to downregulate the chemokine CXCR3, which would otherwise bind antiviral CXCL10 circulating in the central nervous system (CNS). This interaction results in calcium transients that lead to caspase-3 mediated apoptosis in the neurons, an adaptive mechanism to prevent cell death. Smith et al[114] showed an important aspect of WNV infection in human cell culture (HEK293, SK-N-MC) and mouse neuronal tissues - regulation of non-coding microRNAs (miRNAs). Among several miRNAs, Hs_154 is significantly up regulated in infection. Two of its targets, CCCTC-binding factor (CTCF) and epidermal growth factor receptor (EGFR), are associated with cell survival; this accounts for the role of Hs_154 in mediating apoptosis. While this activation has been found to lower viral replication, apoptotic cell death is also the basis for WNV pathogenesis.
As in dengue, both structural and non-structural proteins play a role in cellular survival after infection. WNV capsid (Cp) protein triggers a caspase-dependent apoptosis, leading to inflammation, in mouse brain and muscle[115]. WNV capsid is dependent upon p53 for its apoptotic effects. It has been shown to sequester HDM2, a negative regulator of p53, into the nucleolus. This results in a higher stability of p53, which can then target Bax to induce apoptosis in MEF cells[116]. Inhibitor-based studies on four types of mammalian cells (A549, HEK293T, Vero-76, BHK-21) suggest a role for WNV capsid (C) protein in the inhibition of apoptosis through Phosphatidylinositol-3-kinase (PI3K)- Akt prosurvival pathway[117]. The helicase and protease domains of NS3 protein are instrumental in inducing a caspase-8 dependent apoptosis in three types (Neuro 2a, HeLa, and Vero) of mammalian cells[118].
Our present knowledge does not suggest any significant role of autophagy in WNV pathogenesis, distinguishing it from dengue and Japanese encephalitis virus. Though infection induced autophagy in mice brain slice and several mammalian cells, it was actually PI3K that was involved in viral replication[119,120].
Japanese encephalitis virus
Japanese encephalitis virus (JEV) is extremely important as it is spreading throughout Asia, China, India, Australia, and Pakistan and is responsible for between 12500 to 17500 deaths reported annually. JEV is transmitted by a primary mosquito vector (Culex tritaeniorhynchus) and secondary mosquito vector (Culex gelidus, Culex fuscocephala and Culex vishnui) that primarily target domestic animals and human host[121]. Humans are “dead end host”, since they cannot infect the feeding mosquitoes because of low viremia. Children are at higher risk for an infection with Japanese encephalitis than adults, especially in rural areas. They are also at higher risk for death due to their weaker immune system as compared to the adults. In addition, people who visit Asia and Indonesia are particularly prone to this viral infection since they lack the protective antibodies. Asymptomatic infection depends on host’s age, immunity, general make-up and current health status. Symptoms include headache, fever, tremor, gadtrointestinal comfort as well as severe conditions of encephalitis and Parkinson-like seizures[122].
The means of the entry of the virus entry into the system plays an important role on the progress of the infection. If the carrier, the mosquito, bites directly into the blood vessel, it is easier for the virus to spread directly to the central nervous system.
There have been efforts to make a vaccine against JEV, although its successful implementation has been impeded by frequent climate changes. The spread of Japanese encephalitis virus is assisted by wind-blown mosquitoes, bird migration and people traveling with infected virus, which further spread the disease. Programs in underdeveloped countries are established in order to prevent the increasing number of yearly deaths caused by Japanese encephalitis virus. These programs include mosquito control by using pesticide, mosquito nets, cattle segregation and vaccination of cattle as well as humans[121,123].
Cell death and survival after infection with japanese encephalitis virus
As shown in Figure 1, JEV-induced apoptotic cell death is reliant on endoplasmic reticulum (ER) stress and production of reactive oxygen species (ROS). ER stress-induced activation of UPR factors (CHOP-p38MAPK) is essential for triggering the apoptotic response in fibroblasts (BHK-21) and neuronal cells (N18, NT-2)[124]. Even replication-incompetent strains (UV-JEV), as shown by Lin et al[125], retain their ability to kill neuronal cells (N18, NT-2) by inducing ROS production and activating NF-κB. The structural E protein from JEV-YL induces apoptotic cytotoxicity in HepG2and Vero cells[126]. Earlier studies had pointed to a link between non-structural NS3 protein and induction of apoptosis. Transfection of pEGFP-NS3 1-619 plasmid (whole NS3 protein) into Vero cells caused apoptotic cell death. The same study also evaluated the role of caspases where it was found that NS3 only activates the intrinsic branch (casp -9,-3) of apoptosis[127,128].
Bcl-2 proteins can prevent apoptosis by controlling the release of cytochrome C. Overexpression of bcl-2, however, did not block viral replication and distribution in mouse neuroblastoma N18 cells, though it delayed cell death in BHK-21 cells. Moreover, in BHK-21 and CHO cells, bcl-2 overexpression established persistent infection by virtue of its antiapoptotic property. Thus, bcl-2 was not a fruitful target for preventing infection. It was due to the ability of this virus to activate complex pathways of caspase-dependent apoptosis in some cells. Though JEV induced classical intrinsic pathway in N18 neuroblastoma cells, it activated both caspase-8 (part of the extrinsic pathway) and caspase-9 in a predominantly mitochondria-dependent pathway in MCF cells[129-131].
Japanese Encephalitis virus causes autophagy to facilitate viral replication in certain cell types. Li et al[132] showed induction of autophagy by virulent (RP-9) and attenuated (RP-2ms) JEV strain in human NT-2 cells. They also showed the positive effect of rapamycin induced autophagy on viral infection, and the reversal of that effect on blocking autophagy. Infection with Japanese encephalitis virus triggers innate immune response (through RIG-1/IRF-3 and P13K/NF signaling pathway) and activates inflammatory cytokines, chemokines and IFN-inducible proteins[133]. JEV Infection also induces autophagy in human microglial (CHME-5) cell line, leading to pro-inflammatory cytokine response.
CONCLUSION
Dengue is the worst arboviral human disease and most lethal among all Flavivirus members. It is remarkable how it manipulates the cell survival pathway in many types of cells, ultimately increasing viral load. From the literature, it is evident that dengue triggers different responses in different mammalian cells. Most of the dengue proteins (NS2, NS3, NS5, C, and E) have been reported to trigger extrinsic apoptosis pathway in many cells, including neurons, hepatocytes, immune cells, and endothelial cells. TNF-α and interleukins (IL-1β, 10) play a key role in this mechanism. However, M protein domains induce intrinsic apoptosis in neurons and hepatocytes. The virus may have alternate strategies to kill the cell, in case one of the cell death pathways is nonfunctional. In some cases, the virus has been able to induce different kinds of stress (ER, ROS, NO) conditions that lead to apoptotic cell death (Figure 1). Recent discoveries have shown that dengue can also activate autophagy in epithelial cells, fibroblasts and hepatocytes. It even uses this pathway to increase energy production, which would facilitate viral replication. Nonstructural proteins (NS2, 3, 4) have been involved in this process. The ability of dengue to use cell death or protective autophagy for virus replication in specific cell types is crucial in dengue’s versatility. Antivirals addressing the vast repertoire of the virus will contribute to counteracting dengue pathogenesis.
West Nile virus, though not as versatile as dengue, can trigger apoptosis in the central nervous system (CNS) to establish neuroinvasiveness. With a higher initial WNV dose, necrosis has been observed. An interesting aspect of infection with different strains lies in the differential regulation of ER stress-UPR pathways to achieve increased viral burden. The capsid protein positively interacts with p53 in vivo, activating the intrinsic pathway; however, in mammalian cells, it blocks apoptosis through PI3KI-Akt pathway. NS3 is involved in extrinsic apoptosis in neuroblastoma and cervical cancer cells. However, we need to know more about the effects of individual WNV proteins. A promising facet of WNV research is the attention focused on miRNA regulation, which needs to be extended to the other members of Flavivirus. This approach holds promise for antiviral therapy.
Japanese encephalitis virus, though pathogenetically similar to WNV, manipulates both intrinsic and extrinsic pathways to its advantage (Figure 1). JEV induces apoptosis in many neuronal cells by inducing upstream stress (UPR response, ROS production) events. JEV NS3, in contrast to DENV and WNV, induces the intrinsic pathway of apoptosis. There is also evidence that the virus can infect and replicate even in the absence of caspase-3, as it can induce caspase-6 and activate caspase-8 and -9 in a mitochondria dependent pathway. Moreover, caspase inhibition does not block viral production. Thus this Flavivirus appears to rely more on mitochondrial apoptosis for its pathogenesis. To add to the severity, it also utilizes autophagy to mediate pro-inflammatory cytokine response in neuronal cells.
Under these circumstances, we postulate that the Flavivirus has the ability to manipulate cell survival and innate immune response. The aftermath of viral invasion is dependent on initial dose and cell type. It can also switch to different mechanisms to exert its pathogenic effect in different cells of our body. The current understanding of cell death and survival during Flavivirus infection has not addressed many critical and complicated issues like the role of apoptosis and autophagy in killing infected cells or helping them to survive. Future studies should be aimed at finding out the function of individual viral proteins and the regulation of non-coding RNAs in viral infection. More emphasis needs to be put on studying the signaling pathways by which viruses regulate the cell survival pathways.
Footnotes
Supported by NIAID NIH grant to Zakeri Z, No. 1R15AIO94351-01; the NIH NIGMS (MARC-USTAR), No. T 34 GM070387
P- Reviewers: Gafencu AV, Migliaccio E Rhee DK S- Editor: Song XX L- Editor: A E- Editor: Lu YJ
References
- 1.McLean JE, Ruck A, Shirazian A, Pooyaei-Mehr F, Zakeri ZF. Viral manipulation of cell death. Curr Pharm Des. 2008;14:198–220. doi: 10.2174/138161208783413329. [DOI] [PubMed] [Google Scholar]
- 2.Kuno G, Chang GJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus Flavivirus. J Virol. 1998;72:73–83. doi: 10.1128/jvi.72.1.73-83.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gubler DJ. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 2002;10:100–103. doi: 10.1016/s0966-842x(01)02288-0. [DOI] [PubMed] [Google Scholar]
- 4. Available from: http: //www.who.int/mediacentre/factsheets/fs117/en/
- 5. Available from: http: //www.cdc.gov/westnile/index.html.
- 6. Available from: http: //www.who.int/mediacentre/factsheets/fs100/en/
- 7.Clarke P, Debiasi RL, Goody R, Hoyt CC, Richardson-Burns S, Tyler KL. Mechanisms of reovirus-induced cell death and tissue injury: role of apoptosis and virus-induced perturbation of host-cell signaling and transcription factor activation. Viral Immunol. 2005;18:89–115. doi: 10.1089/vim.2005.18.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fazakerley JK, Allsopp TE. Programmed cell death in virus infections of the nervous system. Curr Top Microbiol Immunol. 2001;253:95–119. doi: 10.1007/978-3-662-10356-2_5. [DOI] [PubMed] [Google Scholar]
- 9.Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol. 2001;2:545–550. doi: 10.1038/35080097. [DOI] [PubMed] [Google Scholar]
- 10.Clarke PG, Clarke S. Nineteenth century research on naturally occurring cell death and related phenomena. Anat Embryol (Berl) 1996;193:81–99. doi: 10.1007/BF00214700. [DOI] [PubMed] [Google Scholar]
- 11.Levraud JP, Adam M, Luciani MF, de Chastellier C, Blanton RL, Golstein P. Dictyostelium cell death: early emergence and demise of highly polarized paddle cells. J Cell Biol. 2003;160:1105–1114. doi: 10.1083/jcb.200212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maghsoudi N, Zakeri Z, Lockshin RA. Programmed cell death and apoptosis--where it came from and where it is going: from Elie Metchnikoff to the control of caspases. Exp Oncol. 2012;34:146–152. [PubMed] [Google Scholar]
- 13.Saunders JW. Death in embryonic systems. Science. 1966;154:604–612. doi: 10.1126/science.154.3749.604. [DOI] [PubMed] [Google Scholar]
- 14.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 16.Vaux DL, Weissman IL, Kim SK. Prevention of programmed cell death in Caenorhabditis elegans by human bcl-2. Science. 1992;258:1955–1957. doi: 10.1126/science.1470921. [DOI] [PubMed] [Google Scholar]
- 17.Ameisen JC, Capron A. Cell dysfunction and depletion in AIDS: the programmed cell death hypothesis. Immunol Today. 1991;12:102–105. doi: 10.1016/0167-5699(91)90092-8. [DOI] [PubMed] [Google Scholar]
- 18.Buttyan R, Zakeri Z, Lockshin R, Wolgemuth D. Cascade induction of c-fos, c-myc, and heat shock 70K transcripts during regression of the rat ventral prostate gland. Mol Endocrinol. 1988;2:650–657. doi: 10.1210/mend-2-7-650. [DOI] [PubMed] [Google Scholar]
- 19.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 20.Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res Ther. 2013;5:53. doi: 10.1186/alzrt217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nossal GJ. Negative selection of lymphocytes. Cell. 1994;76:229–239. doi: 10.1016/0092-8674(94)90331-x. [DOI] [PubMed] [Google Scholar]
- 22.Vaux DL, Korsmeyer SJ. Cell death in development. Cell. 1999;96:245–254. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 23.Greenberg JT. Programmed cell death: a way of life for plants. Proc Natl Acad Sci USA. 1996;93:12094–12097. doi: 10.1073/pnas.93.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinstein AD, Kimchi A. Life in the balance - a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci. 2012;125:5259–5268. doi: 10.1242/jcs.115865. [DOI] [PubMed] [Google Scholar]
- 26.Thomas LR, Johnson RL, Reed JC, Thorburn A. The C-terminal tails of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas receptors have opposing functions in Fas-associated death domain (FADD) recruitment and can regulate agonist-specific mechanisms of receptor activation. J Biol Chem. 2004;279:52479–52486. doi: 10.1074/jbc.M409578200. [DOI] [PubMed] [Google Scholar]
- 27.Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/s1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 28.Barnhart BC, Lee JC, Alappat EC, Peter ME. The death effector domain protein family. Oncogene. 2003;22:8634–8644. doi: 10.1038/sj.onc.1207103. [DOI] [PubMed] [Google Scholar]
- 29.Colombel M, Symmans F, Gil S, O’Toole KM, Chopin D, Benson M, Olsson CA, Korsmeyer S, Buttyan R. Detection of the apoptosis-suppressing oncoprotein bc1-2 in hormone-refractory human prostate cancers. Am J Pathol. 1993;143:390–400. [PMC free article] [PubMed] [Google Scholar]
- 30.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 31.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochim Biophys Acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 32.Caro-Maldonado A, Tait SW, Ramírez-Peinado S, Ricci JE, Fabregat I, Green DR, Muñoz-Pinedo C. Glucose deprivation induces an atypical form of apoptosis mediated by caspase-8 in Bax-, Bak-deficient cells. Cell Death Differ. 2010;17:1335–1344. doi: 10.1038/cdd.2010.21. [DOI] [PubMed] [Google Scholar]
- 33.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 34.Meijer WH, van der Klei IJ, Veenhuis M, Kiel JA. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy. 2007;3:106–116. doi: 10.4161/auto.3595. [DOI] [PubMed] [Google Scholar]
- 35.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J, Yang KH, Joe CO, Kang SS. Formation of distinct inclusion bodies by inhibition of ubiquitin-proteasome and autophagy-lysosome pathways. Biochem Biophys Res Commun. 2011;404:672–677. doi: 10.1016/j.bbrc.2010.12.040. [DOI] [PubMed] [Google Scholar]
- 37.Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL, Wogan GN. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci USA. 2013;110:E2950–E2957. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem. 2002;277:18619–18625. doi: 10.1074/jbc.M111889200. [DOI] [PubMed] [Google Scholar]
- 39.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:859–864. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaminskyy V, Zhivotovsky B. To kill or be killed: how viruses interact with the cell death machinery. J Intern Med. 2010;267:473–482. doi: 10.1111/j.1365-2796.2010.02222.x. [DOI] [PubMed] [Google Scholar]
- 42.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Vanden Berghe T, Grootjans S, Goossens V, Dondelinger Y, Krysko DV, Takahashi N, Vandenabeele P. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods. 2013;61:117–129. doi: 10.1016/j.ymeth.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 44.Lorenz IC, Allison SL, Heinz FX, Helenius A. Folding and dimerization of tick-borne encephalitis virus envelope proteins prM and E in the endoplasmic reticulum. J Virol. 2002;76:5480–5491. doi: 10.1128/JVI.76.11.5480-5491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997;71:8475–8481. doi: 10.1128/jvi.71.11.8475-8481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allison SL, Schalich J, Stiasny K, Mandl CW, Heinz FX. Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J Virol. 2001;75:4268–4275. doi: 10.1128/JVI.75.9.4268-4275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allison SL, Schalich J, Stiasny K, Mandl CW, Kunz C, Heinz FX. Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acidic pH. J Virol. 1995;69:695–700. doi: 10.1128/jvi.69.2.695-700.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corver J, Ortiz A, Allison SL, Schalich J, Heinz FX, Wilschut J. Membrane fusion activity of tick-borne encephalitis virus and recombinant subviral particles in a liposomal model system. Virology. 2000;269:37–46. doi: 10.1006/viro.1999.0172. [DOI] [PubMed] [Google Scholar]
- 49.Lindenbach BD, Rice CM. Molecular biology of flaviviruses. Adv Virus Res. 2003;59:23–61. doi: 10.1016/s0065-3527(03)59002-9. [DOI] [PubMed] [Google Scholar]
- 50.Brinton MA. The molecular biology of West Nile Virus: a new invader of the western hemisphere. Annu Rev Microbiol. 2002;56:371–402. doi: 10.1146/annurev.micro.56.012302.160654. [DOI] [PubMed] [Google Scholar]
- 51.Mukhopadhyay S, Kuhn RJ, Rossmann MG. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 52.La Ruche G, Souarès Y, Armengaud A, Peloux-Petiot F, Delaunay P, Desprès P, Lenglet A, Jourdain F, Leparc-Goffart I, Charlet F, et al. First two autochthonous dengue virus infections in metropolitan France, September 2010. Euro Surveill. 2010;15:19676. [PubMed] [Google Scholar]
- 53.Guha-Sapir D, Schimmer B. Dengue fever: new paradigms for a changing epidemiology. Emerg Themes Epidemiol. 2005;2:1. doi: 10.1186/1742-7622-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cleaves GR, Dubin DT. Methylation status of intracellular dengue type 2 40 S RNA. Virology. 1979;96:159–165. doi: 10.1016/0042-6822(79)90181-8. [DOI] [PubMed] [Google Scholar]
- 55.Amberg SM, Nestorowicz A, McCourt DW, Rice CM. NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: in vitro and in vivo studies. J Virol. 1994;68:3794–3802. doi: 10.1128/jvi.68.6.3794-3802.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cahour A, Falgout B, Lai CJ. Cleavage of the dengue virus polyprotein at the NS3/NS4A and NS4B/NS5 junctions is mediated by viral protease NS2B-NS3, whereas NS4A/NS4B may be processed by a cellular protease. J Virol. 1992;66:1535–1542. doi: 10.1128/jvi.66.3.1535-1542.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Falgout B, Pethel M, Zhang YM, Lai CJ. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol. 1991;65:2467–2475. doi: 10.1128/jvi.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- 59.Martina BE, Koraka P, Osterhaus AD. Dengue virus pathogenesis: an integrated view. Clin Microbiol Rev. 2009;22:564–581. doi: 10.1128/CMR.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gubler DJ. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. 1998;11:480–496. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guzmán MG, Kouri GP, Bravo J, Soler M, Vazquez S, Morier L. Dengue hemorrhagic fever in Cuba, 1981: a retrospective seroepidemiologic study. Am J Trop Med Hyg. 1990;42:179–184. doi: 10.4269/ajtmh.1990.42.179. [DOI] [PubMed] [Google Scholar]
- 62.Harris E, Videa E, Pérez L, Sandoval E, Téllez Y, Pérez ML, Cuadra R, Rocha J, Idiaquez W, Alonso RE, et al. Clinical, epidemiologic, and virologic features of dengue in the 1998 epidemic in Nicaragua. Am J Trop Med Hyg. 2000;63:5–11. doi: 10.4269/ajtmh.2000.63.5. [DOI] [PubMed] [Google Scholar]
- 63.Guilarde AO, Turchi MD, Siqueira JB, Feres VC, Rocha B, Levi JE, Souza VA, Boas LS, Pannuti CS, Martelli CM. Dengue and dengue hemorrhagic fever among adults: clinical outcomes related to viremia, serotypes, and antibody response. J Infect Dis. 2008;197:817–824. doi: 10.1086/528805. [DOI] [PubMed] [Google Scholar]
- 64.Kittigul L, Pitakarnjanakul P, Sujirarat D, Siripanichgon K. The differences of clinical manifestations and laboratory findings in children and adults with dengue virus infection. J Clin Virol. 2007;39:76–81. doi: 10.1016/j.jcv.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 65.Rothman AL. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat Rev Immunol. 2011;11:532–543. doi: 10.1038/nri3014. [DOI] [PubMed] [Google Scholar]
- 66.Wills BA, Oragui EE, Stephens AC, Daramola OA, Dung NM, Loan HT, Chau NV, Chambers M, Stepniewska K, Farrar JJ, et al. Coagulation abnormalities in dengue hemorrhagic Fever: serial investigations in 167 Vietnamese children with Dengue shock syndrome. Clin Infect Dis. 2002;35:277–285. doi: 10.1086/341410. [DOI] [PubMed] [Google Scholar]
- 67.Balsitis SJ, Coloma J, Castro G, Alava A, Flores D, McKerrow JH, Beatty PR, Harris E. Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am J Trop Med Hyg. 2009;80:416–424. [PubMed] [Google Scholar]
- 68.Basílio-de-Oliveira CA, Aguiar GR, Baldanza MS, Barth OM, Eyer-Silva WA, Paes MV. Pathologic study of a fatal case of dengue-3 virus infection in Rio de Janeiro, Brazil. Braz J Infect Dis. 2005;9:341–347. doi: 10.1590/s1413-86702005000400012. [DOI] [PubMed] [Google Scholar]
- 69.Guzmán MG, Alvarez M, Rodríguez R, Rosario D, Vázquez S, Vald s L, Cabrera MV, Kourí G. Fatal dengue hemorrhagic fever in Cuba, 1997. Int J Infect Dis. 1999;3:130–135. doi: 10.1016/s1201-9712(99)90033-4. [DOI] [PubMed] [Google Scholar]
- 70.Jessie K, Fong MY, Devi S, Lam SK, Wong KT. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J Infect Dis. 2004;189:1411–1418. doi: 10.1086/383043. [DOI] [PubMed] [Google Scholar]
- 71.Silveira GF, Meyer F, Delfraro A, Mosimann AL, Coluchi N, Vasquez C, Probst CM, Báfica A, Bordignon J, Dos Santos CN. Dengue virus type 3 isolated from a fatal case with visceral complications induces enhanced proinflammatory responses and apoptosis of human dendritic cells. J Virol. 2011;85:5374–5383. doi: 10.1128/JVI.01915-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Myint KS, Endy TP, Mongkolsirichaikul D, Manomuth C, Kalayanarooj S, Vaughn DW, Nisalak A, Green S, Rothman AL, Ennis FA, et al. Cellular immune activation in children with acute dengue virus infections is modulated by apoptosis. J Infect Dis. 2006;194:600–607. doi: 10.1086/506451. [DOI] [PubMed] [Google Scholar]
- 73.Jaiyen Y, Masrinoul P, Kalayanarooj S, Pulmanausahakul R, Ubol S. Characteristics of dengue virus-infected peripheral blood mononuclear cell death that correlates with the severity of illness. Microbiol Immunol. 2009;53:442–450. doi: 10.1111/j.1348-0421.2009.00148.x. [DOI] [PubMed] [Google Scholar]
- 74.Nagila A, Netsawang J, Srisawat C, Noisakran S, Morchang A, Yasamut U, Puttikhunt C, Kasinrerk W, Malasit P, Yenchitsomanus PT, et al. Role of CD137 signaling in dengue virus-mediated apoptosis. Biochem Biophys Res Commun. 2011;410:428–433. doi: 10.1016/j.bbrc.2011.05.151. [DOI] [PubMed] [Google Scholar]
- 75.Khunchai S, Junking M, Suttitheptumrong A, Yasamut U, Sawasdee N, Netsawang J, Morchang A, Chaowalit P, Noisakran S, Yenchitsomanus PT, et al. Interaction of dengue virus nonstructural protein 5 with Daxx modulates RANTES production. Biochem Biophys Res Commun. 2012;423:398–403. doi: 10.1016/j.bbrc.2012.05.137. [DOI] [PubMed] [Google Scholar]
- 76.Morchang A, Yasamut U, Netsawang J, Noisakran S, Wongwiwat W, Songprakhon P, Srisawat C, Puttikhunt C, Kasinrerk W, Malasit P, et al. Cell death gene expression profile: role of RIPK2 in dengue virus-mediated apoptosis. Virus Res. 2011;156:25–34. doi: 10.1016/j.virusres.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 77.Long X, Li Y, Qi Y, Xu J, Wang Z, Zhang X, Zhang D, Zhang L, Huang J. XAF1 contributes to dengue virus-induced apoptosis in vascular endothelial cells. FASEB J. 2013;27:1062–1073. doi: 10.1096/fj.12-213967. [DOI] [PubMed] [Google Scholar]
- 78.Lin CF, Lei HY, Shiau AL, Liu HS, Yeh TM, Chen SH, Liu CC, Chiu SC, Lin YS. Endothelial cell apoptosis induced by antibodies against dengue virus nonstructural protein 1 via production of nitric oxide. J Immunol. 2002;169:657–664. doi: 10.4049/jimmunol.169.2.657. [DOI] [PubMed] [Google Scholar]
- 79.Catteau A, Kalinina O, Wagner MC, Deubel V, Courageot MP, Desprès P. Dengue virus M protein contains a proapoptotic sequence referred to as ApoptoM. J Gen Virol. 2003;84:2781–2793. doi: 10.1099/vir.0.19163-0. [DOI] [PubMed] [Google Scholar]
- 80.Alonzo MT, Lacuesta TL, Dimaano EM, Kurosu T, Suarez LA, Mapua CA, Akeda Y, Matias RR, Kuter DJ, Nagata S, et al. Platelet apoptosis and apoptotic platelet clearance by macrophages in secondary dengue virus infections. J Infect Dis. 2012;205:1321–1329. doi: 10.1093/infdis/jis180. [DOI] [PubMed] [Google Scholar]
- 81.Duarte dos Santos CN, Frenkiel MP, Courageot MP, Rocha CF, Vazeille-Falcoz MC, Wien MW, Rey FA, Deubel V, Desprès P. Determinants in the envelope E protein and viral RNA helicase NS3 that influence the induction of apoptosis in response to infection with dengue type 1 virus. Virology. 2000;274:292–308. doi: 10.1006/viro.2000.0457. [DOI] [PubMed] [Google Scholar]
- 82.Avirutnan P, Malasit P, Seliger B, Bhakdi S, Husmann M. Dengue virus infection of human endothelial cells leads to chemokine production, complement activation, and apoptosis. J Immunol. 1998;161:6338–6346. [PubMed] [Google Scholar]
- 83.McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J Biol Chem. 2011;286:22147–22159. doi: 10.1074/jbc.M110.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, Lin YS, Yeh TM, Liu CC, Liu HS. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008;374:240–248. doi: 10.1016/j.virol.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Panyasrivanit M, Greenwood MP, Murphy D, Isidoro C, Auewarakul P, Smith DR. Induced autophagy reduces virus output in dengue infected monocytic cells. Virology. 2011;418:74–84. doi: 10.1016/j.virol.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 86.Mateo R, Nagamine CM, Spagnolo J, Méndez E, Rahe M, Gale M, Yuan J, Kirkegaard K. Inhibition of cellular autophagy deranges dengue virion maturation. J Virol. 2013;87:1312–1321. doi: 10.1128/JVI.02177-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA. 2013;310:308–315. doi: 10.1001/jama.2013.8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zou S, Foster GA, Dodd RY, Petersen LR, Stramer SL. West Nile fever characteristics among viremic persons identified through blood donor screening. J Infect Dis. 2010;202:1354–1361. doi: 10.1086/656602. [DOI] [PubMed] [Google Scholar]
- 89.Lim PY, Behr MJ, Chadwick CM, Shi PY, Bernard KA. Keratinocytes are cell targets of West Nile virus in vivo. J Virol. 2011;85:5197–5201. doi: 10.1128/JVI.02692-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schneider BS, Higgs S. The enhancement of arbovirus transmission and disease by mosquito saliva is associated with modulation of the host immune response. Trans R Soc Trop Med Hyg. 2008;102:400–408. doi: 10.1016/j.trstmh.2008.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cho H, Diamond MS. Immune responses to West Nile virus infection in the central nervous system. Viruses. 2012;4:3812–3830. doi: 10.3390/v4123812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lindsey NP, Staples JE, Lehman JA, Fischer M. Surveillance for human West Nile virus disease - United States, 1999-2008. MMWR Surveill Summ. 2010;59:1–17. [PubMed] [Google Scholar]
- 93.Bode AV, Sejvar JJ, Pape WJ, Campbell GL, Marfin AA. West Nile virus disease: a descriptive study of 228 patients hospitalized in a 4-county region of Colorado in 2003. Clin Infect Dis. 2006;42:1234–1240. doi: 10.1086/503038. [DOI] [PubMed] [Google Scholar]
- 94.Klee AL, Maidin B, Edwin B, Poshni I, Mostashari F, Fine A, Layton M, Nash D. Long-term prognosis for clinical West Nile virus infection. Emerg Infect Dis. 2004;10:1405–1411. doi: 10.3201/eid1008.030879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cook RL, Xu X, Yablonsky EJ, Sakata N, Tripp JH, Hess R, Piazza P, Rinaldo CR. Demographic and clinical factors associated with persistent symptoms after West Nile virus infection. Am J Trop Med Hyg. 2010;83:1133–1136. doi: 10.4269/ajtmh.2010.09-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haaland KY, Sadek J, Pergam S, Echevarria LA, Davis LE, Goade D, Harnar J, Nofchissey RA, Sewel CM, Ettestad P. Mental status after West Nile virus infection. Emerg Infect Dis. 2006;12:1260–1262. doi: 10.3201/eid1208.060097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sadek JR, Pergam SA, Harrington JA, Echevarria LA, Davis LE, Goade D, Harnar J, Nofchissey RA, Sewell CM, Ettestad P, et al. Persistent neuropsychological impairment associated with West Nile virus infection. J Clin Exp Neuropsychol. 2010;32:81–87. doi: 10.1080/13803390902881918. [DOI] [PubMed] [Google Scholar]
- 98.Tilley PA, Fox JD, Jayaraman GC, Preiksaitis JK. Nucleic acid testing for west nile virus RNA in plasma enhances rapid diagnosis of acute infection in symptomatic patients. J Infect Dis. 2006;193:1361–1364. doi: 10.1086/503577. [DOI] [PubMed] [Google Scholar]
- 99.Busch MP, Kleinman SH, Tobler LH, Kamel HT, Norris PJ, Walsh I, Matud JL, Prince HE, Lanciotti RS, Wright DJ, et al. Virus and antibody dynamics in acute west nile virus infection. J Infect Dis. 2008;198:984–993. doi: 10.1086/591467. [DOI] [PubMed] [Google Scholar]
- 100.Beasley DW. Vaccines and immunotherapeutics for the prevention and treatment of infections with West Nile virus. Immunotherapy. 2011;3:269–285. doi: 10.2217/imt.10.93. [DOI] [PubMed] [Google Scholar]
- 101.Diamond MS. Progress on the development of therapeutics against West Nile virus. Antiviral Res. 2009;83:214–227. doi: 10.1016/j.antiviral.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zohrabian A, Hayes EB, Petersen LR. Cost-effectiveness of West Nile virus vaccination. Emerg Infect Dis. 2006;12:375–380. doi: 10.3201/eid1203.050782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chu JJ, Ng ML. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J Gen Virol. 2003;84:3305–3314. doi: 10.1099/vir.0.19447-0. [DOI] [PubMed] [Google Scholar]
- 104.Parquet MC, Kumatori A, Hasebe F, Morita K, Igarashi A. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 2001;500:17–24. doi: 10.1016/s0014-5793(01)02573-x. [DOI] [PubMed] [Google Scholar]
- 105.Samuel MA, Morrey JD, Diamond MS. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J Virol. 2007;81:2614–2623. doi: 10.1128/JVI.02311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kleinschmidt MC, Michaelis M, Ogbomo H, Doerr HW, Cinatl J. Inhibition of apoptosis prevents West Nile virus induced cell death. BMC Microbiol. 2007;7:49. doi: 10.1186/1471-2180-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Medigeshi GR, Lancaster AM, Hirsch AJ, Briese T, Lipkin WI, Defilippis V, Früh K, Mason PW, Nikolich-Zugich J, Nelson JA. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J Virol. 2007;81:10849–10860. doi: 10.1128/JVI.01151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ambrose RL, Mackenzie JM. ATF6 signaling is required for efficient West Nile virus replication by promoting cell survival and inhibition of innate immune responses. J Virol. 2013;87:2206–2214. doi: 10.1128/JVI.02097-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Michaelis M, Kleinschmidt MC, Doerr HW, Cinatl J. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. J Antimicrob Chemother. 2007;60:981–986. doi: 10.1093/jac/dkm307. [DOI] [PubMed] [Google Scholar]
- 110.Kobayashi S, Orba Y, Yamaguchi H, Kimura T, Sawa H. Accumulation of ubiquitinated proteins is related to West Nile virus-induced neuronal apoptosis. Neuropathology. 2012;32:398–405. doi: 10.1111/j.1440-1789.2011.01275.x. [DOI] [PubMed] [Google Scholar]
- 111.Ma DY, Suthar MS, Kasahara S, Gale M, Clark EA. CD22 is required for protection against West Nile virus Infection. J Virol. 2013;87:3361–3375. doi: 10.1128/JVI.02368-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shrestha B, Pinto AK, Green S, Bosch I, Diamond MS. CD8+ T cells use TRAIL to restrict West Nile virus pathogenesis by controlling infection in neurons. J Virol. 2012;86:8937–8948. doi: 10.1128/JVI.00673-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang B, Patel J, Croyle M, Diamond MS, Klein RS. TNF-alpha-dependent regulation of CXCR3 expression modulates neuronal survival during West Nile virus encephalitis. J Neuroimmunol. 2010;224:28–38. doi: 10.1016/j.jneuroim.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Smith JL, Grey FE, Uhrlaub JL, Nikolich-Zugich J, Hirsch AJ. Induction of the cellular microRNA, Hs_154, by West Nile virus contributes to virus-mediated apoptosis through repression of antiapoptotic factors. J Virol. 2012;86:5278–5287. doi: 10.1128/JVI.06883-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang JS, Ramanathan MP, Muthumani K, Choo AY, Jin SH, Yu QC, Hwang DS, Choo DK, Lee MD, Dang K, et al. Induction of inflammation by West Nile virus capsid through the caspase-9 apoptotic pathway. Emerg Infect Dis. 2002;8:1379–1384. doi: 10.3201/eid0812.020224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang MR, Lee SR, Oh W, Lee EW, Yeh JY, Nah JJ, Joo YS, Shin J, Lee HW, Pyo S, et al. West Nile virus capsid protein induces p53-mediated apoptosis via the sequestration of HDM2 to the nucleolus. Cell Microbiol. 2008;10:165–176. doi: 10.1111/j.1462-5822.2007.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Urbanowski MD, Hobman TC. The West Nile virus capsid protein blocks apoptosis through a phosphatidylinositol 3-kinase-dependent mechanism. J Virol. 2013;87:872–881. doi: 10.1128/JVI.02030-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ramanathan MP, Chambers JA, Pankhong P, Chattergoon M, Attatippaholkun W, Dang K, Shah N, Weiner DB. Host cell killing by the West Nile Virus NS2B-NS3 proteolytic complex: NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway. Virology. 2006;345:56–72. doi: 10.1016/j.virol.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 119.Beatman E, Oyer R, Shives KD, Hedman K, Brault AC, Tyler KL, Beckham JD. West Nile virus growth is independent of autophagy activation. Virology. 2012;433:262–272. doi: 10.1016/j.virol.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vandergaast R, Fredericksen BL. West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS One. 2012;7:e45800. doi: 10.1371/journal.pone.0045800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Unni SK, Růžek D, Chhatbar C, Mishra R, Johri MK, Singh SK. Japanese encephalitis virus: from genome to infectome. Microbes Infect. 2011;13:312–321. doi: 10.1016/j.micinf.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 122.Misra UK, Kalita J. Overview: Japanese encephalitis. Prog Neurobiol. 2010;91:108–120. doi: 10.1016/j.pneurobio.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 123.Impoinvil DE, Ooi MH, Diggle PJ, Caminade C, Cardosa MJ, Morse AP, Baylis M, Solomon T. The effect of vaccination coverage and climate on Japanese encephalitis in Sarawak, Malaysia. PLoS Negl Trop Dis. 2013;7:e2334. doi: 10.1371/journal.pntd.0002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Su HL, Liao CL, Lin YL. Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J Virol. 2002;76:4162–4171. doi: 10.1128/JVI.76.9.4162-4171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lin RJ, Liao CL, Lin YL. Replication-incompetent virions of Japanese encephalitis virus trigger neuronal cell death by oxidative stress in a culture system. J Gen Virol. 2004;85:521–533. doi: 10.1099/vir.0.19496-0. [DOI] [PubMed] [Google Scholar]
- 126.Chen SO, Chang TJ, Stone G, Chen CH, Liu JJ. Programmed cell death induced by Japanese encephalitis virus YL vaccine strain or its recombinant envelope protein in varied cultured cells. Intervirology. 2006;49:346–351. doi: 10.1159/000095154. [DOI] [PubMed] [Google Scholar]
- 127.Yang TC, Shiu SL, Chuang PH, Lin YJ, Wan L, Lan YC, Lin CW. Japanese encephalitis virus NS2B-NS3 protease induces caspase 3 activation and mitochondria-mediated apoptosis in human medulloblastoma cells. Virus Res. 2009;143:77–85. doi: 10.1016/j.virusres.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 128.Yiang GT, Chen YH, Chou PL, Chang WJ, Wei CW, Yu YL. The NS3 protease and helicase domains of Japanese encephalitis virus trigger cell death via caspase-dependent and -independent pathways. Mol Med Rep. 2013;7:826–830. doi: 10.3892/mmr.2013.1261. [DOI] [PubMed] [Google Scholar]
- 129.Liao CL, Lin YL, Wang JJ, Huang YL, Yeh CT, Ma SH, Chen LK. Effect of enforced expression of human bcl-2 on Japanese encephalitis virus-induced apoptosis in cultured cells. J Virol. 1997;71:5963–5971. doi: 10.1128/jvi.71.8.5963-5971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liao CL, Lin YL, Shen SC, Shen JY, Su HL, Huang YL, Ma SH, Sun YC, Chen KP, Chen LK. Antiapoptotic but not antiviral function of human bcl-2 assists establishment of Japanese encephalitis virus persistence in cultured cells. J Virol. 1998;72:9844–9854. doi: 10.1128/jvi.72.12.9844-9854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tsao CH, Su HL, Lin YL, Yu HP, Kuo SM, Shen CI, Chen CW, Liao CL. Japanese encephalitis virus infection activates caspase-8 and -9 in a FADD-independent and mitochondrion-dependent manner. J Gen Virol. 2008;89:1930–1941. doi: 10.1099/vir.0.2008/000182-0. [DOI] [PubMed] [Google Scholar]
- 132.Li JK, Liang JJ, Liao CL, Lin YL. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect. 2012;14:159–168. doi: 10.1016/j.micinf.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 133.Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W, Xiao G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS One. 2013;8:e52909. doi: 10.1371/journal.pone.0052909. [DOI] [PMC free article] [PubMed] [Google Scholar]