

Abstract
Clear cell renal cell carcinoma (ccRCC) is a highly aggressive and common pathological subtype of renal cancer. This cancer is characterized by biallelic inactivation of the von Hippel–Lindau (VHL) tumor suppressor gene, which leads to the accumulation of hypoxia-inducible factors (HIFs). Although therapies targeted at HIFs can significantly improve survival, nearly all patients with advanced ccRCC eventually succumb to the disease. Thus, additional oncogenic events are thought to be involved in the development of ccRCC tumors. In this study, we investigated the role of RASSF6 in ccRCC. Downregulation of RASSF6 was commonly observed in primary tumors relative to matched adjacent normal tissues. Moreover, functional studies established that ectopic re-expression of RASSF6 in ccRCC cells inhibited cell proliferation, clonogenicity, and tumor growth in mice, whereas silencing of RASSF6 dramatically enhanced cell proliferation in vitro and in vivo. Mechanistic investigation suggested that RASSF6 triggers p21Cip1/Waf1 accumulation to induce G1 cell cycle arrest and promote apoptosis upon exposure to pro-apoptotic agents, and both of these mechanisms appear to be mediated by activated JNK signaling. Together, these findings suggest that RASSF6 may play a tumor suppressor role in the progression of ccRCC.
Keywords: RASSF6, ccRCC, cell cycle, apoptosis, p21Cip1/Waf1, JNK signaling
Introduction
Renal cell carcinoma (RCC) is the most common malignant renal tumor, and the occurrence of RCC has increased worldwide in recent years. Clear cell RCC (ccRCC), which accounts for approximately 80% of all RCCs, is characterized by inactivation of the von Hippel–Lindau (VHL) tumor suppressor gene. Loss of this gene leads to the stabilization of hypoxia-inducible factors (HIFs), which mediate the effects of hypoxia and play a central role in tumorigenesis.1 Drugs targeting the HIF axis (including mTOR inhibitors) have been approved for advanced RCC.2,3 However, the efficacy is thought to be limited, and treatment response is not long-standing.4 Therefore the overall survival of ccRCC remains poor.5-7 The pathogenesis of ccRCC is quite complicated, and it is unrealistic to expect that any single mechanism will uncover the truth. Additional tumorigenic events was supposed to contribute to the genesis and development of ccRCCs.8-10
In recent years, the aberrant activation of Ras-MAPK signaling has been reported in renal cell carcinoma.11,12 Serve as RAS effector, Ras-association domain family (RASSF) genes 6 harbor Ras-association (RA) domains in their C-terminal region and have the ability to bind to RAS.13 Ras superfamily proteins are activated by various extracellular stimuli and play important roles in regulating intracellular MAPK signaling pathways that control gene expression, mitosis, motility, and cell survival and differentiation. Recently, RASSF6 was reported to be frequently lost in primary tumors (including in a relatively small number of kidney cancers) and to play an important role in regulating several physiological processes, including cell proliferation, apoptosis, and tumor development.14,15 These findings suggest that RASSF6 may function as a tumor suppressor gene in cancers. However, the role of RASSF6 in ccRCC has not been previously investigated. In the present study, we investigated RASSF6 expression status in ccRCC samples, and found that it was significantly downregulated in renal cancer tissues and cultured cells. Both in vitro and in vivo functional studies were also performed to characterize the growth-inhibiting effects of RASSF6 in ccRCC. Moreover, the biological role of RASSF6 in cell cycle arrest and the promotion of apoptosis was mechanistically associated with the activation of JNK/SAPK signaling. These results collectively indicate a suppressive role for RASSF6 in ccRCC tumorigenesis.
Results
RASSF6 is frequently downregulated in archived ccRCC tissues and cell lines
RASSF6 mRNA expression levels were initially measured in 20 pairs of primary ccRCC samples and their corresponding non-tumor tissues by real-time quantitative PCR (qPCR). The relative expression level of RASSF6 was significantly lower in tumor tissues compared with the non-tumor counterparts (Fig. 1A, P < 0.01, paired t test). Western blotting further showed that downregulation of RASSF6 protein occurred in 5/8 randomly selected pairs of ccRCC and normal tissues (Fig. 1B). Downregulation of RASSF6 was also observed in all tested ccRCC cell lines compared with HK-2 immortalized human renal proximal epithelial tubular cells (Fig. 1C and D). These findings indicate that a reduction in the RASSF6 expression level is associated with the development of ccRCC.

Figure 1. Downregulation of RASSF6 expression in ccRCC tissues and cell lines. (A) RASSF6 mRNA expression levels in 20 matched primary ccRCC tissues (T) and adjacent noncancerous tissues (N) were determined by qPCR assays. GAPDH and 18S were used as reference genes. P < 0.01, paired t test. (B) Western blotting analysis of RASSF6 protein levels in another randomly selected 8 pairs of matched ccRCC tissues (T) and adjacent noncancerous tissues (N). (C and D) qPCR (C) and western blotting (D) analysis of RASSF6 expression in ccRCC cell lines and HK-2 immortalized renal proximal epithelial tubular cells.
RASSF6 demonstrates tumor suppressive ability in vitro and in vivo
To evaluate the function of RASSF6 in ccRCC development, RASSF6 was stably overexpressed in 2 ccRCC cell lines, 786-O and SKRC-39 (786-O-RF6 and SKRC39-RF6). Empty vector-transfected 786-O and SKRC-39 (786-O-Vec and SKRC-39-Vec) cells were used as controls. The expression of RASSF6 in these cells was confirmed by western blot analysis (Fig. 2A). In vitro assays revealed that ectopic expression of RASSF6 effectively inhibited cell proliferation, resulting in a significant inhibition of the cell growth rate (Fig. 2B, P < 0.01, Student t test) and a reduction in colony formation ability (Fig. 2C, P < 0.01, Student t test). To further explore the tumor suppressive role of RASSF6 in vivo, 786-O-RF6 and 786-O-Vec cells were subcutaneously injected into nude mice, and their capacity for tumorigenesis was evaluated. Tumor growth was significantly suppressed in mice injected with RASSF6-expressing 786-O cells compared with controls (Fig. 2D, P < 0.05, Student t test). We next stably suppressed RASSF6 expression in ACHN cells using 2 different shRNAs (ACHN-KD1 and ACHN-KD3, Fig. 3A). Suppression of RASSF6 led to a significant increase in cell viability, as analyzed by MTS and colony-formation assays (Fig. 3B and C). In vivo study further revealed that tumors formed from RASSF6 deplection ACHN cells presented significantly increased growth and weight compared with tumors formed from vector-transfected ACHN cells. These results strongly suggest that RASSF6 plays a tumor suppressor role in the development of ccRCC.

Figure 2. Overexpression of RASSF6 inhibits the proliferation of ccRCC cells in vitro and in vivo. (A–C) 786-O and SKRC39 cells stably overexpressing RASSF6 (RF6) or transfected with empty vector (Vec) were analyzed as follows. (A) RASSF6 protein expression levels were determined by western blot analysis; β-actin was used as a loading control. (B) Cell proliferation was determined by the MTS assay; *P < 0.05, **P < 0.01, Student t test. (C) Colony formation ability; representative micrographs (left) and quantification (right) of crystal violet-stained cells from 3 independent experiments; *P < 0.05, **P < 0.01, Student t test. (D) Control or RASSF6-overexpressing 786-O cells were inoculated subcutaneously into nude mice (n = 5/group). Tumor volumes were measured (left) and weighed (right) on the last day of the experiment. Representative images of isolated tumors (middle) are presented; *P < 0.05, Student t test; scale bar in picture: 1 cm.

Figure 3. RASSF6 knockdown promotes cell growth in vitro and tumor growth in vivo. ACHN cells were stably transfected with one of 2 RASSF6 shRNAs (KD1, KD3) or negative control shRNA (NC). (A) Western blotting analysis of RASSF6 expression; β-actin was used as a loading control. (B) RASSF6 depletion enhanced ACHN cell proliferation compared with control cells as determined by an MTS assay; *P < 0.05, **P < 0.01 for KD1 cells compared with NC cells; #P < 0.05, ##P < 0.01 for KD3 cells compared with NC cells. (C) The colony-formation ability of RASSF6-depleted ACHN cells was increased compared with vector control cells; *P < 0.05 for KD1 cells compared with NC cells; #P < 0.05 for KD3 cells compared with NC cells. (D) ACHN cells transfected with RASSF6 shRNA (KD1) or empty vector (NC) were inoculated subcutaneously into nude mice (n = 5/group). Tumor volumes were measured (left) and weighed (right) on the last day of the experiment. Representative images of isolated tumors (middle) are presented; *P < 0.05, Student t test; scale bar: 1 cm.
RASSF6 regulates the G1/S phase transition of the cell cycle through p21Cip1/Waf1
Ectopic expression of RASSF6 in 786-O and SKRC39 cells significantly increased the proportion of cells in G0/G1 phase and decreased the proportion in S phase (Fig. 4A). Conversely, RASSF6 depletion in ACHN cells decreased the proportion of cells in G0/G1 phase and increased the proportion in S phase (Fig. 4B). Cell cycle progression from G1/S phase is orchestrated by a tightly regulated interplay between cyclins D, E and A and their inhibitors. To confirm the role of RASSF6 in the G1/S-phase transition, these regulators were further evaluated. As shown in Figure 4C, RASSF6-expressing 786-O and SKRC39 cells displayed a reduction in cyclin D1 levels and an accumulation of p21Cip1/Waf1. In contrast, RASSF6 depletion in ACHN cells resulted in cyclin D1 upregulation and p21Cip1/Waf1 downregulation. Transient silencing of p21Cip1/Waf1 in RASSF6-expressing 786-O cells partially prevented the G0/G1 phase accumulation caused by RASSF6 (Fig. 4D, upper panel, and 4E; Fig. S1). Restoration of p21Cip1/Waf1 expression in ACHN-KDs and ACHN-NC cells (Fig. 4D, lower panel) restored the G0/G1-phase reduction mediated by RASSF6 depletion (Fig. 4F; Fig. S1). To further confirm the relationship between RASSF6 and p21Cip1/Waf1, we measured expression of both of them in ccRCC samples by using quantitative PCR, and found out p21Cip1/Waf1 expression was positively associated with RASSF6 expression in clinical samples (Fig. 4E). These results indicate the RASSF6-mediated cell cycle arrest is dependent on p21Cip1/Waf1.

Figure 4. RASSF6 promotes p21-dependent arrest at the G1/S-phase transition.(A) 786-O or SKRC39 cells stably overexpressing RASSF6 (RF6) or transfected with empty vector (Vec) were subjected to cell cycle analysis by flow cytometry. Images and qualification of the cell cycle distribution in 3 experiments are shown; *P < 0.05, **P < 0.01, Student t test. (B) ACHN cells stably transfected with one of 2 RASSF6 shRNAs (KD1, KD3) or with negative control shRNA (NC) were subjected to cell cycle distribution analysis by flow cytometry; *P < 0.05, **P < 0.01 for KD1 cells compared with NC cells; #P < 0.05 for KD3 cells compared with NC cells. (C) Western blotting analysis of cyclin A, cyclin D, cyclin E, and their inhibitors p21 and p27 in the indicated cell lines. β-tubulin was used as a loading control. (D and E) RASSF6-expressing and control 786-O cells were transiently transfected with p21 siRNA (Sip21) or scrambled control siRNA (SiNC). p21 expression levels were evaluated by western blot analysis (D, upper panel), and cells were subjected to cell cycle analysis (E); *P < 0.05, **P < 0.01, ns, not significant. (D and F) ACHN cells stably transfected with RASSF6 shRNA (KD1, KD3) or negative control shRNA (NC) were transiently transfected with a p21 plasmid (p21) or empty vector (Vec). p21 expression levels were evaluated by western blot analysis (D,lower panel), and cells were subjected to cell cycle analysis (F); *P < 0.05, **P < 0.01, ns, not significant. (G) RASSF6 mRNA levels were positively correlated with p21 expression in ccRCC tissues as determined by qPCR (GAPDH and 18S were used as reference genes. Pearson analysis, r = 0.712; P < 0.001).
RASSF6 promotes apoptosis in renal cancer cells
The potential role of RASSF6 in apoptosis was studied by treating RASSF6-expressing or empty vector control 786-O and SKRC39 cells with cisplatin, a strong and widely used chemotherapy drug that can induce apoptosis in a wide variety of cancer cells. As measured by Annexin-V/7-AAD staining, the apoptotic index (total number of Annexin-V-positive/PI-negative and Annexin-V-positive/PI-positive cells) was similar between RASSF6-expressing cells and vector-transfected cells without cisplatin treatment. After cisplatin exposure, the apoptotic index was significantly increased in RASSF6-expressing 786-O and SKRC39 cells (Fig. 5A). Conversely, RASSF6 silencing in ACHN cells resulted in a reduced amount of apoptosis upon cisplatin treatment (Fig. 5B). The apoptotic markers cleaved-PARP and Bax were dramatically increased in RASSF6-expressing cells after cisplatin treatment compared with control cells, whereas the converse results were observed in ACHN cells with RASSF6 depletion. However, no obvious alterations in p53 or Bcl-2 levels were detected in the ccRCC cells with altered RASSF6 expression (Fig. 5C).

Figure 5. RASSF6 has a pro-apoptotic role in ccRCC cells. (A) 786-O and SKRC39 cells stably overexpressing RASSF6 (RF6) or transfected with empty vector (Vec) were treated with or without cisplatin (6 μM for 786-O cells and 10 μM for SKRC39 cells) and then subjected to apoptosis analysis by Annexin-V and 7-AAD double staining. The error bars represent the mean ± SD from 3 independent experiments; *P < 0.05, **P < 0.01. (B) ACHN cells stably transfected with RASSF6 shRNA (KD1, KD3) or negative control shRNA (NC) were treated with or without cisplatin and then subjected to apoptosis analysis by Annexin-V and 7-AAD double staining. The error bars represent the mean ± SD from 3 independent experiments; **P < 0.01 for KD1 cells compared with NC cells; #P < 0.05 for KD3 cells compared with NC cells. (C) Western blot analysis of cleaved PARP, p53, Bax, and Bcl-2 in the indicated cells; GAPDH was used as a loading control.
RASSF6 mediates p21Cip1/Waf1-dependent cell cycle arrest and apoptosis by modulating the JNK/SAPK pathway
To elucidate the molecular basis of the cell cycle arrest and apoptosis promoted by RASSF6, we analyzed the effects of RASSF6 on Ras-MAPK signaling. Ectopically expressed RASSF6 specifically enhanced the phosphorylation of JNK (except for SKRC39 cells, indicating that phosphorylation of C-jun was independent of JNK phosphorylation in SKRC39 cells) and c-Jun, but not that of p38 kinase or ERK. The phosphorylation of JNK and c-Jun was conversely inhibited in RASSF6-depleted ACHN cells.
As mentioned above, RASSF6-induced G0/G1 arrest depended on increased expression of p21Cip1/Waf1 (Fig. 4), which indicated either a direct effect of RASSF6 on p21Cip1/Waf1 or an indirect mode of action via another unknown modulatory event. In fact, JNK activation has been reported to contribute to cell cycle arrest through p21Cip1/Waf1.16-18 We therefore explored whether the RASSF6-mediated upregulation of p21Cip1/Waf1 proceeds via activation of the JNK pathway. RASSF6-expressing 786-O or SKRC39 cells were treated with SP600125, a selective inhibitor of JNK, and the efficacy of the treatment was confirmed by reduced phosphorylation of c-Jun. This treatment effectively blocked the significant increase in p21Cip1/Waf1 expression induced by RASSF6 (Fig. 6B, upper panel). Moreover, SP600125 also reduced the RASSF6-induced G0/G1 cell cycle arrest (Fig. 6C; Fig. S2), which mimicked the effect of transient silencing of p21 in RASSF6-expressing cells. These data support a model wherein RASSF6 mediates p21Cip1/Waf1-dependent G0/G1 accumulation through JNK signaling in ccRCC.

Figure 6. RASSF6 inhibits the G1/S phase transition and promotes apoptosis though the JNK/SAPK pathway.(A) 786-O or SKRC39 cells stably overexpressing RASSF6 (RF6) or transfected with empty vector (Vec) and ACHN cells stably transfected with RASSF6 shRNAs (KD1, KD3) or negative control shRNA (NC) were analyzed for protein expression of phosphorylated JNK, total JNK, phosphorylated c-Jun (ser63), phosphorylated c-Jun (ser73), total c-Jun, phosphorylated ERK, total ERK, phosphorylated p38, and total p38. GAPDH or β-tubulin was used as the loading control. (B) RASSF6-expressing and control 786-O or SKRC39 cells were cultured in normal conditions (upper panel) or exposed to cisplatin treatment (lower panel) with or without SP600125 (5 μM for 786-O cells and 8 μM for SKRC39 cells). Levels of phosphorylated c-Jun, total c-Jun, p21, and cleaved-PARP (Cl−PARP) were measured by western blot. (C) RASSF6-expressing and control 786-O or SKRC39 cells were incubated in the presence or absence of SP600125, and their cell cycle distribution was assessed by flow cytometry; *P < 0.05; ns, not significant. (D) RASSF6-expressing and control 786-O or SKRC39 cells were incubated in the presence or absence of SP600125 (8 μM). Cisplatin was added as indicated, and the cells were incubated for an additional 48 h. Induction of apoptosis was determined by flow cytometry. The levels of apoptosis are presented as the mean ± SD from 3 independent experiments; *P < 0.05, **P < 0.01; ns, not significant.
We next sought to clarify whether RASSF6-induced apoptosis depended on the JNK signaling pathway. We co-treated RASSF6-expressing and vector-transfected cells with cisplatin in the absence or presence of the specific JNK inhibitor SP600125. This experiment showed that treatment with SP600125 resulted in an inhibition of RASSF6-induced PARP activation (Fig. 6B, lower panel) and a significant increase in cell viability after cisplatin treatment compared with controls (Fig. 6D; Fig. S3). These results indicate that activation of JNK is required for the pro-apoptotic function of RASSF6.
Discussion
RASSF family proteins are potential mediators of the growth-inhibitory effects of Ras.19-22 RASSF6, which demonstrates a different range of identity relative to other C-terminal RASSF proteins (RASSF1–5), is frequently lost in primary tumors, including breast, liver, colon, pancreas, stomach, and thyroid gland cancers. Therefore, a tumor suppressor role has been speculated for RASSF6.14 Consistent with previous reports, we found, for the first time, that RASSF6 was downregulated at both the mRNA and protein levels in ccRCC primary tumors and cell lines. CpG islands are predicted in the RASSF6 promoter region, and studies have shown that heavy methylation of these RASSF6 CpG islands is associated with expression silencing in leukemia cell lines23 and neuroblastoma.24 Based on these findings, we treated ccRCC cell lines exhibiting low levels of RASSF6 (786-O, SKRC-39, Caki-1) with 5-Aza-dC, an inhibitor of DNA methylation. However, RASSF6 expression was not significantly altered in the presence of 5-Aza-dC (Fig. S4), indicating that RASSF6 is not epigenetically regulated in ccRCC. Allen et al.14 also have reported nonmethylation-based mechanisms are involved in the downregulation of RASSF6, as 30~60% of primary tumor showed reduced levels of RASSF6 mRNA but only 1/7 of the tumor cell lines examined demonstrated partial promoter methylation of the promoter of RASSF6. RASSF6 is located at 4q13.3 in the genome, which has been reported to suffer from deletions during tumor development.25 A high number of chromosomal imbalances and losses of 4q are involved in the progression of multiple myeloma,26 hepatocellular carcinoma,27 and pancreatic adenocarcinoma.28 Thus, the association of RASSF6 silencing with 4q loss warrants further investigation.
RASSF6 reduces cell viability in specific tumor cell lines via a poorly understood mode of action.14,15 In the present study (the experimental design was presented in Fig. S5), we revealed the role of RASSF6 in ccRCC to involve G1/S-phase arrest as well as a pro-apoptotic effect that could be triggered by stimulation with apoptotic agents such as cisplatin. A model for the effect of RASSF6 on cell cycle and apoptosis in ccRCC is shown in Figure 7. RASSF6 inhibits the growth of both VHL-deficient (786-O) and VHL wild-type (SKRC39, ACHN)29-31 cells, suggesting that RASSF6 may not depend on VHL status in ccRCC. The treatment strategy to enhance the inhibitory function of RASSF6 is promising in ccRCC, especially after treatment failure of targeting VHL.
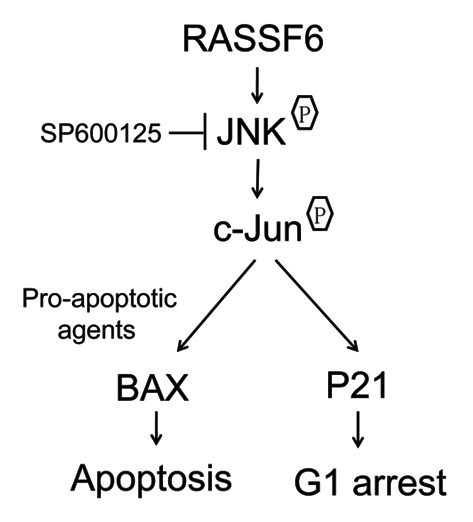
Figure 7. A model for the effect of RASSF6 on cell cycle and apoptosis in ccRCC. When RASSF6 is restored in ccRCC cells, RASSF6 activated JNK/SAPK pathway. Activated JNK/SAPK pathway then promotes apoptosis when exposed to pre-apoptotic agents and regulates p21 accumulation that leads to G1 arrest.
Cell cycle regulation is a common characteristic of the RASSF family,32-37 and RASSF1 and RASSF5 cause G1/S arrest through p21, a regulator of cell cycle progression at G1 and S phase.38,39 We observed that reintroduction of RASSF6 stimulated p21 protein expression, whereas inhibition of RASSF6 reduced p21 protein levels. This pattern was consistent with arrest in G0/G1 phase, which can be mediated by the CDK inhibitor p21Cip1/Waf1. The fact that transient silencing of p21Cip1/Waf1 blocked the G1 arrest caused by RASSF6, and that overexpression of p21Cip1/Waf1 restored the G1 phase further confirmed that RASSF6-mediated cell cycle arrest is closely associated with the activation of p21Cip1/Waf1.
RASSF family proteins, including RASSF6, have also been shown to participate in pro-apoptotic programs.14,20,22,40,41 For example, RASSF1A mediates some of its apoptotic functions by directly binding the pro-apoptotic Bax activator MOAP-1. RASSF6 was also previously reported to activate Bax and interact with MOAP-1, although MOAP-1 knockdown partially suppressed RASSF6-induced Bax activation,15,42 suggesting that other mechanisms may regulate the apoptosis mediated by RASSF6.43-45
p53 has also been reported to be involved in the cell cycle regulation and apoptosis mediated by RASSF proteins.37,38,46 In the latest published finding, Iwasa showed that RASSF6 regulates apoptosis and the cell cycle via stabilization of p53.47 Interestingly, in our study, we did not observe any changes in p53 expression related to RASSF6 in ccRCC, and we therefore suspect that RASSF6-induced cell cycle arrest and apoptosis in ccRCC is not dependent on p53 levels.
Loss or altered expression of RASSFs has also been associated with altered activity of the Ras-MAPK signaling axis.39,48,49 Like other RASSFs, RASSF6 preferentially associates with activated K-Ras,14 and Ras transduces extracellular information through a multitude of signaling cascades, primarily the ERK, JNK/SAPK, and p38 pathways. RASSF1A mediates cell cycle arrest by modulating the Raf-MEK-ERK pathway39 or blocking the JNK pathway,50 and RASSF1C participates in DNA damage through activation of JNK/SAPK.49 In our study, we found that RASSF6 overexpression resulted in activation of JNK signaling, but not ERK or p38 signaling. Therefore, the cell cycle arrest and apoptosis promotion induced by RASSF6 most likely resulted from activation of the JNK pathway. Moreover, inhibition of the JNK pathway in RASSF6-expressing cells decreased RASSF6-mediated upregulation of p21 and reduced G1 arrest, mimicking the effect of p21 silencing on cell cycle regulation. The relationship between JNK activation and p21Cip1/Waf1 in the cell cycle was also reported by Densham and colleagues, who found that JNK activation contributed to cell cycle arrest through the stabilization of p21Cip1/ Waf1 via phosphorylation of Thr57.16,17 All of these results support a model wherein RASSF6-mediated upregulation of p21Cip1/Waf1 in ccRCC occurs through enhancement of JNK signaling. We confirmed that the JNK pathway was required for RASSF6-mediated apoptosis in ccRCC by showing that JNK inhibition partially abolished the apoptosis induced by RASSF6. Based on these findings, we conclude that the RASSF6-mediated pro-apoptosis effect is partially dependent on the JNK/SAPK pathway.
Taken together, our findings demonstrate that RASSF6 is a novel tumor suppressor that plays an important role in the pathogenesis of ccRCC. In particular, RASSF6 activates JNK signaling to promote p21Cip1/Waf1-dependent inhibition of S-phase progression and apoptosis upon exposure to apoptotic agents. Thus, our findings reveal a central role for the RASSF6 tumor suppressor in the inhibition of survival signals in ccRCC.
Materials and Methods
Cell line selection, cell culture, cellular growth curve, and colony-formation assays
To rule out the effect of VHL, we chose VHL-deficient cells lines (786-O), as well as wide-type VHL cell lines (SKRC39, ACHN). All of 3 cell lines are well-established lines and suitable for transfection. Moreover, their abilities of tumor formation in nude mice have been repeatedly reported.29-31
The ccRCC cell lines 786-O, SKRC39, and ACHN were maintained in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) at 37 °C and 5% CO2. To plot the cellular growth curve, 3 × 103 cells suspended in 200 μl of medium were seeded into a 96-well plate (Corning) and cultured under normal conditions. At various time points after seeding, the cells in each well were stained with MTS (Promega, G5421), and the OD490 was determined with a microplate reader. For the colony-formation assays, cells were counted and plated at 500 cells per well in a 6-well plate (Corning). After 2 wk, the cells were washed with phosphate-buffered saline (PBS), fixed with methanol for 15 min at room temperature, and stained with crystal violet for 30 min. The colony was counted when it had more than 50 cells.
Paired tumor and tumor-adjacent tissues
Pairs of ccRCC tissues and matched tumor-adjacent morphologically normal tissues were obtained from Sun Yat-Sen University Cancer Center and frozen and stored in liquid nitrogen until used for comparing RASSF6 mRNA and protein expression levels.
Real-time quantitative PCR (qPCR)
Total RNA was extracted from cultured cell lines using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions and then reverse-transcribed using a cDNA Synthesis Kit (Takara, 6111A). Real-time qPCR was performed using a SYBR Green PCR Kit (Bio-Rad, 172–5200). The expression of each target gene was normalized to the endogenous levels of GAPDH or 18S. The relative mRNA levels are shown as 2ΔCt values. The sequences of the PCR primers used for amplification were as follows: GAPDH forward, 5′-AAGGTCATCC CTGAGCTGAA-3′; GAPDH reverse, 5′- TGACAAAGTG GTCGTTGAGG-3′; 18S forward, 5′-CATGGCCGTT CTTAGTTGGT-3′; 18S reverse, 5′-CGCTGAGCCA GTCAGTGTAG-3′; RASSF6 forward, 5′-GGGGGAATTT GACGATCTCT-3′; RASSF6 reverse, 5′-TAGAGCACTG GGGAGTCTGG -3′; p21 forward, 5′-GGATGTCCGT CAGAACCCAT-3′; p21 reverse, 5′-GTGGGAAGGT AGAGCTTGGG-5′.
Plasmid transfection experiment
Plasmid construction and transfection were performed as previously described.51 Briefly, full-length human p21 cDNA were cloned into pcDNA3.1. Growing ccRCC cells seeded at 2 × 105 cells per well of a 6-well tissue culture dish were transfected with 2 μg plasmid DNA or the corresponding empty control vector plasmid using Lipofectamine 2000 (Invitrogen, 11668–027) according to the manufacturer’s instructions.
RNAi treatment
The sequence of the small-interfering RNA (siRNA) targeting p21 has been reported52 and was synthesized by GenePharma. Growing cells were seeded at 2 × 105 cells per well in a 6-well tissue culture dish, and siRNAs were added 24 h later at a concentration of 80 nM using the RNAiMAX reagent (Invitrogen, 13778-075). The transfected cells were incubated for 6 h and then supplied with fresh medium containing serum.
Lentivirus transduction studies
A RASSF6 expression construct was generated by subcloning PCR-amplified full-length human RASSF6 cDNA into the pCDH-CMV-MCS-EF1-RFP plasmid. Cells stably expressing either RASSF6 short hairpin RNA (shRNAs) or a scrambled non-target shRNA were established using the LV3 plasmid according to the manufacturer's instructions. The targets of RASSF6 shRNA-1 and shRNA-3 were 5′- GAACAAAGAC GACTAAAGA-3′ and 5′- GGAATTTGAC GATCTCTAT-3′, respectively. Retroviral production and infection were performed as previously described.53 Stable cell lines expressing the shRNAs were selected with 5 μg/ml puromycin for 7 d.
Cell cycle analysis
Cells were harvested by trypsinization, washed in ice-cold PBS, and fixed in 75% ice-cold ethanol at −20 °C for at least 2 h. Before staining, cells were spun down and resuspended in PBS. RNase (Sigma, R3629) was added at a final concentration of 2 mg/ml, and the cells were incubated at 37 °C for 15 min, followed by incubation in 15 mg/ml propidium iodide (PI, Sigma, P4170) for 15 min at room temperature. At least 10 000 cells per sample were collected and analyzed on a flow cytometer (Beckman Coulter, Cytomics FC 500).
Apoptosis assays
After treating cells with or without cisplatin, both floating and adherent cells were harvested, washed with PBS, and stained with Annexin-V-Phycoerythrin/FITC (BD Biosciences, 559763/556547) and 7-AAD (BD Biosciences). The cells then were subjected to flow cytometry analysis.
Immunoblotting
Western blotting was performed according to standard methods as previously described.54 The primary antibodies used included RASSF6 (Sigma, HPA037711), β-tubulin (Sigma, T3952), p53 (Santa Cruz, sc-126), and Bax (Proteintech, 50599-2), as well as a number of antibodies from Cell Signaling Technology, including human cleaved PARP (6525), caspase 3 (9662), SAPK/JNK (9258), phospho-SAPK/JNK (Thr183/Tyr185, 4668), c-Jun (60A8), phospho-c-Jun (Ser63, 9261), phospho-c-Jun (Ser73, 3270), ERK (4780), phospho-ERK1/2 (5726), p38 (8690), phospho-p38 (Thr180/Tyr182, 4511), β-actin (4970), and GAPDH (2118). Anti-mouse and anti-rabbit peroxidase-conjugated secondary antibodies were purchased from Promega.
Animal experiments
All animal experiments were approved by the Sun Yat-Sen University Cancer Center Institutional Animal Care and Usage Committee. Mice were housed under standard conditions and cared for according to institutional guidelines. BALB/c (nu/nu) nude mice (4 wk of age, 15–18 g) were randomly divided into groups of 5 mice each. Cells (1 × 106 for 786 cells with stable RASSF6 overexpression or transfected with an empty vector; 8 × 106 for ACHN cells transfected with RASSF6 shRNA (KD1) or negative control shRNA (NC) were suspended in 150 μl RPMI 1640 medium and injected subcutaneously into the flank of each mouse. Tumor diameters were measured, and the volume (length × width2 × 0.5236) was calculated every week. Finally, the mice were euthanized, and the primary tumors were isolated and weighed.
Statistical analyses
All statistical analyses were performed using the SPSS 17.0 statistical software package. Data from 3 independent experiments are presented as the mean values with standard deviations. The differences between groups were evaluated using two-tailed Student t tests. The correlation between RASSF6 and p21Cip1/Waf1 levels in tissues was evaluated using 2-tailed Pearson’s correlation analysis. P values < 0.05 were considered statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81030043 and No. 81272340) and the National High Technology Research and Development Program of China (863 Program) (No. 2012AA02A501).
Glossary
Abbreviations:
- ccRCC
clear cell renal cell carcinoma
- VHL
von Hippel-Lindau
- HIFs
hypoxia-inducible factors
- RASSF
Ras-association domain family
- PBS
phosphate-buffered saline
- qPCR
quantitative real-time polymerase chain reaction
References
- 1.Vogelzang NJ, Stadler WM. Kidney cancer. Lancet. 1998;352:1691–6. doi: 10.1016/S0140-6736(98)01041-1. [DOI] [PubMed] [Google Scholar]
- 2.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277–85. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 4.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010;1:530–43. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motzer RJ, Russo P. Systemic therapy for renal cell carcinoma. J Urol. 2000;163:408–17. doi: 10.1016/S0022-5347(05)67889-5. [DOI] [PubMed] [Google Scholar]
- 6.Yang L, Yu SY, Hu GY. Pituitary metastasis from a renal cell carcinoma progressed after sorafenib treatment. Chin J Cancer. 2013;32:353–6. doi: 10.5732/cjc.012.10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang ZL, Li YH, Xiong YH, Hou GL, Yao K, Dong P, Liu ZW, Han H, Qin ZK, Zhou FJ. Oncological outcome of surgical treatment in 336 patients with renal cell carcinoma. Chin J Cancer. 2010;29:995–9. doi: 10.5732/cjc.010.10383. [DOI] [PubMed] [Google Scholar]
- 8.Sjölund J, Johansson M, Manna S, Norin C, Pietras A, Beckman S, Nilsson E, Ljungberg B, Axelson H. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J Clin Invest. 2008;118:217–28. doi: 10.1172/JCI32086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 10.Majid S, Saini S, Dahiya R. Wnt signaling pathways in urological cancers: past decades and still growing. Mol Cancer. 2012;11:7. doi: 10.1186/1476-4598-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang D, Ding Y, Luo WM, Bender S, Qian CN, Kort E, Zhang ZF, VandenBeldt K, Duesbery NS, Resau JH, et al. Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer Res. 2008;68:81–8. doi: 10.1158/0008-5472.CAN-07-5311. [DOI] [PubMed] [Google Scholar]
- 12.Furge KA, MacKeigan JP, Teh BT. Kinase targets in renal-cell carcinomas: reassessing the old and discovering the new. Lancet Oncol. 2010;11:571–8. doi: 10.1016/S1470-2045(09)70380-8. [DOI] [PubMed] [Google Scholar]
- 13.Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta. 2009;1796:114–28. doi: 10.1016/j.bbcan.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Allen NP, Donninger H, Vos MD, Eckfeld K, Hesson L, Gordon L, Birrer MJ, Latif F, Clark GJ. RASSF6 is a novel member of the RASSF family of tumor suppressors. Oncogene. 2007;26:6203–11. doi: 10.1038/sj.onc.1210440. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda M, Hirabayashi S, Fujiwara N, Mori H, Kawata A, Iida J, Bao Y, Sato Y, Iida T, Sugimura H, et al. Ras-association domain family protein 6 induces apoptosis via both caspase-dependent and caspase-independent pathways. Exp Cell Res. 2007;313:1484–95. doi: 10.1016/j.yexcr.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Coleman ML, Densham RM, Croft DR, Olson MF. Stability of p21Waf1/Cip1 CDK inhibitor protein is responsive to RhoA-mediated regulation of the actin cytoskeleton. Oncogene. 2006;25:2708–16. doi: 10.1038/sj.onc.1209322. [DOI] [PubMed] [Google Scholar]
- 17.Densham RM, O’Neill E, Munro J, König I, Anderson K, Kolch W, Olson MF. MST kinases monitor actin cytoskeletal integrity and signal via c-Jun N-terminal kinase stress-activated kinase to regulate p21Waf1/Cip1 stability. Mol Cell Biol. 2009;29:6380–90. doi: 10.1128/MCB.00116-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolomeichuk SN, Bene A, Upreti M, Dennis RA, Lyle CS, Rajasekaran M, Chambers TC. Induction of apoptosis by vinblastine via c-Jun autoamplification and p53-independent down-regulation of p21WAF1/CIP1. Mol Pharmacol. 2008;73:128–36. doi: 10.1124/mol.108.039750. [DOI] [PubMed] [Google Scholar]
- 19.Pfeifer GP, Yoon JH, Liu L, Tommasi S, Wilczynski SP, Dammann R. Methylation of the RASSF1A gene in human cancers. Biol Chem. 2002;383:907–14. doi: 10.1515/BC.2002.097. [DOI] [PubMed] [Google Scholar]
- 20.Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J Biol Chem. 2003;278:28045–51. doi: 10.1074/jbc.M300554200. [DOI] [PubMed] [Google Scholar]
- 21.Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005;65:3497–508. doi: 10.1158/0008-5472.CAN-04-4088. [DOI] [PubMed] [Google Scholar]
- 22.Eckfeld K, Hesson L, Vos MD, Bieche I, Latif F, Clark GJ. RASSF4/AD037 is a potential ras effector/tumor suppressor of the RASSF family. Cancer Res. 2004;64:8688–93. doi: 10.1158/0008-5472.CAN-04-2065. [DOI] [PubMed] [Google Scholar]
- 23.Hesson LB, Dunwell TL, Cooper WN, Catchpoole D, Brini AT, Chiaramonte R, Griffiths M, Chalmers AD, Maher ER, Latif F. The novel RASSF6 and RASSF10 candidate tumour suppressor genes are frequently epigenetically inactivated in childhood leukaemias. Mol Cancer. 2009;8:42. doi: 10.1186/1476-4598-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Djos A, Martinsson T, Kogner P, Carén H. The RASSF gene family members RASSF5, RASSF6 and RASSF7 show frequent DNA methylation in neuroblastoma. Mol Cancer. 2012;11:40. doi: 10.1186/1476-4598-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diep CB, Teixeira MR, Thorstensen L, Wiig JN, Eknaes M, Nesland JM, Giercksky KE, Johansson B, Lothe RA. Genome characteristics of primary carcinomas, local recurrences, carcinomatoses, and liver metastases from colorectal cancer patients. Mol Cancer. 2004;3:6. doi: 10.1186/1476-4598-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng SH, Ng MH, Lau KM, Liu HS, Chan JC, Hui AB, Lo KW, Jiang H, Hou J, Chu RW, et al. 4q loss is potentially an important genetic event in MM tumorigenesis: identification of a tumor suppressor gene regulated by promoter methylation at 4q13.3, platelet factor 4. Blood. 2007;109:2089–99. doi: 10.1182/blood-2006-04-018770. [DOI] [PubMed] [Google Scholar]
- 27.Chan KY, Lai PB, Squire JA, Beheshti B, Wong NL, Sy SM, Wong N. Positional expression profiling indicates candidate genes in deletion hotspots of hepatocellular carcinoma. Mod Pathol. 2006;19:1546–54. doi: 10.1038/modpathol.3800674. [DOI] [PubMed] [Google Scholar]
- 28.Luebke AM, Baudis M, Matthaei H, Vashist YK, Verde PE, Hosch SB, Erbersdobler A, Klein CA, Izbicki JR, Knoefel WT, et al. Losses at chromosome 4q are associated with poor survival in operable ductal pancreatic adenocarcinoma. Pancreatology. 2012;12:16–22. doi: 10.1016/j.pan.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science. 1995;269:1444–6. doi: 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]
- 30.An J, Liu H, Magyar CE, Guo Y, Veena MS, Srivatsan ES, Huang J, Rettig MB. Hyperactivated JNK is a therapeutic target in pVHL-deficient renal cell carcinoma. Cancer Res. 2013;73:1374–85. doi: 10.1158/0008-5472.CAN-12-2362. [DOI] [PubMed] [Google Scholar]
- 31.Gemmill RM, Zhou M, Costa L, Korch C, Bukowski RM, Drabkin HA. Synergistic growth inhibition by Iressa and Rapamycin is modulated by VHL mutations in renal cell carcinoma. Br J Cancer. 2005;92:2266–77. doi: 10.1038/sj.bjc.6602646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, Baier K, Dammann R, Pfeifer GP. The tumor suppressor RASSF1A does not interact with Cdc20, an activator of the anaphase-promoting complex. Cell Cycle. 2007;6:1663–5. doi: 10.4161/cc.6.13.4435. [DOI] [PubMed] [Google Scholar]
- 33.Shivakumar L, Minna J, Sakamaki T, Pestell R, White MA. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol Cell Biol. 2002;22:4309–18. doi: 10.1128/MCB.22.12.4309-4318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rong R, Jiang LY, Sheikh MS, Huang Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene. 2007;26:7700–8. doi: 10.1038/sj.onc.1210575. [DOI] [PubMed] [Google Scholar]
- 35.Deng ZH, Wen JF, Li JH, Xiao DS, Zhou JH. Activator protein-1 involved in growth inhibition by RASSF1A gene in the human gastric carcinoma cell line SGC7901. World J Gastroenterol. 2008;14:1437–43. doi: 10.3748/wjg.14.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song SJ, Song MS, Kim SJ, Kim SY, Kwon SH, Kim JG, Calvisi DF, Kang D, Lim DS. Aurora A regulates prometaphase progression by inhibiting the ability of RASSF1A to suppress APC-Cdc20 activity. Cancer Res. 2009;69:2314–23. doi: 10.1158/0008-5472.CAN-08-3984. [DOI] [PubMed] [Google Scholar]
- 37.Kudo T, Ikeda M, Nishikawa M, Yang Z, Ohno K, Nakagawa K, Hata Y. The RASSF3 candidate tumor suppressor induces apoptosis and G1-S cell-cycle arrest via p53. Cancer Res. 2012;72:2901–11. doi: 10.1158/0008-5472.CAN-12-0572. [DOI] [PubMed] [Google Scholar]
- 38.Calvisi DF, Donninger H, Vos MD, Birrer MJ, Gordon L, Leaner V, Clark GJ. NORE1A tumor suppressor candidate modulates p21CIP1 via p53. Cancer Res. 2009;69:4629–37. doi: 10.1158/0008-5472.CAN-08-3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thaler S, Hähnel PS, Schad A, Dammann R, Schuler M. RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer Res. 2009;69:1748–57. doi: 10.1158/0008-5472.CAN-08-1377. [DOI] [PubMed] [Google Scholar]
- 40.Baksh S, Tommasi S, Fenton S, Yu VC, Martins LM, Pfeifer GP, Latif F, Downward J, Neel BG. The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol Cell. 2005;18:637–50. doi: 10.1016/j.molcel.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Park SJ, Lee D, Choi CY, Ryu SY. Induction of apoptosis by NORE1A in a manner dependent on its nuclear export. Biochem Biophys Res Commun. 2008;368:56–61. doi: 10.1016/j.bbrc.2008.01.044. [DOI] [PubMed] [Google Scholar]
- 42.Ikeda M, Kawata A, Nishikawa M, Tateishi Y, Yamaguchi M, Nakagawa K, Hirabayashi S, Bao Y, Hidaka S, Hirata Y, et al. Hippo pathway-dependent and -independent roles of RASSF6. Sci Signal. 2009;2:ra59. doi: 10.1126/scisignal.2000300. [DOI] [PubMed] [Google Scholar]
- 43.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kashkar H, Wiegmann K, Yazdanpanah B, Haubert D, Krönke M. Acid sphingomyelinase is indispensable for UV light-induced Bax conformational change at the mitochondrial membrane. J Biol Chem. 2005;280:20804–13. doi: 10.1074/jbc.M410869200. [DOI] [PubMed] [Google Scholar]
- 45.Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005;12:783–95. doi: 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- 46.Song MS, Song SJ, Kim SY, Oh HJ, Lim DS. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 2008;27:1863–74. doi: 10.1038/emboj.2008.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwasa H, Kudo T, Maimaiti S, Ikeda M, Maruyama J, Nakagawa K, Hata Y. The RASSF6 tumor suppressor protein regulates apoptosis and the cell cycle via MDM2 protein and p53 protein. J Biol Chem. 2013;288:30320–9. doi: 10.1074/jbc.M113.507384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moshnikova A, Frye J, Shay JW, Minna JD, Khokhlatchev AV. The growth and tumor suppressor NORE1A is a cytoskeletal protein that suppresses growth by inhibition of the ERK pathway. J Biol Chem. 2006;281:8143–52. doi: 10.1074/jbc.M511837200. [DOI] [PubMed] [Google Scholar]
- 49.Kitagawa D, Kajiho H, Negishi T, Ura S, Watanabe T, Wada T, Ichijo H, Katada T, Nishina H. Release of RASSF1C from the nucleus by Daxx degradation links DNA damage and SAPK/JNK activation. EMBO J. 2006;25:3286–97. doi: 10.1038/sj.emboj.7601212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whang YM, Kim YH, Kim JS, Yoo YD. RASSF1A suppresses the c-Jun-NH2-kinase pathway and inhibits cell cycle progression. Cancer Res. 2005;65:3682–90. doi: 10.1158/0008-5472.CAN-04-2792. [DOI] [PubMed] [Google Scholar]
- 51.Xu S, Feng Z, Zhang M, Wu Y, Sang Y, Xu H, Lv X, Hu K, Cao J, Zhang R, et al. hSSB1 binds and protects p21 from ubiquitin-mediated degradation and positively correlates with p21 in human hepatocellular carcinomas. Oncogene. 2011;30:2219–29. doi: 10.1038/onc.2010.596. [DOI] [PubMed] [Google Scholar]
- 52.Miyazaki M, Sakaguchi M, Akiyama I, Sakaguchi Y, Nagamori S, Huh NH. Involvement of interferon regulatory factor 1 and S100C/A11 in growth inhibition by transforming growth factor beta 1 in human hepatocellular carcinoma cells. Cancer Res. 2004;64:4155–61. doi: 10.1158/0008-5472.CAN-03-2750. [DOI] [PubMed] [Google Scholar]
- 53.Li XJ, Ong CK, Cao Y, Xiang YQ, Shao JY, Ooi A, Peng LX, Lu WH, Zhang Z, Petillo D, et al. Serglycin is a theranostic target in nasopharyngeal carcinoma that promotes metastasis. Cancer Res. 2011;71:3162–72. doi: 10.1158/0008-5472.CAN-10-3557. [DOI] [PubMed] [Google Scholar]
- 54.Li XJ, Peng LX, Shao JY, Lu WH, Zhang JX, Chen S, Chen ZY, Xiang YQ, Bao YN, Zheng FJ, et al. As an independent unfavorable prognostic factor, IL-8 promotes metastasis of nasopharyngeal carcinoma through induction of epithelial-mesenchymal transition and activation of AKT signaling. Carcinogenesis. 2012;33:1302–9. doi: 10.1093/carcin/bgs181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.