

Abstract
Malignant transformation of the endothelium is rare, and hemangiosarcomas comprise only 1% of all sarcomas. For this reason and due to the lack of appropriate mouse models, the genetic mechanisms of malignant endothelial transformation are poorly understood. Here, we describe a hemangiosarcoma mouse model generated by deleting p53 specifically in the endothelial and hematopoietic lineages. This strategy led to a high incidence of hemangiosarcoma, with an average latency of 25 weeks. To study the in vivo roles of autocrine or endothelial cell autonomous VEGF signaling in the initiation and/or progression of hemangiosarcomas, we genetically deleted autocrine endothelial sources of VEGF in this mouse model. We found that loss of even a single conditional VEGF allele results in substantial rescue from endothelial cell transformation. These findings highlight the important role of threshold levels of autocrine VEGF signaling in endothelial malignancies and suggest a new approach for hemangiosarcoma treatment using targeted autocrine VEGF inhibition.
Keywords: hemangiosarcoma, endothelial cell, VEGF, p53, autocrine, mouse model
Introduction
Angiosarcoma is a rare but highly malignant type of mesenchymal neoplasm that arises from endothelial cells lining the walls of blood vessels (hemangiosarcoma; HSA) or lymphatic vessels (lymphangiosarcoma).1 These tumors are composed of poorly differentiated endothelial cells that form diffuse tubular vascular networks, which are highly hemorrhagic.2 Hemangiosarcomas are distributed similarly in males and females. They can develop spontaneously at any age, but are more common in the elderly.3 Malignant transformation can also occur within pre-existing benign vascular lesions. In addition, several predisposing conditions and treatments have been linked to the development of hemangiosarcomas, including chronic lymphoedema, radiotherapy, and exposure to various chemicals such as vinyl chloride, arsenic, radium, and anabolic steroids.4
Hemangiosarcomas principally spread hematogenously to lungs, liver, bone, and lymph nodes, and are typically known to express endothelial markers, including von Willebrand factor, CD34, CD31, and autocrine vascular endothelial growth factor (VEGF). However, progressive tumor dedifferentiation can lead to loss of some or all of these markers.4,5 In addition, hemangiosarcomas are believed be heterogeneous tumors consisting of multiple cell populations, including endothelial cells,6 and have been proven difficult to study in vitro because of their cellular heterogeneity and variability in their differentiation status.6,7
Vascular endothelial growth factor (VEGF) is an important mitogen and survival factor for endothelial cells and is essential for the generation of new blood vessels de novo (vasculogenesis) or from pre-existing vasculature (angiogenesis). Besides the major role of paracrine VEGF signaling, autocrine or endothelial cell-derived VEGF is also required for homeostasis of blood vessels, particularly in the adult endothelium, when paracrine (non-endothelial) sources of VEGF become more limiting. Genetic endothelial-specific deletion of VEGF causes vessel degeneration and leads to sudden death in 55% of mice by the age of 25 wk. This effect is endothelial cell-autonomous, as paracrine sources of VEGF do not compensate for the absence of endothelial VEGF.8 High VEGF levels have been implicated in the poor prognosis of various solid tumors, and high autocrine levels have been observed in hemangiosarcoma patients.9,10 Excessively high levels of VEGF also result in dysfunctional vessels11 and cytogenetically abnormal endothelial cells with centrosome overduplication and aneuploidy.12,13 This suggests that increased VEGF levels may play important roles in the malignant transformation of endothelial cells and the initiation and/or progression of hemangiosarcomas in addition to tumor vasculature abnormalization.14
Abrogation of p53 signaling also plays a critical role in the development of neoplasms of the vascular endothelium. Both p53 mutations and upregulation of the proto-oncogene Mdm2, which is an upstream negative regulator of p53, have been observed in two-thirds of angiosarcoma patients.15 Most somatic p53 mutations occur between exons 5 to 9, and the mutational frequency and pattern differ at various tumor anatomical sites.1 Although p53-deficient mice are primarily predisposed to developing thymic lymphomas (>70%), they also develop hemangiosarcoma with relatively low frequency (8–23%).16-18 These findings suggest that alteration in the Trp53 gene could be associated with tumorigenesis of the blood vessel endothelium. In addition, p53 inhibits VEGF expression via SP1,19 E2F,20 and p21/RB-dependent21 processes. Overall then, we hypothesized that decreased p53 expression and upregulated VEGF levels might act synergistically to transform the vascular endothelium.
In order to test this hypothesis and avoid the previously published pitfalls of in vitro mechanistic studies,6,7 we generated an in vivo hemangiosaroma mouse model in which p53 is specifically deleted in both the endothelial and hematopoietic lineages. We did this by crossing the Tie2–Cre line22 to mice carrying the conditional p53fl/fl allele.23 The resultant mice spontaneously developed hemangiosarcomas with high incidence by the age of 25 wk on average. To elucidate the role of autocrine or endothelial cell autonomous VEGF signaling in the development of hemangiosarcoma, we crossed these mice with conditional VEGFfl/fl loss-of-function mice.24 Genetic deletion of VEGF resulted in significantly reduced hemangiosarcoma incidence, even by single allelic deletion. These findings highlight the fundamental role of autocrine VEGF signaling in hemangiosarcoma development, and imply that targeting the endothelial cell-intrinsic pathway might be useful for hemangiosarcoma therapy.
Results
Tie2-Cre, p53fl/fl mice spontaneously develop hemangiosarcoma with high frequency, as well as precursor T-cell lymphoblastic lymphoma/leukemia
Absence of the Trp53 gene in mice predisposes them to neoplastic disease before the age of 6 mo, particularly sarcomas and lymphomas.17,27 To more specifically study the effects of p53 deletion on the endothelium, Tie2-Cre transgenic mice,22 in which Cre recombinase is active in both the endothelium and the hematopoietic system,28 were crossed with conditional p53fl/fl mice.23 The resultant Tie2-Cre, p53fl/fl mice developed tumors by the average age of 25 wk (Fig. 1A). Detailed pathological analysis was performed on 21 of these mice. Comprehensive immunohistological examination using a panel of endothelial and lymphatic as well as T- and B-cell-specific markers (Table S1) showed 2 main tumor types: (1) T-cell lymphomas/leukemia (46.9% of total neoplasm) developed in 71.4% of Tie2-Cre, p53fl/fl mice; and (2) hemangiosarcomas (40.6% of total neoplasms) developed in 61.9% of Tie2-Cre, p53fl/fl mice (Fig. 1B). For a more detailed overview of tumor spectrum and frequency in this mouse cohort, see Table 1 and Supplemental Material online describing the tumor categories.

Figure 1. Hematopoietic/endothelial deletion of p53 in mice results in T-cell lymphoma/leukemia and hemangiosarcoma development with an average latency of 25 wk. (A) Kaplan–Meier tumor-free survival curve for Cre negative control vs. Tie2-Cre, p53fl/+ and Tie2-Cre, p53fl/fl mice. ***P < 0.0001 (B) Tumor spectrum observed in 21 Tie2-Cre,p53fl/fl mice. Pie chart displaying percentage of T-cell lymphoma/leukemia and hemangiosarcoma observed. (C) Gross presentation of precursor T-cell lymphoblastic lymphoma (PTCLL) arising from the thymus. (D) Microscopically PTCLL consists of dense monomorphic sheets of neoplastic lymphoid cells displaying the typical “starry sky” pattern; H&E stain. (E) Immunohistochemically PTCLL diffusely expresses the T-cell marker CD3. (F) Neoplastic T cells are also variably positive for cKit. (G) PTCLL, negative control for CD3 and cKit immunohistochemistry. (H) Neoplastic lymphocytes are negative for the B-cell marker CD45R/B220. (I) Gross presentation of splenic HSA. (J) Microscopically HSA consists of anaplastic spindle cells lining irregular blood-filled vascular spaces; H&E stain. (K) Immunohistochemically HSA is positive for the endothelial cell marker CD31. (L) HSA, negative control for CD31 immunohistochemistry. (M) CD31-positive emboli of HSA metastasizing to the lung. (N) Metastatic HSA, negative control for CD31 immunohistochemistry. Scale bar for is 100 μm for (J–L) and 200 μm for (D–H, M,and N).
Table 1. Tumor spectrum and frequency in the different cohorts examined.
| Tumor phenotype | Tie2-Cre;p53fl/fl (total no. of mice = 21) | Tie2-Cre;p53fl/fl;Vegffl/+(total no. of mice = 12) | Tie2-Cre;p53fl/fl;Vegffl/fl(total no. of mice = 11) |
|---|---|---|---|
| Lymphoid hematopoietic neoplasms | 16 | 12 | 11 |
| Pre-T LL/L | 11 | 12 | 9 |
| (thymic) | (7) | (5) | (6) |
| (generalized/systemic) | (4) | (7) | (3) |
| Uncharacterised (non-thymic) T L | 4 | 0 | 1 |
| (multicentric) | (4) | (0) | (0) |
| (diffuse ACT tropism) | (0) | (0) | (1) |
| SMZL | 1 | 0 | 1 |
| Vascular neoplasms | 15 | 1 | 1 |
| HMA | 2 | 0 | 0 |
| HSA | 13 | 1 | 1 |
| (solitary) | (2) | (0) | (0) |
| (multicentric) | (10) | (1) | (1) |
| (multicentric with metastases) | (1) | (0) | (0) |
| Miscellaneous neoplasms | 1 | 0 | 0 |
| PA | 1 | 0 | 0 |
| Total number of tumors | 32 | 13 | 12 |
Pre-T LL/L, precursor T-cell lymphoblastic lymphoma/leukemia; T L, T cell lymphoma; ACT tropism, neoplastic infiltrates confined to adipose connective tissues; SMZL, splenic marginal zone lymphoma; HMA, hemangioma; HSA, hemangiosarcoma; PA, solitary pulmonary adenoma.
T-cell lymphomas/leukemia was by far the most common lymphoid neoplasm observed (Fig. 1C–H). As determined by immunohistochemistry, precursor T-cell lymphoblastic leukemia (Pre-T LL/L) and otherwise non-characterized T-cell lymphomas (T L) exhibited a CD45/CLA+; CD3+; CD45/B220− and IBA-1− immunophenotype. These are classical markers to differentiate between T- and B-cell lymphomas. Pre-T LL/L was characterized by primary thymic involvement with different degrees of local invasion (e.g., mediastinum, lungs, and heart) and distant spread to or involvement of extrathoracic organs/tissues, including spleen, superficial, and internal lymph nodes, bone marrow, liver, and kidneys. Otherwise non-characterized T-cell lymphomas displayed primary involvement of superficial and internal lymph nodes, Gut-associated lymphoid tissue (GALT), and spleen with different degrees of local invasion and massive spread to liver, kidneys, lungs, pancreas, gonads, and CNS. In these cases, thymus and bone marrow were distinctively spared. Several T-cell lymphoma cell lines were established from this mouse model that were capable of forming secondary T-cell lymphoma/leukemia when transplanted into NOD/SCID recipient mice (data not shown).
Hemangiosarcoma (HSA) was by far the most common vascular neoplasm observed (Fig. 1I–N). HSA were characterized as being positive for the endothelial marker CD31 but negative for the lymphatic marker Lyve1 and consisted of anaplastic spindle cells lining irregular blood-filled vascular spaces (Fig. 1J). These tumors were either solitary, arising from the retroperitoneal space or subcutis or muscular fascia of the trunk, or multicentric, arising simultaneously from 2 or more of the following locations: subcutis or muscular fascia of the trunk, heart, liver, spleen, or gonads. In one multicentric case there was evidence of metastatic spread to the lungs (Fig. 1M and N). In total, more than 60% of the mice developed multicentric and aggressive hemangiosarcomas, making these mice the first model to develop spontaneous hemangiosarcoma at such a high frequency.
Next, we tried to establish stable hemangiosarcoma cell lines from various Tie2-Cre, p53fl/fl primary hemangiosarcoma tumors. Therefore, we cultured either pure populations of CD31+ve tumor endothelial cells or the whole tumor mass and performed subsequent transplants into atymic nude mice. However, after in vitro expansion, none of these early passage primary cultures gave rise to secondary CD31+ hemangiosarcoma in atymic nude recipient mice (data not shown). Therefore, despite our inability to establish biologically relevant in vitro cell models of this complex disease, we then tested the usefulness of our model for studying genetic aspects of endothelial transformation and tumorigenesis.
Threshold levels of autocrine VEGF expression are involved in hemangiosarcoma formation in Tie2-Cre, p53fl/fl mice
There is increasing evidence that the VEGF–VEGFR autocrine loop is altered in endothelial and hematopoietic-related pathologies.9,10,29 Clinical studies demonstrated upregulated levels of VEGF and its receptors VEGFR1-R3, particularly in human angiosarcomas.9 Therefore, to genetically determine the role of autocrine VEGF sources in endothelial and hematopoietic-related tumorigenesis in vivo, we generated Tie2-Cre, p53fl/fl, VEGFfl/+ and Tie2-Cre, p53fl/fl, VEGFfl/fl mice to conditionally delete either one or both VEGF alleles, respectively.
Although VEGF deletion did not alter the survival rate and tumor incidence (Fig. 2A), it drastically changed the tumor spectrum (Fig. 2B). Deletion of a single conditional VEGF allele in Tie2-Cre, p53fl/fl, VEGFfl/+ mice significantly lowered the hemangiosarcoma incidence from 40.6% to 7.7% (n = 13, P ≤ 0.005). A similar decrease of the hemangiosarcoma incidence was obtained upon deletion of both VEGF alleles (8.3%; n = 12, P ≤ 0.005) (Fig. 2B; Table 1 for a detailed description of tumor spectrum and frequency in these 2 mouse lines). These results highlight the central role and importance of threshold levels of autocrine or cell autonomous VEGF–VEGFR signaling in hemangiosarcoma in this mouse model. However, neither mono- nor bi-allelic deletion of VEGF altered the latency or progression of T-cell lymphoma/leukemia incidence, suggesting that neither endothelial nor hematopoietic VEGF sources are essential in this particular tumor setting.
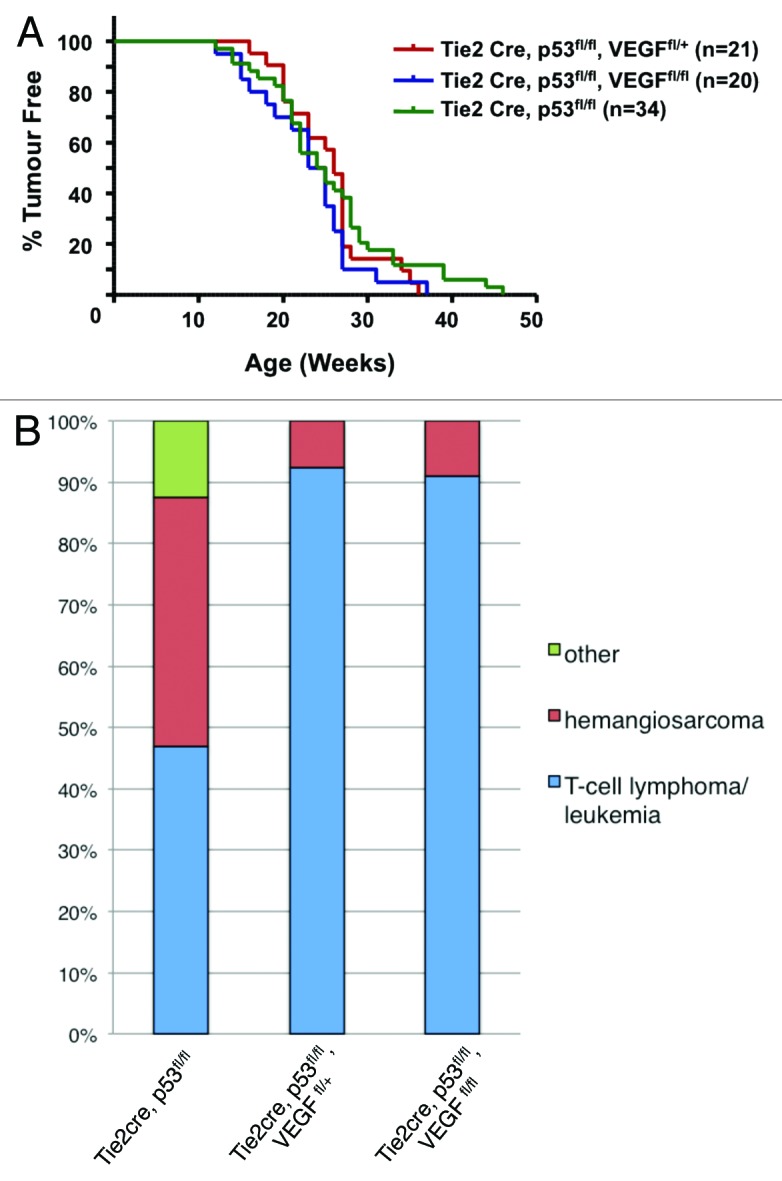
Figure 2. Simultaneous deletion of VEGF and p53 in the hematopoietic/endothelial lineage reduces hemangiosarcoma incidence. (A) Kaplan–Meier tumor-free survival curve for Tie2-Cre, p53fl/+, VEGFfl/fl; Tie2-Cre, p53fl/fl, VEGFfl/+, and Tie2-Cre, p53fl/fl, VEGFfl/fl mice. (B) Bar chart comparing tumor spectrum before and after VEGF deletion. Decrease in hemangiosarcoma development is observed in Tie2-Cre, p53fl/fl, VEGFfl/+ mice (n = 12; P ≤ 0.005) and Tie2-Cre, p53fl/fl, VEGFfl/fl mice (n = 11; P ≤ 0.005) vs. Tie2-Cre, p53fl/fl mice (n = 21).
Discussion
Although the endothelium is one of the largest cellular compartments of the body and can at any time shift between quiescence and growth phases, it rarely transforms to a malignant state.15 Therefore, angiosarcomas are very rare, and the genetic and molecular mechanisms involved in the malignant transformation of the endothelium are not well understood.1 The significance of p53 in endothelial malignancies can be appreciated from the observation that transgenic mice with global homozygous or heterozygous deletion of p53 are predisposed to hemangiosarcomas by up to 23%.16-18 To develop a mouse model that is prone to even higher incidences of spontaneous hemangiosarcoma, we generated transgenic Tie2-Cre, p53fl/fl mice with conditional deletion of p53 in the endothelial and hematopoietic lineages. More than 60% of these mice developed spontaneous (mostly multicentric) hemangiosarcoma by the average age of 25 wk. To further restrict p53 loss to the vascular endothelial lineages, other endothelial-specific Cre lines that have temporal control of Cre activity, such as VE-cadherin-CreER(T2)30 or PDGFB-iCreER(T2),31 could be used to avoid Cre activity in the hemogenic endothelium during development: this would thereby restrict p53 loss to the neonatal or adult endothelium and avoid p53 loss in the hematopoietic system.32 We expect that such mice will only develop hemangiosarcomas. However, as these inducible Cre mouse models often show mosaicism in tamoxifen-induced Cre activity, they might not develop angiosarcomas with such a high frequency, but with more variability than reported here.
It has been shown that p53 is a negative regulator of VEGF expression. This inhibition is achieved via different mechanisms.19,20 p21 is a well-known downstream target of p53 and is also a negative regulator of VEGF.21 Intriguingly, the second most common tumors in p21-null mice are of vascular origin.33 Therefore, the occurrence of hemangiosarcoma is most likely the result of a series of defects in cell cycle regulation and angiogenesis,34 which ultimately lead to malignant transformation of the endothelial cells. One of the challenges facing mechanistic studies on angiosarcomas at the cellular level is that these tumors are composed of multiple cell populations that are prone to the loss of some endothelial markers upon in vitro culturing.35,36 Therefore, establishing a primary murine angiosarcoma cell line that can give rise to secondary tumors in allotransplantation assays has been proven difficult.6,7 Using our mouse model, we experienced similar difficulties in establishing a hemangiosarcoma cell line that would reliably reflect this disease in vitro. After culturing, all cells lost their ability to develop hemangiosarcomas in allotransplant settings (data not shown). The use of this novel hemangiosarcoma mouse model that develops disease at such a high frequency partially circumvents these problems and allows further in vivo therapeutic and genetic studies concerning the initiation and progression of this rare but highly malignant tumor type. In this way, and for the first time, we have proven the in vivo significance of autocrine or endothelial cell-derived VEGF signaling in hemangiosarcoma development. Single allelic deletion of VEGF is enough to significantly decrease hemangiosarcoma tumor incidence from 40.6% in Tie2-Cre, p53fl/fl to 7.7% in Tie2-Cre, p53fl/fl, VEGFfl/+ mice, thereby demonstrating that this tumor type is dependent upon threshold levels of autocrine VEGF for efficient hemangiosarcoma initiation and/or progression. Specific inhibition of autocrine VEGF may therefore represent a very useful target for the development and refinement of anti-tumor strategies. Anti-VEGF-signaling drugs can affect tumor growth via different mechanisms, depending on the tumor type, which is why some patients do not benefit from these treatments.37 In addition, some studies of solid tumors have shown that treatment with VEGF inhibitors reduces primary tumor growth but increases tumor invasiveness and metastasis.38,39 Consistently, targeting VEGF and VEGFR to treat patients with angiosarcoma is promising but incomplete. Phase II trials using bevacizumab as a single agent to treat unresectable angiosarcoma and epithelioid hemangioendothelioma have demonstrated the deficiency of this therapeutic strategy.10 However, most of the previous strategies for anti-VEGF therapies are based on targeting paracrine or extracellular signaling, whereas our data argues that targeting local autocrine (or even intracellular) signaling may be a more powerful approach when treating hemangiosarcomas, such as through the use of intracellular suppressors of the VEGFR2 activation, like inhibitor SU4312.8
Although loss of a single VEGF allele caused a shift in tumor spectrum, it did not change tumor latency, as all mice died around 25 wk of age due to lymphoma development. Surprisingly, no sudden deaths were observed as reported before (55% by 25 wk of age8) upon deletion of autocrine VEGF signaling in our conditional p53 mice. This suggests that lymphoma development was the predominant cause of lethality at this time, precluding cardiovascular-related deaths. A more interesting possibility, however, is that p53 signaling may be involved in blood vessel homeostasis, and that upon decreased threshold levels of VEGF, endothelial cells may undergo a p53-dependent apoptotic process. These results suggest a complex cross-talk between the p53 and VEGF signaling pathways to ensure normal vessel homeostasis in the quiescent vasculature.21
Seventy percent of the Tie2-Cre p53fl/fl mice developed another tumor type, T-cell lymphoma/leukemia (46.9% of total neoplasms). We expected the development of lymphomas, as the Tie2-Cre is active in both the endothelium and hematopoietic lineages,40 and the most common tumor type reported in p53-null mice is T-cell lymphomas.18 Upon Tie2-Cre-mediated VEGF loss, these mice still developed these lymphomas at the same rate, indicating that endothelial and/or hematopoietic sources of VEGF do not play an essential role in the predisposition of mice to develop T-cell lymphoma. This does not, however, exclude the importance of other paracrine sources of VEGF in T-ALL development and spread of this disease.41
In conclusion, our study contributes to elucidating the genetic mechanisms involved in hemangiosarcoma development and emphasizes the important role of threshold autocrine or endothelial cell autonomous (intracellular) VEGF signaling in endothelial malignancy. Given the challenges facing anti-VEGF treatment by using paracrine (extracellular) inhibitors such as Bevacizumab, our study provides a useful target for development and refinement of future anti-angiogenic strategies based on autocrine (intracellular) inhibition of VEGF for more effective treatment of endothelial malignancies. Moreover, this mouse model of p53 deficiency should prove useful for in vivo screening of novel small-molecule inhibitors that may prove effective for treatment of hemangiosarcomas.
Materials and Methods
Transgenic mouse strains
The generation of transgenic Tie2-Cre,22 floxed-p53,23 and floxed-VEGF24 mice has been described. The ethics committee of Ghent University approved all animal experiments. The mouse cohorts used in these experiments were sibling littermates and were maintained on a mixed CD1 outbred genetic background to help eliminate strain-dependent phenotypes.
Mouse pathology
In the longitudinal survival study, a complete necropsy was performed on each mouse. Samples of gross lesions, brain, skin, salivary glands, trachea, esophagus, thyroids, lungs, heart, spleen, liver, kidneys, pancreas, gastrointestinal tract, thymus, lymph nodes (cervical, pancreatic, and mesenteric), urogenital tract, adrenals, and lumbar vertebrae (bone marrow) were formalin-fixed, paraffin-embedded, sectioned at 4 µm, and stained with hematoxylin and eosin for histopathological examination. Additional immunohistochemical analyses were also performed in order to better characterize the origin of the neoplastic lesions. Details of the immunohistochemical stains are in Table S1. The different categories of lymphoid and vascular tumors were defined according to the diagnostic criteria proposed by Morse25 and Rehg and Ward,26 respectively, and are listed in Supplemental Material.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dagmar Bonnier and Celine Steyt for help with mice breedings. We also thank Dr Castiglioni Vittoria and Recordati Camilla for aiding some of the necropsies of the mice and Dr Pieter De Bleser for help with statistical analysis. The VIB International PhD program provided M.F.G funding. S.G. is a postdoctoral fellow of the Basic Science Research Foundation-Flanders (FWO). L.H. funding was provided by the agency for Innovation by Science and Technology (IWT). This study was supported by the Belgium Federation against Cancer (Stichting tegen Kanker) and is part of the IUAP-Belspo network DevRepair (P7/07).
Glossary
Abbreviations:
- VEGF
vascular endothelial growth factor
- HSA
hemangiosarcoma
- Pre-T LL/L
precursor T-cell lymphoblastic leukemia
- TL
T-cell lymphoma
References
- 1.Naka N, Tomita Y, Nakanishi H, Araki N, Hongyo T, Ochi T, Aozasa K. Mutations of p53 tumor-suppressor gene in angiosarcoma. Int J Cancer. 1997;71:952–5. doi: 10.1002/(SICI)1097-0215(19970611)71:6<952::AID-IJC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 2.Rathmell WK, Acs G, Simon MC, Vaughn DJ. HIF transcription factor expression and induction of hypoxic response genes in a retroperitoneal angiosarcoma. Anticancer Res. 2004;24:167–9. [PubMed] [Google Scholar]
- 3.Fury MG, Antonescu CR, Van Zee KJ, Brennan MF, Maki RG. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer J. 2005;11:241–7. doi: 10.1097/00130404-200505000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. Lancet Oncol. 2010;11:983–91. doi: 10.1016/S1470-2045(10)70023-1. [DOI] [PubMed] [Google Scholar]
- 5.Ohsawa M, Naka N, Tomita Y, Kawamori D, Kanno H, Aozasa K. Use of immunohistochemical procedures in diagnosing angiosarcoma. Evaluation of 98 cases. Cancer. 1995;75:2867–74. doi: 10.1002/1097-0142(19950615)75:12<2867::AID-CNCR2820751212>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Zindy F, Nilsson LM, Nguyen L, Meunier C, Smeyne RJ, Rehg JE, Eberhart C, Sherr CJ, Roussel MF. Hemangiosarcomas, medulloblastomas, and other tumors in Ink4c/p53-null mice. Cancer Res. 2003;63:5420–7. [PubMed] [Google Scholar]
- 7.Krump-Konvalinkova V, Bittinger F, Olert J, Bräuninger W, Brunner J, Kirkpatrick CJ. Establishment and characterization of an angiosarcoma-derived cell line, AS-M. Endothelium. 2003;10:319–28. doi: 10.1080/714007546. [DOI] [PubMed] [Google Scholar]
- 8.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tokuyama W, Mikami T, Masuzawa M, Okayasu I. Autocrine and paracrine roles of VEGF/VEGFR-2 and VEGF-C/VEGFR-3 signaling in angiosarcomas of the scalp and face. Hum Pathol. 2010;41:407–14. doi: 10.1016/j.humpath.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 10.Park MS, Ravi V, Araujo DM. Inhibiting the VEGF-VEGFR pathway in angiosarcoma, epithelioid hemangioendothelioma, and hemangiopericytoma/solitary fibrous tumor. Curr Opin Oncol. 2010;22:351–5. doi: 10.1097/CCO.0b013e32833aaad4. [DOI] [PubMed] [Google Scholar]
- 11.Taylor SM, Nevis KR, Park HL, Rogers GC, Rogers SL, Cook JG, Bautch VL. Angiogenic factor signaling regulates centrosome duplication in endothelial cells of developing blood vessels. Blood. 2010;116:3108–17. doi: 10.1182/blood-2010-01-266197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akino T, Hida K, Hida Y, Tsuchiya K, Freedman D, Muraki C, Ohga N, Matsuda K, Akiyama K, Harabayashi T, et al. Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am J Pathol. 2009;175:2657–67. doi: 10.2353/ajpath.2009.090202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, Klagsbrun M. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64:8249–55. doi: 10.1158/0008-5472.CAN-04-1567. [DOI] [PubMed] [Google Scholar]
- 14.De Bock K, Cauwenberghs S, Carmeliet P. Vessel abnormalization: another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr Opin Genet Dev. 2011;21:73–9. doi: 10.1016/j.gde.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 15.Zietz C, Rössle M, Haas C, Sendelhofert A, Hirschmann A, Stürzl M, Löhrs U. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Pathol. 1998;153:1425–33. doi: 10.1016/S0002-9440(10)65729-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donehower LA, Harvey M, Vogel H, McArthur MJ, Montgomery CA, Jr., Park SH, Thompson T, Ford RJ, Bradley A. Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinog. 1995;14:16–22. doi: 10.1002/mc.2940140105. [DOI] [PubMed] [Google Scholar]
- 17.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 18.Harvey M, McArthur MJ, Montgomery CA, Jr., Bradley A, Donehower LA. Genetic background alters the spectrum of tumors that develop in p53-deficient mice. FASEB J. 1993;7:938–43. doi: 10.1096/fasebj.7.10.8344491. [DOI] [PubMed] [Google Scholar]
- 19.Pal S, Datta K, Mukhopadhyay D. Central role of p53 on regulation of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in mammary carcinoma. Cancer Res. 2001;61:6952–7. [PubMed] [Google Scholar]
- 20.Qin G, Kishore R, Dolan CM, Silver M, Wecker A, Luedemann CN, Thorne T, Hanley A, Curry C, Heyd L, et al. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc Natl Acad Sci U S A. 2006;103:11015–20. doi: 10.1073/pnas.0509533103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farhang Ghahremani M, Goossens S, Nittner D, Bisteau X, Bartunkova S, Zwolinska A, Hulpiau P, Haigh K, Haenebalcke L, Drogat B, et al. p53 promotes VEGF expression and angiogenesis in the absence of an intact p21-Rb pathway. Cell Death Differ. 2013;20:888–97. doi: 10.1038/cdd.2013.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kisanuki YYHR, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 23.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 24.Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–59. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- 25.Morse HC, 3rd, Anver MR, Fredrickson TN, Haines DC, Harris AW, Harris NL, Jaffe ES, Kogan SC, MacLennan IC, Pattengale PK, et al. Hematopathology subcommittee of the Mouse Models of Human Cancers Consortium Bethesda proposals for classification of lymphoid neoplasms in mice. Blood. 2002;100:246–58. doi: 10.1182/blood.V100.1.246. [DOI] [PubMed] [Google Scholar]
- 26.Rehg JE, Ward JM. Morphological and immunohistochemical characterization of sarcomatous tumors in wild-type and genetically engineered mice. Vet Pathol. 2012;49:206–17. doi: 10.1177/0300985811429813. [DOI] [PubMed] [Google Scholar]
- 27.Harvey M, McArthur MJ, Montgomery CA, Jr., Butel JS, Bradley A, Donehower LA. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet. 1993;5:225–9. doi: 10.1038/ng1193-225. [DOI] [PubMed] [Google Scholar]
- 28.Goossens S, Janzen V, Bartunkova S, Yokomizo T, Drogat B, Crisan M, Haigh K, Seuntjens E, Umans L, Riedt T, et al. The EMT regulator Zeb2/Sip1 is essential for murine embryonic hematopoietic stem/progenitor cell differentiation and mobilization. Blood. 2011;117:5620–30. doi: 10.1182/blood-2010-08-300236. [DOI] [PubMed] [Google Scholar]
- 29.Aguayo A, Kantarjian H, Manshouri T, Gidel C, Estey E, Thomas D, Koller C, Estrov Z, O’Brien S, Keating M, et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood. 2000;96:2240–5. [PubMed] [Google Scholar]
- 30.Monvoisin A, Alva JA, Hofmann JJ, Zovein AC, Lane TF, Iruela-Arispe ML. VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev Dyn. 2006;235:3413–22. doi: 10.1002/dvdy.20982. [DOI] [PubMed] [Google Scholar]
- 31.Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- 32.Zovein AC, Hofmann JJ, Lynch M, French WJ, Turlo KA, Yang Y, Becker MS, Zanetta L, Dejana E, Gasson JC, et al. Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell. 2008;3:625–36. doi: 10.1016/j.stem.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martín-Caballero J, Flores JM, García-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61:6234–8. [PubMed] [Google Scholar]
- 34.Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–4. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 35.Masuzawa M, Fujimura T, Tsubokawa M, Nishiyama S, Katsuoka K, Terada E, Kunita S, Sakurai Y, Kato H. Establishment of a new murine-phenotypic angiosarcoma cell line (ISOS-1) J Dermatol Sci. 1998;16:91–8. doi: 10.1016/S0923-1811(97)00032-7. [DOI] [PubMed] [Google Scholar]
- 36.Masuzawa M, Fujimura T, Hamada Y, Fujita Y, Hara H, Nishiyama S, Katsuoka K, Tamauchi H, Sakurai Y. Establishment of a human hemangiosarcoma cell line (ISO-HAS) Int J Cancer. 1999;81:305–8. doi: 10.1002/(SICI)1097-0215(19990412)81:2<305::AID-IJC22>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 37.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumor activity. Nat Rev Cancer. 2008;8:579–91. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 38.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zape JP, Zovein AC. Hemogenic endothelium: origins, regulation, and implications for vascular biology. Semin Cell Dev Biol. 2011;22:1036–47. doi: 10.1016/j.semcdb.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Hayashibara T, Fujimoto T, Miyanishi T, Yamada Y, Maita T, Kamihira S, Tomonaga M. Vascular endothelial growth factor at high plasma levels is associated with extranodal involvement in adult T cell leukemia patients. Leukemia. 1999;13:1634–5. doi: 10.1038/sj.leu.2401546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.