

Abstract
FANCD2 is required for the repair of DNA damage by the FA (Fanconi anemia) pathway, and, consequently, FANCD2-deficient cells are sensitive to compounds such as cisplatin and formaldehyde that induce DNA:DNA and DNA:protein crosslinks, respectively. The DNA2 helicase/nuclease is required for RNA/DNA flap removal from Okazaki fragments during DNA replication and for the resection of DSBs (double-strand breaks) during HDR (homology-directed repair) of replication stress-induced damage. A knockdown of DNA2 renders normal cells as sensitive to cisplatin (in the absence of EXO1) and to formaldehyde (even in the presence of EXO1) as FANCD2−/− cells. Surprisingly, however, the depletion of DNA2 in FANCD2-deficient cells rescues the sensitivity of FANCD2−/− cells to cisplatin and formaldehyde. We previously showed that the resection activity of DNA2 acts downstream of FANCD2 to insure HDR of the DSBs arising when replication forks encounter ICL (interstrand crosslink) damage. The suppression of FANCD2−/− by DNA2 knockdowns suggests that DNA2 and FANCD2 also have antagonistic roles: in the absence of FANCD2, DNA2 somehow corrupts repair. To demonstrate that DNA2 is deleterious to crosslink repair, we used psoralen-induced ICL damage to trigger the repair of a site-specific crosslink in a GFP reporter and observed that “over-resection” can account for reduced repair. Our work demonstrates that excessive resection can lead to genome instability and shows that strict regulatory processes have evolved to inhibit resection nucleases. The suppression of FANCD2−/− phenotypes by DNA2 depletion may have implications for FA therapies and for the use of ICL-inducing agents in chemotherapy.
Keywords: DNA recombination, DNA replication, DNA2, FANCD2, Fanconi anemia, formaldehyde DNA damage, interstrand crosslinks
Introduction
Fanconi anemia (FA) is a rare hereditary disease characterized by chromosomal instability, developmental abnormalities, bone marrow failure, and predisposition to cancer.1-5 FA can be classified into 16 complementation groups, each associated with a defective FA gene. The FA gene products participate in the FA/breast cancer allele (FA/BRCA) DNA repair pathway, which is responsible for genome maintenance after DNA:DNA crosslinks, DNA:protein crosslinks, and S-phase replication stress.4 While the study of the repair of interstrand crosslinks has been instrumental in defining the players in the FA/BRCA network, recent work is focusing on defining the endogenous causes of DNA damage underlying FA pathogenesis. These studies suggest that the FA/BRCA pathway has evolved as a central surveillance mechanism for DNA replication fork problems.6,7
The FA proteins themselves coordinate an array of enzymes from different repair pathways. FANCM/FAAP24 and the FA core complex function early. FANCM/FAAP24, an ATP-dependent translocase, has multiples roles, one of which may be the recognition of DNA lesions.1,8-13 The FA core complex, in turn, ubiquitylates the downstream FANCD2 and FANCI factors. The FA core complex consists of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, and FANCL, an E3 ubiquitin ligase together with an associated 100-kDa protein FAAP100.4 During the intermediate steps of the pathway, in S-phase cells, the monoubiquitylated FANCD2:I complex stabilizes replication forks stalled at the sites of DNA damage.6,7 FANCD2:I also coordinates the actions of SLX4(FANCP) and multiple nucleases (MUS81:EME1, XPF:ERCC1, SLX1, and FAN1) to incise and excise the lesions in an incompletely delineated sequence of steps that somehow correctly feeds one intermediate into the next. FANCD2:I also regulates translesion polymerases and the NER (nucleotide excision repair) machinery that collaborate to remove damaged bases from the strand that is not incised. At a stalled replication fork or stalled converging forks, these nucleolytic events give rise to DSBs (double-strand breaks). During the late stages of repair, the DSBs are repaired by HDR (homology-directed repair), requiring BRCA2 (FANCD1), RAD51C (FANCO), BRIP1, BRCA1, PALB2, and FANCJ. Alternatively, if repair is mediated by a competing pathway of NHEJ (non-homologous end joining), chromosomal instability can ensue.14-16 The proteins involved in the critical DSB resection step in HDR have not yet been unequivocally identified.
In addition to comprising a complex network in and of itself, this FA/BRCA pathway integrates with many additional repair pathways. Bloom syndrome has many symptoms and cytological defects similar to FA. The gene mutated in Bloom syndrome, BLM, and FANCD2 are localized to the ultrafine bridges that form in mitosis and are associated with chromosome instability and aneuploidy.17,18 These bridges are thought to be due to the failure to resolve incomplete replication at common fragile sites. BLM and FANCD2 also interact and cooperate to promote the restart of stalled replication forks during replication stress.19,20 FANCM is an additional FA protein that interacts with the BLM complex (BLM:TOPOIIIα:RMI1:RMI2).11,21,22 Moreover, the MRE11, RAD50, and NBS1 (MRN) complex and C-terminal-interacting protein (CtIP) stabilize FANCD2 at sites of DSBs, allowing for its localization to DNA damage and subsequent ubiquitylation.23 Thus, CtIP and MRN complex play a role early in the FA/BRCA pathway in addition to their critical role in the HDR step required for the completion of FA/BRCA repair. In summary, the interaction of FA/BRCA factors with so many additional DNA repair factors and pathways suggests that the FA pathway may be the central sensor and/or regulator for most cellular DNA damage responses.
The links between the BLM and MRN complexes and FA proteins suggested to us that the DNA2 helicase/nuclease, which partners with BLM and MRN in resection, might be critical for the completion of the FA pathway.24 Consistent with this hypothesis, DNA2 knockdown cells accumulate in G2 and show mitotic defects, such as aneuploidy, increased internuclear chromatin bridges, increased micronuclei, and pulverized chromosomes,25,26 similar—though not identical—to phenotypes of FA and BLM cell lines. The DNA2 nuclease functions in flap removal during Okazaki fragment processing, in protection against unwanted fork regression at stalled forks, and is one of 2 major nucleases, the other being EXO1, required for the critical step of resection in HDR to produce the 3′ overhang essential for recombination.27-33 In addition, in both yeast and human we have shown that DNA2 physically interacts with the BLM helicase to affect resection at DSBs.31,32,34 Human BLM protein suppresses the temperature-sensitive growth defects and the DNA damage sensitivity of dna2–1 mutant yeast.34 The FANCM ortholog in yeast is MPH1. Interestingly, yeast MPH1 overexpression suppresses the replication-defective phenotype of dna2 mutants, and purified Mph1 stimulates the exo-endonuclease activities of Dna2 during Okazaki fragment processing.35
In investigating a role for DNA2 in the HDR step of the FA/BRCA pathway, we have found that the double knockdown of both DNA2 and EXO1, but not of either nuclease alone, leads to hypersensitivity to cisplatin.24 Mechanistically, DNA2 or EXO1 appear to act in a 5′ to 3′ resection event. Breaks accumulate in metaphase chromosomes in DNA2/EXO1 knockdowns, and knockdowns fail to produce single-stranded DNA at sites of cisplatin-induced crosslinks, as measured by reduced RPA phosphorylation and fewer RAD51 foci.24 DNA2 immunoprecipitates reproducibly contain FANCD2, though the reverse is observed only after overexpression of DNA2, probably due to the low levels of nuclear DNA2 in human cells.24,25,36 In our current work we show that, unexpectedly, depletion of DNA2 can suppress the sensitivity of PD20 FANCD2−/− cells to cisplatin and to formaldehyde. Similarly to DNA2 depletion, the deletion of the key NHEJ factor Ku also suppresses the ICL sensitivity of FANCD2−/− cells.14,15 It has been proposed that in the absence of FANCD2, Ku corrupts repair by funneling the repair events toward error-prone NHEJ instead of error-free HDR.14,15 To explain the suppression of FANCD2−/− by depletion of DNA2, we suggest that unregulated DNA2 also corrupts repair. In the case of DNA2, this occurs due to over-resection, either of flaps or the ends of DSBs. To support this hypothesis, we present evidence to show that DNA2 can inhibit faithful FA/BRCA-dependent HR by unregulated resection. Demonstration that DNA2 can be deleterious in FA repair is important, since over-resection has been implicated in the production of single-stranded DNA, which may be involved in increased clustered mutagenesis and the massive genome rearrangements occurring in a single step in many cancer genomes.37-40 More specifically, suppression of FANCD2−/− phenotypes by DNA2 depletion may have therapeutic impact on survival of FA patients and in the use of ICL-inducing agents in chemotherapy.
Results
Cisplatin and formaldehyde sensitivity of FANCD2-deficient cells are rescued after DNA2 depletion
We examined the genetic interaction between FANCD2 and DNA2 in the repair of cisplatin- or formaldehyde-induced damage. Using PD20 FANCD2−/− cells complemented with wild-type FANCD2 or an empty vector, we depleted DNA2 using shRNA techniques (Fig. 1A). DNA2 was reduced to levels undetectable by western blotting. The cell lines were exposed to cisplatin, and a clonogenic assay was performed (Fig. 1A). As expected, the FANCD2−/− cells were very sensitive to cisplatin, whereas the FANCD2−/− cells complemented with FANCD2 were resistant. However, in shDNA2 and FANCD2−/− doubly deficient cells, instead of increased ICL sensitivity, we found significant (P < 0.05) resistance to cisplatin damage compared with FANCD2-deficient cells alone (Fig. 1A). This rescue is stronger than we previously reported, consistent with lower residual DNA2 levels detected by western blotting in the knockdowns.24

Figure 1. Depletion of DNA2 in FANCD2−/− cells rescues both cisplatin and formaldehyde sensitivity. (A) DNA2 depletion was performed using shSCR (scrambled) or shDNA2 targeted to exon 22 in PD20 FANCD2−/− cells (PD20:EV) or in PD20 FANCD2−/− complemented with FANCD2 (PD20:FD2). Knockdown was confirmed by western blot analysis. Clonogenic survival assays were performed after exposure to low doses of cisplatin. FANCD2−/− refers to the PD20 cells, FANCD2+/+ refers to the FANCD2-complemented PD20 cells. For these experiments, cells were transduced with virus containing shSCR or shDNA2 for 24 h, selected with puromycin for 48 h, then seeded in 12- or 24-well plates for treatment with cisplatin or formaldehyde. See “Materials and Methods” for details. (B) PD20 FANCD2−/− shDNA2 cells are resistant to formaldehyde. Cells were treated as in (A). (C) DNA2 depleted A549 cells are sensitive to formaldehyde. DNA2 was depleted in A549 lung cancer cells by targeting endogenous DNA2 at exon 22 (shDNA2) or the 3′ UTR (shDNA2*). As a control, a scrambled shRNA was also used (shSCR). Survival was determined after high-dose 4-h exposure or low-dose 6-d exposure. (D) DNA2-complemented A549 cells were resistant to formaldehyde exposure for 48 h. Western analysis confirms DNA2 depletion and complementation with wild-type, shRNA-resistant DNA2 in A549 cells treated as in (C), see “Materials and Methods”. (E) DNA2 depletion also sensitizes HeLa cells to formaldehyde. DNA2 was depleted in HeLa cells with shDNA2 or shDNA2* and treated with low-dose formaldehyde as in panel (C). Error bars indicate the mean ± SEM for n ≥ 2 independent experiments.
In addition to DNA:DNA crosslinks generated by clastogenic agents such as cisplatin, the FA/BRCA pathway is also implicated in the repair of DNA:protein crosslinks (DPCs), such as those produced by either endogenous or exogenous formaldehyde (HCHO).41 FANCD1/BRCA2- and FANCD2-deficient cells are hypersensitive to endogenous levels of formaldehyde, and in chicken DT40 cells, mutations in the ADH5 gene, encoding a major formaldehyde-detoxifying enzyme, are synthetically lethal with FANCD2−/−, implying that the FA pathway is vital for DPC repair.41,42 We next tested whether depletion of DNA2 also suppressed formaldehyde sensitivity in PD20 FANCD2−/−. Cells were exposed to formaldehyde, grown for 48 h, and the surviving fraction was determined. As with cisplatin, we found that the double DNA2 knockdown:FANCD2−/− cells were significantly less sensitive to formaldehyde than the single FANCD2-deficient cells (Fig. 1B). Thus, depletion of DNA2 suppresses the sensitivity FANCD2−/− cells to formaldehyde, as well as to cisplatin.
DNA2-depleted cells are hypersensitive to formaldehyde
To further implicate DNA2 in the FA/BRCA network, we determined if DNA2 might be required for the repair of DPC lesions, by testing whether DNA2 deficiency results in elevated formaldehyde sensitivity. We depleted DNA2 in FANCD2-proficient cells prior to exposure to formaldehyde and measured cell survival after a short exposure to high doses of formaldehyde (Fig. 1C). We found that DNA2-depleted lung cancer cells were hypersensitive to formaldehyde (Fig. 1C, center). We performed similar studies for chronic exposure to low doses of formaldehyde (Fig. 1C; right). We included in these studies 2 independent shDNA2 constructs, shDNA2 and shDNA2*, which target exon 22 and the 3′UTR of DNA2, respectively, to ascertain whether the results were DNA2-specific. The cells were exposed to low doses of formaldehyde, within the range of physiological concentration in human plasma,43 for 6 d and assayed for survival. We found a significant increase in cellular sensitivity to formaldehyde after DNA2 depletion (Fig. 1C). To further confirm that the formaldehyde sensitivity was due to DNA2 depletion and not off-target effects, we performed complementation experiments where shDNA2* was co-introduced into cells that expressed recombinant, RNAi-resistant DNA2. The cells expressing an empty vector were hypersensitive to formaldehyde, but the cells expressing RNAi resistant-DNA2 were able to repair the DPCs (Fig. 1D). DNA2-depleted HeLa cells were as sensitive to formaldehyde as the lung cancer line (Fig. 1E). We conclude that depletion of DNA2 leads to sensitivity to ICLs and DPCs in human cell lines.
FANCD2 competent cells lacking DNA2 are resistant to cisplatin in the presence of EXO1. However, such cells were sensitive to formaldehyde, even in the presence of EXO1 (Fig. 1C–E). Interestingly, depletion of FANCD2 reduces the need for DNA2 in formaldehyde-induced damage repair (Fig. 1B) (see “Discussion”).
Although we have successfully depleted DNA2 using shRNA technologies to study its role in DNA replication and repair, the cells cease to express the targeting construct after ~5 d in culture, and we generally only achieved 70–80% depletion. To examine the response of cells to the complete absence of DNA2, we generated a conditional knockout line where exon 2 of the DNA2 gene is deleted (Lee et al. manuscript in preparation). The colorectal carcinoma HCT116 cell line that is otherwise diploid44 carries 3 copies of DNA2 due to a small duplication on one copy of chromosome 10. Two chromosomal copies were disrupted using rAAV-mediated gene targeting technology,44,45 and exon 2 of the third allele was replaced with a conditional exon, where the exon was flanked by loxP sites (DNA2flox/−/−). These cells were additionally engineered to express a tamoxifen (4-OHT)-inducible Cre recombinase. Thus, the cell line is viable and can be propagated, but the addition of tamoxifen to the culture media leads to excision of the endogenous DNA2 and the generation of a true DNA2-null cell (Fig. 2A). DNA2-proficient cells (treated with ethanol, EtOH) actively replicated, while DNA2-deficient cells did not proliferate, but remained viable for the course of 8 d (Fig. 2A and B). We examined the sensitivity of these tamoxifen (4OHT)-treated cells to formaldehyde. The cells were treated with tamoxifen for 48 h and then exposed to formaldehyde for a further 48 h before fixation, staining, and quantitation using Li-Cor Odyssey scanner. DNA2-deficient cells were significantly more sensitive to formaldehyde than the controls (Fig. 2C). At the higher exposure concentration, heterozygotes were also more sensitive than cells carrying 3 wild-type copies of DNA2.
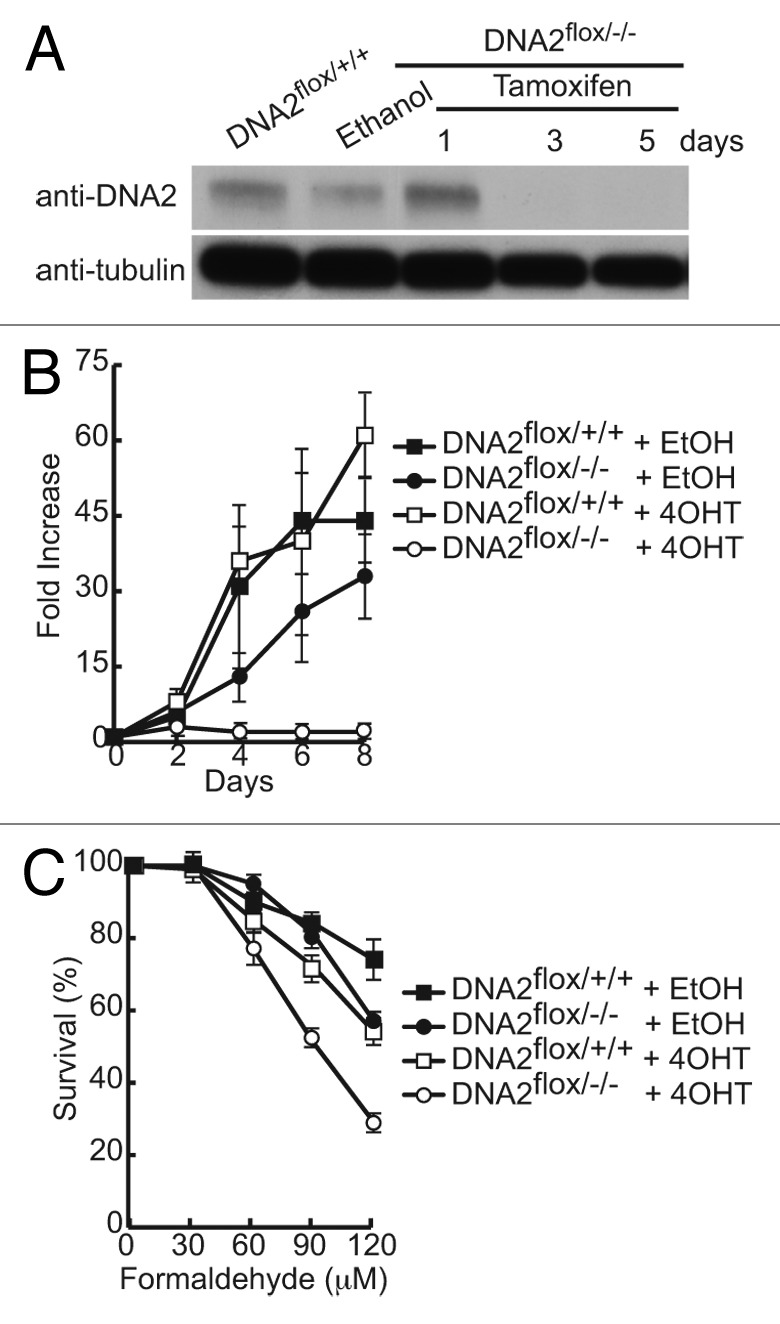
Figure 2. HCT116 cells without endogenous DNA2 expression are sensitive to formaldehyde. (A) Western blot showing DNA2 depletion in HCT116 DNA2flox/−/− cells after addition of tamoxifen (4-OHT). (B) Cell proliferation is impaired after knockdown of endogenous DNA2. (C) DNA2-knockout cells are sensitive to formaldehyde. Error bars indicate mean ± SEM for n ≥ 2 independent experiments.
Over-resection can inhibit crosslink-triggered repair
The experiments in this section support the hypothesis that DNA2 knockdown can reduce unwanted resection at an early step in the FA pathway, thus increasing HDR. To investigate if DNA2 depletion can increase ICL- or DPC-induced repair, we studied various repair-specific reporter constructs. We monitored ICL repair using a plasmid-encoded TR-oriP-GFP with a conjugated psolaren moiety (pso-TFO) to deliver the psolaren to a specific site within the GFP gene in the construct.46 After exposure to UV, a DNA:DNA crosslink is formed, which, when efficiently repaired, leads to GFP expression. The plasmid carrying the reporter also has an Epstein–Barr virus (EBV) origin of replication, oriP, and the U2OS cells in which these experiments were performed express the Epstein–Barr nuclear antigen (EBNA1) replication initiation protein. Thus, repair of the resulting crosslink is coupled to DNA replication. This repair is dependent on both early (FANCA) and late (HDR proteins such as BRCA2, RAD51) acting components of the FA pathway and is a specific measure of ICL-induced repair.47 We depleted DNA2 in cells containing the TR-ori-GFP construct, and unlike cells with BRCA2 depletion, which showed a reduction of GFP+ cells,47 we found a significant (P < 0.03) increase in GFP+ cells using shDNA2 depletion (16%) compared with shSCR controls (6%) (Fig. 3A). We conclude that in wild-type (shSCR-treated) cells, DNA2 was responsible for excess resection, resulting in reduced repair of this specific construct. After depletion, the cells were better able to repair the ICL damage and yield an increase in GFP+ cells (using EXO1 or residual DNA2).
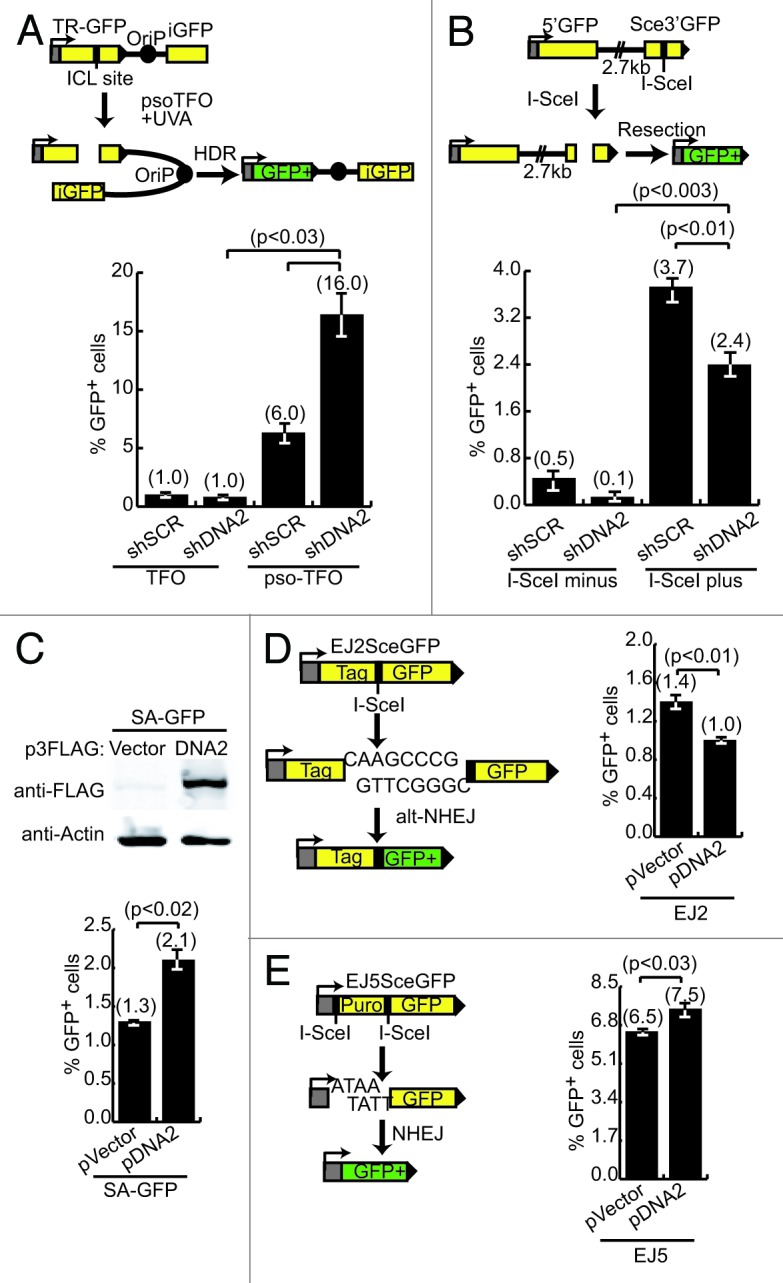
Figure 3. Suppression of FANCD2 deficiency correlates with a decrease in resection after DNA2 depletion. (A) Top panel: diagram of the U2OS TR-oriP-GFP reporter used to examine repair after ICL generation. TFO, triplex forming oligonucleotide without psoralen; pso-TFO, oligonucleotide with psoralen. Bottom panel: DNA2-depleted cells have a significantly higher percentage of GFP+ cells. Repair efficiency is defined as %GFP+ cells relative to transfection frequency in panels (A–E). (B) Top panel: diagram of the U2OS SA-GFP reporter used to examine resection after I-SceI-induced DSB. Bottom panel: DNA2-depleted cells have significantly lower percentage of GFP+ cells. (C) The top panel shows high-level expression of DNA2 in U2OS cells with SA-GFP reporter. Bottom panel: cells with high-level expression of DNA2 have a significantly higher percentage of GFP+ cells. (D) Left panel: diagram of the EJ2-GFP alt-NHEJ reporter used in U2OS cells to examine resection of microhomology DNA after I-SceI expression. High-level expression of DNA2 caused a significant reduction in GFP+ cells. (E) Left panel: diagram of the EJ5-GFP total-NHEJ reporter in U2OS cells used to examine resection after I-SceI expression. High-level expression of DNA2 caused a significant increase in GFP+ cells. Error bars indicate mean ± SEM for n ≥ 2 independent experiments.
To support this interpretation, we used a different GFP reporter that monitors single-strand annealing (SSA). SSA involves the repair of DSBs between 2 repeated sequences.48 SSA requires resection but does not require strand invasion,48 as the 2 3′-overhangs simply align and anneal. SSA was measured with the SA-GFP reporter system.49 GFP+ cells can only arise after extensive resection of a 2.7-kb intervening region between 2 incomplete but overlapping GFP gene segments. After induction of a DSB by I-SceI, and resection, the 2 segments can align and anneal generating GFP+ cells (Fig. 3B). If our model is correct, this reporter should have decreased production of GFP+ cells in the absence of DNA2. Indeed, a significant reduction (P < 0.01) in GFP+ cells in shDNA2-treated cells compared with shSCR controls was observed (Fig. 3B; ref. 24). Using the same SA-GFP reporter, we overexpressed recombinant DNA2 and measured repair after I-SceI expression. In this assay we reasoned that increasing the cellular pool of DNA2 would improve the efficiency of resection. We achieved a robust expression of DNA2 (Fig. 3C; top panel) that resulted in a significant increase (P < 0.02) in GFP+ cells (Fig. 3C; bottom panel). We conclude that DNA2 catalyzes extensive resection in these reporter assays.
FANCD2-deficient cells have increased chromosomal aberrations attributed to NHEJ.14,15 We next tested for the ability of DNA2 to resect DNA at DSB ends designed to be repaired by NHEJ using additional GFP expression reporters.49 First, the EJ2SceGFP construct measures alternative NHEJ (alt-NHEJ), a process that utilizes microhomology immediately adjacent to DSB ends for repair (Fig. 3D). Resection by DNA2 of the microhomology regions after DSB induction would be expected to decrease the efficiency of alt-NHEJ. To test the effect of DNA2, we overexpressed the protein and enumerated GFP+ cells by flow cytometry. We observed a significant (P < 0.01) reduction in GFP+ cells among cells overexpressing DNA2 in comparison to controls (Fig. 3D). Next, the EJ5SceGFP construct (Fig. 3E) measures total NHEJ that takes into account DSBs repaired by both alt-NHEJ and canonical NHEJ. In this reporter, trimming at the sites of an I-SceI-induced DSB is required to efficiently generate substrates for canonical NHEJ. Impairment of alt-NHEJ does not affect the readout (because removal of microhomology regions produces a substrate for canonical NHEJ). We hypothesized that since overexpression of DNA2 reduced alt-NHEJ (presumably because of over-resection), then using a construct that favors resection for repair should increase GFP+ cells. In keeping with our other results, we saw a small but significant (P < 0.03) increase in GFP+ cells after expression of DNA2 compared with vector control (Fig. 3E). We conclude that DNA2 upregulation increases resection at I-SceI-induced breaks, regardless of their configuration.
FANCD2 foci formation is not affected by DNA2 depletion
FANCD2 localizes to chromatin at sites of ICL damage, where it interacts with BRCA2 and forms immuno-detectable foci.45,50 CtIP or Mre11 appear to be involved in an early step in ICL repair, because CtIP or Mre11 depletion prevents FANCD2 recruitment to sites of damage and ubiquitylation of FANCD2 in the presence of ICL damage.23,42 Since CtIP and Mre11 are implicated in resection just upstream of DNA2,29,31,32,51 it was of interest to test whether DNA2 was also required at an early step for FANCD2 activation. Previously, we showed that the depletion of endogenous DNA2 did not affect FANCD2 monoubiquitination after cisplatin treatment.24 Here we examined whether DNA2 knockdown affected FANCD2 foci formation after treatment with ICL-generating cisplatin. Consistent with our results with the ubiquitylation of FANCD2, DNA2-depleted cells were able to accumulate FANCD2 foci after cisplatin treatment (Fig. 4A and B). DNA2 was similarly dispensable for FANCD2 ubiquitylation after treatment with formaldehyde (Fig. 4C and D). The control in these experiments was a FANCD2 K561R that cannot be ubiquitylation (Fig. 4D). Interestingly, FANCD2 foci also accumulated in DNA2-depleted cells without cisplatin exposure (Fig. 4A and B). This accumulation is also seen in cells lacking the DNA2 partner, BLM helicase.17 A consequence of DNA2 depletion is the accumulation of endogenous DNA replication fork stress26 that, like nucleotide deprivation,7 may recruit FANCD2 to stalled or damaged DNA replication forks. We conclude that DNA2 is required downstream of MRE11 but not for FANCD2 ubiquitylation and focus formation. These observations support the proposed role of DNA2 in the HDR step.

Figure 4. DNA2 depletion does not affect formation of FANCD2 foci after cisplatin exposure. (A) U2OS cells with endogenous DNA2 depletion were treated with cisplatin (15 μM, 24 h) and stained with anti-FANCD2 or γH2Ax antibodies. A representative immunofluorescence image of FANCD2 foci is shown. (B) Quantification of FANCD2 foci-positive cells that are also positive for γH2Ax foci. (C) FANCD2 is monoubiquitylated in DNA2-depleted A549 cells treated with or without formaldehyde. (D) PD20 FANCD2−/− complemented with vector, FANCD2 wild-type, or FANCD2-K561R cDNAs were treated with formaldehyde and western blots prepared. Formaldehyde-treated cells showed FANCD2 monoubiquitylation, and FANCD2-K561R mutants were not monoubiquitylated. Error bars indicate mean ± SEM for n ≥ 2 independent experiments.
Discussion
We previously reported that human DNA2 helicase/nuclease and EXO1 nuclease are involved in DSB resection during repair of cisplatin-induced ICLs. We proposed that these nucleases function during late steps in the FA/BRCA pathway that requires HDR proteins.24 In this report, we provide additional evidence that DNA2 coordinates with the FA/BRCA network. Our study suggests that the activity of DNA2 at DSBs, while ultimately participating in repair, must be tightly regulated to ensure that the nuclease does not abnormally resect the intermediate DNA substrates needed for efficient DNA damage repair. Our major finding is that depletion of DNA2 rescues the cisplatin and formaldehyde sensitivity of PD20 FANCD2−/− cells, rather than increasing their sensitivity, as might have been expected. We went on to show that this suppression correlated with an increase in the FA-directed repair of a single, site-specific psoralen crosslink in cells depleted of DNA2. Moreover, we observed a decrease in resection of DSBs, which suggested that the lethality in FANCD2−/− cells may be in part due to over-resection by DNA2. Since FANCD2 activates multiple nucleases to incise next to crosslinks, and this repair is coupled to replication and occurs at stalled replication forks, we propose that FANCD2 is needed to negatively regulate DNA2 nuclease activity, directly or indirectly. Otherwise, DNA2 may carry out excessive and/or premature clipping/resection, for example, before the translesion synthesis (TLS) and nucleotide excision repair (NER) machinery can repair the sister chromatid. Such repair must occur before the sister chromatid can provide intact donor information for faithful HDR repair of putative DSB intermediates.
DNA2 over-resection might also play a negative role in the later HDR steps in the absence of FANCD2. Over-resection could inhibit HDR, and we have previously demonstrated that depletion of DNA2 actually increases HDR in GFP-reporter-based assays.24 In a similar manner, depletion of the PCNA-associated recombination inhibitor (PARI) abrogates the HDR defect and sensitivity to PARP1 inhibitor of FANCD1/BRCA2-deficient cells.52 Importantly, unregulated resection, both at DSBs and at stalled replication forks, has recently been implicated in the highly clustered mutations frequently arising in cancer genomes, and, thus, our results implicate DNA2 as one possible culprit in this mutagenic process37,38,53,54 as well as in other chromosomal catastrophes in cancer cells.55
Like depletion of DNA2, depletion of Ku or DNA-PKcs also suppresses the sensitivity of FANCD2−/− to cisplatin in chicken, worm, and human cells.14,15 It has been suggested that in the absence of FANCD2 or core FANC proteins, NHEJ proteins, including Ku70 and DNA-PKcs, achieve access to the DSBs and disrupt the desired error-free HDR-mediated genome repair.14,15 Supporting this idea, DNA-PKcs was found to inappropriately accumulate at sites of replication stress in the absence of FANCD2.15 DNA2 might also have increased access to ends in the absence of FANCD2. In the case of DNA2, the FA/BRCA proteins may normally limit the access of DNA2 to DNA ends arising during the multi-stage processing that occurs before the positive function of DNA2 is required in HDR of DSBs arising in the pathway, i.e., after crosslink removal, TLS, and NER repair of the sister chromosome to provide the donor for faithful DSB healing.
An attractive alternative interpretation of our results derives from consideration of the recently established role of FANCD2 in protection of replication forks stalled in the presence of reduced levels of nucleotide precursors by HU (hydroxyurea).6,7 DNA-damaging agents cause cellular stress that is especially amplified during DNA replication. DNA replication stress leads to replication fork stalling and, eventually, fork collapse. Stalled forks are susceptible to instability, and cellular mechanisms have evolved to stabilize the forks, remove the perturbation, and restart DNA replication.56 Compelling evidence suggests that FA proteins are involved in the stabilization of replication forks independent of their ICL damage repair functions. For example, FANCA- and FANCD2-deficient cells, like FANCD1/BRCA2-deficient cells, show extensive nascent DNA degradation after HU treatment.6,57 FANCD2/I are 2 of the major proteins recruited to replication forks stalled by nucleotide depletion, further supporting a major role in fork protection,7,58 and recruitment requires FANCD2 monoubiquitylation.7 Overexpression of RAD51 in FANCD2-deficient cells is enough to suppress DNA degradation, and this appears to be solely due to single-stranded DNA:RAD51 filament stabilization, rather than stimulation of HDR.6,57
MRE11 nuclease inhibition reduces nascent DNA degradation at stalled replication forks.57,59 The prevailing model for DSB resection presumes that the MRN/CtIP nuclease complex licenses resection of DSBs with limited exo-endonuclease action.60,61 Resection to produce the overhangs for strand invasion then proceeds using either EXO1/BLM or DNA2/BLM.24,31,61,62 Thus, it is unlikely that the Mre11 nuclease is solely responsible for the nascent DNA degradation and extensive single-stranded DNA observed in the absence of FANCD2,6,7 and DNA2 might also be involved in nascent DNA degradation in the absence of FANCD2. Supporting this sequence of events, we have shown that DNA2 depletion, unlike Mre11 or CtIP depletion, does not affect FANCD2 monoubiquitylation24 or FANCD2 DNA damage focus formation (Fig. 4A). Thus, DNA2 seems to act downstream of CtIP and MRN in the FA/BRCA pathway, since depletion of either MRN complex or CtIP, unlike depletion of DNA2, reduces FANCD2 monoubiquitylation and foci accumulation.23,63 DNA2 removal may suppress the genome instability in FANCD2−/− cells if DNA2 is involved in fork degradation in FANCD2−/− cells.
A model for antagonistic DNA2:FANCD2 interactions is summarized in Figure 5. Our previous model for DNA2 function in ICL repair is shown on the right (Fig. 5, C2–F2). On the left is a model for a general role for DNA2 at stalled replication forks (Fig. 5, C1 to E1). We envision that the substrate for fork degradation at a fork stalled by many types of barriers, including but not limited to crosslinks, is a reversed fork (Fig. 5, C1). This might participate in recombinational fork restart, as proposed by others (Fig. 5 C1; arrow).6,24,57,64 Alternatively, this recombinogenic intermediate may lead to genome instability. DNA2 is an intrinsic replication fork protein and might be poised to recognize reversed forks.26,62 In fact, in fission yeast, DNA2 is proposed to prevent deleterious recombinogenic replication fork reversal by cleaving nascent ssDNA at stalled replication forks.33 In normal cells, FANCD2 may downregulate the resection of reversed forks. In FANCD2−/−cells, however, DNA2 catalyzes excessive resection resulting in genome instability (Fig. 5, D1 and E2). Thus, a role for DNA2 at the replication fork, either in preventing or degrading reversed forks, may account for the apparent resistance to ICLs and DPCs. It will be interesting to analyze the genome status of the cells surviving without FANCD2 and DNA2. It is possible that the cells still accumulate gross chromosomal rearrangements, but at a lower frequency, thus allowing them to better survive. This will be important to establish, as it might affect tumorigenesis in such cells.

Figure 5. Models depicting the roles of DNA2 and FANCD2 during DNA repair and replication stress. (A) Replication fork is stalled by an ICL or DPC or other impediment. (B) Replication halts at the obstacle, and various proteins are recruited, including FANCM/FAAP24 and the FA core complex. Left: Fork protection, (C1) Fork regression may occur to form a chicken foot structure. FANCD2 is required to prevent licensing of resection of nascent DNA by MRE11 and perhaps regulate the nuclease activity of DNA2. (D1) In FANCD2−/− cells, DNA2 and other nucleases excessively resect the nascent DNA. (E1) In FANCD2−/− cells, depletion of DNA2 prevents excessive resection allowing cells to survive. Right: FA/BRCA repair, (C2) during ICL repair, FANCD2/FANCI recruits nucleases to unhook the crosslink. (D2) FANCD2 regulates resection by nucleases to generate 3′ DNA ends suitable for HDR. (E2) In FANCD2−/− cells DNA2 and other nucleases excessively resect the DNA at the DSB. (F2) In FANCD2−/− cells, depletion of DNA2 prevents excessive resection allowing the cells to survive.
Because the FA/BRCA proteins have also been implicated in the resolution of DNA:protein crosslinks,41,42,65 it is interesting that DNA2-deficient cells are sensitive to formaldehyde, and that the formaldehyde sensitivity of PD20 FANCD2−/− cells is suppressed by DNA2 depletion. Formaldehyde is thought to induce DNA:protein crosslinks. It is not immediately clear why DNA2-depleted cells are sensitive to formaldehyde, because there is conflicting evidence as to whether other HDR pathway proteins are involved in the repair of formaldehyde-induced DNA damage.41,42 However, since replication forks are predicted to stall at DNA:protein crosslinks, DNA2 may play a role protecting the stalled forks, in either preventing or resolving reversed forks that arise due to the stall or in removal of flaps that arise during repair of the adducts (Fig. 5). We were surprised that in the absence of FANCD2, the DNA2 knockdowns were no longer sensitive to formaldehyde. It is possible that another formaldehyde-repair pathway is activated in the absence of both DNA2 and FANCD2. However, we have no evidence as to what the pathway involves.
Based on our findings, it is conceivable that the interplay between DNA2 and FANCD2 is not limited to the repair of ICLs, but rather is a part of an evolutionarily conserved mechanism for addressing genome perturbation. Although the suppression of FANC-deficient chicken, worm, and human cells had suggested that FA phenotypes might be relieved by inhibition of NHEJ proteins, the contradictory finding in mouse, that Ku−/−:FANCD2−/− mice showed enhanced sensitivity to crosslinking agents, brings the usefulness of NHEJ inhibitors into question.16 The improvement in survival of FA cells after DNA2 depletion may be advantageous to FA patients, offering a chance for better survival.
Materials and Methods
Cell culture and transfections
A549, U2OS, U2OS-DR-GFP66 U2OS-SA-GFP, EJ2, and EJ548 cells were gifts from Dr P Dervan, Dr W Dunphy, California Institute of Technology, Dr M Jasin, Memorial Sloan-Kettering Cancer Center, and Dr J Stark, City of Hope, respectively. PD20 FANCD2−/− lines were obtained from the Fanconi Anemia Research Fund. Cells were maintained in Dulbecco modified Eagle medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in 5% CO2.
Virus production and infection
Virus was produced in HEK293T cells as described.25,26,67 Briefly, cells were transfected with pLKO.1.shSCR, pLKO.1.shDna2, pLKO.1.shDna2, or pLKO.1.shDna2* and pCMVΔR8.2 and pCMV-VSV-G using BioT (Bioland Scientific).25,26 Virus was recovered 48 h post-transfection, and infections were performed in cells overnight in the presence of 10 μg/ml of protamine sulfate. Transduced cells were selected with 2 μg/ml of puromycin for 48 h. The following sequences were used for the hDNA2 short hairpins: 5′-CATAGCCAGT AGTATTCGAT G-3′ for shDna2, 5′-CCGGCCAGCT TTGAAGATGG ATTAACTCGA GTTAATCCAT CTTCAAAGCT GGTTTTTG-3′ for shDna2*.
Construction of human conditional DNA2-null somatic cell lines
The conditional DNA2flox/−/− null HCT116 cell line was constructed with the aid of rAAV (recombinant adeno-associated virus)-mediated gene-targeting technology.44,45 DNA2 is triploid in the HCT116 cell line, and all 3 alleles were sequentially targeted with a rAAV targeting vector designed with 3 loxP sites flanking the exon and a drug-resistance marker. Subsequently, the cell line was also modified to express Cre recombinase under a tamoxifen-inducible promoter. As a control, DNA2flox/+/+ cells, which were generated after one round of targeting, were used. Details of the cell line construction and additional phenotypes will be described elsewhere (Lee et al., manuscript in preparation).
Knockdown rescue experiment
For complementation studies, virus was produced as described above using plasmids with pLKO.1.shDna2*. After transduction and selection, cells were transfected with pCMV7.1–3XFLAG-DNA2 wild-type, pCMV7.1–3XFLAG-DNA2-nuclease-dead, or pCMV7.1–3XFLAG-empty vector.68
Clonogenic and survival assays
Briefly for survival assays, 48 h after depletion of DNA2, 50 000 cells were seeded into 24-well plates the day before treatment with drugs. For acute exposure, formaldehyde (0, 40, 80, 160, 320 μM) was added for 4 h in PBS. Cells were washed in PBS and grown for 5 d. For chronic exposure, cells were continuously grown in formaldehyde (0, 30, 60, 90, 120 μM) for 6 d. To determine viability, cells were fixed and stained with crystal violet and scanned with a Li-Cor Odyssey scanner (Li-Cor Biosciences). The surviving fraction was determined by comparing treated with the non-treated controls. A clonogenic assay was also performed for cisplatin sensitivity by seeding 1000, 2000, or 3000 PD20 cells per well, which were then exposed to cisplatin (0, 0.62, 1.25, 2.5 μM) for 24 h, washed, and cultured. Colonies were allowed to form for 14 d prior to fixation, staining with crystal violet, and enumeration of visible colonies. Surviving fraction was determined by comparing treated with the non-treated controls.
Immunofluoresence microscopy
U2OS cells were grown on poly-L-lysine-coated coverslips and treated with 15 μM cisplatin for 1 h. Cells were pre-extracted with cytoskeletal buffer (10 mM HEPES/KOH pH7.4, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100, 1 mM PMSF, protease, and phosphatase inhibitors) and fixed with 4% formaldehyde for 25 min at room temperature. After fixation, cells were washed and blocked in 10% FBS/PBS before addition of primary antibodies (α-FANCD2, α-γH2Ax) diluted at 1:1000 in 10% FBS in PBS. Cells were incubated overnight at 4 °C, washed in 10% FBS/PBS, and stained with secondary antibodies anti-rabbit IgG-Alexa Fluor 488 or anti-mouse IgG-Alex Fluor 594 for 1 h at room temperature. DNA was counterstained with 4’ 6’-diamidino-2-phenylindole (DAPI, 0.3 μM), and coverslips were mounted on slides with Vectashield mounting agent (Vector Laboratories). Images were acquired using Zeiss Axio epifluoresent microscope and processed with AxioVision Rel. 4.8 (Carl Zeiss) and Adobe Photoshop (Adobe) software.
Resection assay using GFP constructs
DNA2 was depleted or expressed from pCMV7.1-3XFLAG-DNA2 vector in TR-oriP-GFP, DR-GFP, SA-GFP, EJ2, or EJ5 containing U2OS cells. Cells were transfected with an empty plasmid, a plasmid expressing the I-SceI endonuclease, or a plasmid expressing GFP for 48 h. GFP+ cells were determined by flow cytometry.
Statistical analysis
Student t tests were performed at P < 0.05 for samples with n > 2.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Bing-hui Shen for access to the DNA2 expression vectors before publication, and Shelley Diamond for expert performance and analysis of GFP by flow cytometry. We thank the Fanconi Anemia Research Fund for the PD20 cell lines. This work was supported by a Breast Cancer grant from Congressionally Directed Medical Research Programs (J.L.C.), GM100186, the Ellison Foundation, and the Ross Fellowship from the Biology Division, California Institute of Technology (K.K.K).
References
- 1.Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–94. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Constantinou A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma. 2012;121:21–36. doi: 10.1007/s00412-011-0349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–63. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11:467–80. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–16. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Décaillet C, Gari K, Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell. 2013;51:678–90. doi: 10.1016/j.molcel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 8.Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010;29:806–18. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collis SJ, Ciccia A, Deans AJ, Horejsí Z, Martin JS, Maslen SL, Skehel JM, Elledge SJ, West SC, Boulton SJ. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell. 2008;32:313–24. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani H, Joenje H, McDonald N, de Winter JP, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell. 2007;25:331–43. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Deans AJ, West SC. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol Cell. 2009;36:943–53. doi: 10.1016/j.molcel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Kim JM, Kee Y, Gurtan A, D’Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–22. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell. 2013;52:434–46. doi: 10.1016/j.molcel.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 2010;329:219–23. doi: 10.1126/science.1192277. [DOI] [PubMed] [Google Scholar]
- 15.Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;39:25–35. doi: 10.1016/j.molcel.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 16.Bunting SF, Callén E, Kozak ML, Kim JM, Wong N, López-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell. 2012;46:125–35. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–60. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 18.Naim V, Rosselli F. The FANC pathway and mitosis: a replication legacy. Cell Cycle. 2009;8:2907–11. doi: 10.4161/cc.8.18.9538. [DOI] [PubMed] [Google Scholar]
- 19.Chaudhury I, Sareen A, Raghunandan M, Sobeck A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013;41:6444–59. doi: 10.1093/nar/gkt348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pichierri P, Franchitto A, Rosselli F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 2004;23:3154–63. doi: 10.1038/sj.emboj.7600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang YH, Lee CH, Seo YS. Dna2 on the road to Okazaki fragment processing and genome stability in eukaryotes. Crit Rev Biochem Mol Biol. 2010;45:71–96. doi: 10.3109/10409230903578593. [DOI] [PubMed] [Google Scholar]
- 22.Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. 2003;23:3417–26. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roques C, Coulombe Y, Delannoy M, Vignard J, Grossi S, Brodeur I, Rodrigue A, Gautier J, Stasiak AZ, Stasiak A, et al. MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO J. 2009;28:2400–13. doi: 10.1038/emboj.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karanja KK, Cox SW, Duxin JP, Stewart SA, Campbell JL. DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle. 2012;11:3983–96. doi: 10.4161/cc.22215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duxin JP, Dao B, Martinsson P, Rajala N, Guittat L, Campbell JL, Spelbrink JN, Stewart SA. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–82. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duxin JP, Moore HR, Sidorova J, Karanja K, Honaker Y, Dao B, Piwnica-Worms H, Campbell JL, Monnat RJ, Jr., Stewart SA. Okazaki fragment processing-independent role for human Dna2 enzyme during DNA replication. J Biol Chem. 2012;287:21980–91. doi: 10.1074/jbc.M112.359018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Budd ME. Cox Ls, Campbell JL. Coordination of Nucleases and Helicases during DNA Replication and Double-strand Break Repair. In: Cox LS, ed. Molecular Themes in DNA Replication. London: RSC Publishing, 2009. [Google Scholar]
- 28.Budd ME, Campbell JL. Interplay of Mre11 nuclease with Dna2 plus Sgs1 in Rad51-dependent recombinational repair. PLoS One. 2009;4:e4267. doi: 10.1371/journal.pone.0004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–94. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–4. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–62. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010;467:112–6. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, Zhang M, Hu Y, Wang Q, Xu W, et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell. 2012;149:1221–32. doi: 10.1016/j.cell.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 34.Imamura O, Campbell JL. The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast dna2 mutants. Proc Natl Acad Sci U S A. 2003;100:8193–8. doi: 10.1073/pnas.1431624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang YH, Kang MJ, Kim JH, Lee CH, Cho IT, Hurwitz J, Seo YS. The MPH1 gene of Saccharomyces cerevisiae functions in Okazaki fragment processing. J Biol Chem. 2009;284:10376–86. doi: 10.1074/jbc.M808894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng P, Fay DS, Burton J, Xiao H, Pinkham JL, Stern DF. SPK1 is an essential S-phase-specific gene of Saccharomyces cerevisiae that encodes a nuclear serine/threonine/tyrosine kinase. Mol Cell Biol. 1993;13:5829–42. doi: 10.1128/mcb.13.9.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012;46:424–35. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Setlur SR, Lee C. Tumor archaeology reveals that mutations love company. Cell. 2012;149:959–61. doi: 10.1016/j.cell.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat Struct Mol Biol. 2011;18:1432–4. doi: 10.1038/nsmb.2173. [DOI] [PubMed] [Google Scholar]
- 42.Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, Buerstedde JM, Gillespie DA, Sale JE, Yamazoe M, et al. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007;67:11117–22. doi: 10.1158/0008-5472.CAN-07-3028. [DOI] [PubMed] [Google Scholar]
- 43.Heck Hd, Casanova M. The implausibility of leukemia induction by formaldehyde: a critical review of the biological evidence on distant-site toxicity. Regul Toxicol Pharmacol. 2004;40:92–106. doi: 10.1016/j.yrtph.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Fattah FJ, Lichter NF, Fattah KR, Oh S, Hendrickson EA. Ku70, an essential gene, modulates the frequency of rAAV-mediated gene targeting in human somatic cells. Proc Natl Acad Sci U S A. 2008;105:8703–8. doi: 10.1073/pnas.0712060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russell DW, Hirata RK. Human gene targeting by viral vectors. Nat Genet. 1998;18:325–30. doi: 10.1038/ng0498-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakanishi K, Cavallo F, Perrouault L, Giovannangeli C, Moynahan ME, Barchi M, Brunet E, Jasin M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat Struct Mol Biol. 2011;18:500–3. doi: 10.1038/nsmb.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakanishi K, Cavallo F, Brunet E, Jasin M. Homologous recombination assay for interstrand cross-link repair. Methods Mol Biol. 2011;745:283–91. doi: 10.1007/978-1-61779-129-1_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gunn A, Bennardo N, Cheng A, Stark JM. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. J Biol Chem. 2011;286:42470–82. doi: 10.1074/jbc.M111.309252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Andreassen PR, D’Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 2004;24:5850–62. doi: 10.1128/MCB.24.13.5850-5862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang SH, Zhou R, Campbell J, Chen J, Ha T, Paull TT. The SOSS1 single-stranded DNA binding complex promotes DNA end resection in concert with Exo1. EMBO J. 2013;32:126–39. doi: 10.1038/emboj.2012.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, D’Andrea AD. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell. 2012;45:75–86. doi: 10.1016/j.molcel.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. Breast Cancer Working Group of the International Cancer Genome Consortium The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan K, Sterling JF, Roberts SA, Bhagwat AS, Resnick MA, Gordenin DA. Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet. 2012;8:e1003149. doi: 10.1371/journal.pgen.1003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu P, Erez A, Nagamani SC, Dhar SU, Kołodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–19. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 57.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–42. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem. 2013;288:31458–67. doi: 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17:1305–11. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 61.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peng G, Dai H, Zhang W, Hsieh HJ, Pan MR, Park YY, Tsai RY, Bedrosian I, Lee JS, Ira G, et al. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res. 2012;72:2802–13. doi: 10.1158/0008-5472.CAN-11-3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duquette ML, Zhu Q, Taylor ER, Tsay AJ, Shi LZ, Berns MW, McGowan CH. CtIP is required to initiate replication-dependent interstrand crosslink repair. PLoS Genet. 2012;8:e1003050. doi: 10.1371/journal.pgen.1003050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–8. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 66.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–8. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saharia A, Guittat L, Crocker S, Lim A, Steffen M, Kulkarni S, Stewart SA. Flap endonuclease 1 contributes to telomere stability. Curr Biol. 2008;18:496–500. doi: 10.1016/j.cub.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin W, Sampathi S, Dai H, Liu C, Zhou M, Hu J, Huang Q, Campbell J, Shin-Ya K, Zheng L, et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013;32:1425–39. doi: 10.1038/emboj.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]