

Abstract
Glioblastoma are the most frequent and malignant human brain tumors, having a very poor prognosis. The enhanced radio- and chemoresistance of glioblastoma and the glioblastoma stem cells might be the main reason why conventional therapies fail. The second messenger cyclic AMP (cAMP) controls cell proliferation, differentiation, and apoptosis. Downregulation of cAMP sensitizes tumor cells for anti-cancer treatment. Opioid receptor agonists triggering opioid receptors can activate inhibitory Gi proteins, which, in turn, block adenylyl cyclase activity reducing cAMP. In this study, we show that downregulation of cAMP by opioid receptor activation improves the effectiveness of anti-cancer drugs in treatment of glioblastoma. The µ-opioid receptor agonist D,L-methadone sensitizes glioblastoma as well as the untreatable glioblastoma stem cells for doxorubicin-induced apoptosis and activation of apoptosis pathways by reversing deficient caspase activation and deficient downregulation of XIAP and Bcl-xL, playing critical roles in glioblastomas’ resistance. Blocking opioid receptors using the opioid receptor antagonist naloxone or increasing intracellular cAMP by 3-isobutyl-1-methylxanthine (IBMX) strongly reduced opioid receptor agonist-induced sensitization for doxorubicin. In addition, the opioid receptor agonist D,L-methadone increased doxorubicin uptake and decreased doxorubicin efflux, whereas doxorubicin increased opioid receptor expression in glioblastomas. Furthermore, opioid receptor activation using D,L-methadone inhibited tumor growth significantly in vivo. Our findings suggest that opioid receptor activation triggering downregulation of cAMP is a promising strategy to inhibit tumor growth and to improve the effectiveness of anti-cancer drugs in treatment of glioblastoma and in killing glioblastoma stem cells.
Keywords: opioid receptor; cAMP; D,L-methadone; chemoresistance; glioblastoma; glioblastoma stem cells; chemotherapy; apoptosis
Introduction
Glioblastoma multiforme is the most common brain tumor in adults.1,2 Despite aggressive treatment protocols, including surgical resection followed by radiochemotherapy, glioblastoma patients have a median overall survival expectancy of 14 mo.1 The high progression and recurrence rate after therapy in focal masses result from a subset of tumor cells, so-called tumor-stem cells, with a very high tumorigenic activity.3,4 The enhanced chemo- and radioresistance of glioblastoma and their tumor-initiating stem cells might be the main reason why conventional therapies are considered a palliative venture with no hope of cure.5,6 Therefore improving therapeutic success and outcome novel treatment strategies are needed.
Cyclic AMP (cAMP) is a second messenger generated by adenylyl cyclases representing an important signal transducer in several physiologic and pathologic settings, as it can activate kinases and is responsible for actions like ion channel regulation.7 cAMP-related signaling can control apoptosis induction and cell growth.7,8 Recently, it was found that cAMP inhibits doxorubicin as well as DNA damage-induced apoptosis.9,10
Agonists triggering opioid receptors can activate inhibitory Gi-proteins, which, in turn, block adenylyl cyclase activity, reducing cAMP.11 D,L-methadone is an µ-opioid receptor agonist inducing apoptosis in leukemia cells in vitro and in vivo.12-14 In addition, D,L-methadone sensitizes leukemia cells for doxorubicin treatment via downregulation of cAMP triggered by opioid receptor activation.13 Apoptosis plays a critical role in the regulation of various physiological or pathological conditions and has also been implied to mediate therapy-induced cytotoxicity following chemotherapy and radiation.15,16 Two principle apoptosis signaling pathways have been delineated, the death receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway.17 Activation of either pathway leads to activation of caspases.18
Resistances to current treatment regimens remain a major concern in oncology and may be caused by defects in apoptosis program.15 Apoptosis signaling may be disrupted by deregulated expression and/or function of anti-apoptotic such as Bcl-xL or proapoptotic molecules like Bax. The majority of human cancers harbor high levels of inhibitor of apoptosis proteins (IAPs), e.g., the well-characterized X-linked IAP (XIAP).15,19 Targeting these proteins by inhibition or downregulation are novel approaches in glioma therapy.20,21
In this study we demonstrate that downregulation of cAMP induced by opioid receptor activation using the µ-opioid receptor agonist D,L-methadone sensitizes chemo- and radioresistant glioblastoma cells for doxorubicin-induced apoptosis and reverses deficient activation of apoptosis pathways by doxorubicin. Interestingly, glioblastoma stem cells, which seem to be the reason for glioblastomas resistances, were strongly killed by activation of opioid receptors using D,L-methadone in addition to doxorubicin.
Results
Opioid receptor activation increases doxorubicin-induced apoptosis in glioblastoma cells
Glioblastomas are hard to kill with anti-cancer drugs or radiation.22 We recently published that the µ-opioid receptor agonist D,L-methadone breaks chemo- and radioresistance in leukemia cells expressing opioid receptors and sensitizes leukemia cells for doxorubicin treatment.12,13 Doxorubicin is an effective substance in treatment of malignant gliomas in in vitro studies and animal models.22 To enhance penetration of doxorubicin and to improve doxorubicin delivery across the blood–brain barrier, doxorubicin is used as liposomal encapsulated formulation (Caelyx) in vivo.22,23 Glioblastoma cells (A172, U118MG) express opioid receptors (Fig. 1A). Therefore, we analyzed if the opioid D,L-methadone can also induce cell death in glioblastoma cells. A172 and U118MG glioblastoma cells were treated with doxorubicin (0.1 µg/mL), D,L-methadone (10, 3, 1 µg/mL) alone or in combination (Fig. 1B), or A172 and U118MG glioblastoma cells were treated with doxorubicin (0.3 µg/mL) or D,L-methadone (10, 3, 1 µg/mL) alone or in combination (Fig. 1C). A strong induction of cell death was observed by co-treatment of D,L-methadone and doxorubicin in A172 and U118MG glioblastoma cells (Fig. 1B and C). This suggests that opioid receptor activation using D,L-methadone strongly potentiates doxorubicin-induced apoptosis in glioblastoma cells.

Figure 1. Opioid receptor activation using D,L-methadone sensitizes glioblastoma cell lines for doxorubicin treatment. (A) Opioid receptor expression on glioblastoma cells. The glioblastoma cell lines U118MG and A172 were stained with naloxone fluorescein measuring opioid receptor expression (OR, thick black curve) by flow cytometry. Controls (Co, unstained cells) are exhibited as thin black curves. (B) A172 and U118MG glioblastoma cells were treated with different concentrations of D,L-methadone (10, 3, 1 µg/mL) alone (- Doxo, white columns), with 0.1 µg/mL doxorubicin (+ Doxo, black columns) alone or with different concentrations of D,L-methadone (10, 3, 1 µg/mL) in addition to 0.1 µg/mL doxorubicin (+ Doxo, black columns). After 120 h and 144 h, the percentages of apoptotic cells were measured by hypodiploid DNA analysis. The percentage of specific apoptosis was calculated as follows: 100 x [experimental dead cells (%) - spontaneous dead cells in medium (%)] / [100% - spontaneous dead cells in medium(%)]. Columns, mean of triplicates; bars, SD < 10%. Similar results were obtained in 3 independent experiments. (C) A172 and U118MG glioblastoma cells were treated with different concentrations of D,L-methadone (10, 3, 1 µg/mL) alone (-Doxo, white columns), with 0.3 µg/mL doxorubicin (+Doxo, black columns) alone or with different concentrations of D,L-methadone (10, 3, 1 µg/mL) in addition to 0.3 µg/mL doxorubicin (+ Doxo, black columns). After 120 h, the percentages of apoptotic cells were measured by hypodiploid DNA analysis. The percentage of specific apoptosis was calculated as described in Figure 1B. Columns, mean of triplicates; bars, SD < 10%. Similar results were obtained in 3 independent experiments.
Opioid receptor activation sensitizes primary human glioblastoma cells and glioblastoma stem cells for doxorubicin-induced cell death
To test the clinical relevance of opioid receptor activation and whether glioblastoma are sensitized for doxorubicin-induced apoptosis, we extended our studies to primary human glioblastoma cells and glioblastoma stem cells. Primary human glioblastoma cells expressing opioid receptors (Fig. 2A) were treated with D,L-methadone (3, 1 µg/mL) or doxorubicin (0.1 µg/mL) alone or in combination (Fig. 2B). Therapeutic concentrations of D,L-methadone (3, 1 µg/mL) in addition to doxorubicin kill strongly primary human glioblastoma cells after 120 h and 144 h (Fig. 2B).

Figure 2. Opioid receptor activation using D,L-methadone sensitizes primary human glioblastoma cells and glioblastoma stem cells for doxorubicin treatment. (A) Primary human glioblastoma cells were stained with naloxone fluorescein measuring opioid receptor expression (OR, thick black curve) by flow cytometry. Control (Co, unstained cells) is exhibited as thin black curve. (B) Primary human glioblastoma cells were treated with different concentrations of D,L-methadone (3, 1 µg/mL) alone (−Doxo, white columns) with 0.1 µg/mL doxorubicin (+Doxo, black columns) alone or with D,L-methadone (3, 1 µg/mL) in addition to 0.1 µg/mL doxorubicin (+Doxo, black columns). After 120 h and 144 h, the percentages of apoptotic cells were measured by hypodiploid DNA analysis. The percentage of specific apoptosis was calculated as described in Figure 1B. Columns, mean of triplicates, bars, SD < 10%. Similar results were obtained in 3 independent experiments. (C) Glioblastoma stem cells were stained with naloxone fluorescein measuring opioid receptor expression (OR, thick black curve) and analyzed by flow cytometry. Control (Co, unstained cells) is exhibited as thin black curve. (D) Glioblastoma stem cells were treated with different concentrations of D,L-methadone (10, 3, 1 µg/mL) alone (−Doxo, white columns), with 0.1 µg/mL doxorubicin (+ Doxo, black columns) alone or with D,L-methadone (3, 1 µg/mL) in addition to 0.1 µg/mL doxorubicin (+ Doxo, black columns). After 144 h, the percentages of apoptotic cells were measured by hypodiploid DNA analysis. The percentage of specific apoptosis was calculated as described in Figure 1B. Columns, mean of triplicates; bars, SD < 10%. Similar results were obtained in 3 independent experiments.
Human glioblastoma stem cells are known for their resistances toward radiation and chemotherapeutic drugs responsible for failure of conventional therapies.5,24 Glioblastoma stem cells expressing opioid receptors (Fig. 2C), were treated with D,L-methadone (10, 3, 1 µg/mL), doxorubicin (0.1 µg/mL), or in combination (Fig. 2D). We found a strong induction of apoptosis using the combination therapy of D,L-methadone and doxorubicin after 144 h (Fig. 2D). This demonstrates that opioid receptor activation by D,L-methadone sensitizes primary human glioblastoma and glioblastoma stem cells for doxorubicin-induced apoptosis.
Opioid receptor activation reverses deficient caspase activation by doxorubicin in glioblastoma cells
Deficient caspase activation was observed in chemo- and radioresistant glioblastoma cells treated with anti-cancer drugs or radiation.25 To clarify the involvement of caspases activation in combination therapy of D,L-methadone and doxorubicin-induced apoptosis in chemo- and radioresistant glioblastomas, we treated A172 glioblastoma cells with D,L-methadone (3, 1 µg/mL) or doxorubicin (0.1 µg/mL) alone or in combination. After combination treatment, we observed strong caspase activation in glioblastoma cells by activation of caspase-3, -9, -2, and -10 and cleavage of Poly-(ADP-ribose)-polymerase (PARP) (Fig. 3A). To investigate the critical role of caspases in opioid receptor activation-induced apoptosis, we pre-incubated A172 cells with the broad-spectrum inhibitor of caspases zVAD.fmk (Fig. 3B). Incubation with zVAD.fmk almost completely inhibited apoptosis in glioblastoma cells induced by D,L-methadone in addition to doxorubicin, suggesting that caspases are central for opioid receptor activation-mediated sensitization of glioblastoma cells for doxorubicin treatment (Fig. 3B). This demonstrates that opioid receptor activation using D,L-methadone reverses deficient activation of caspases by doxorubicin in glioblastoma cells.

Figure 3. Opioid receptor activation using D,L-methadone sensitizes glioblastoma cell for doxorubicin-induced activation of caspases. (A) D,L-methadone restored deficient caspases activation by doxorubicin in glioblastoma cells. A172 were treated with different concentrations of D,L-methadone (3, 1 µg/mL, -Doxo) alone, with 0.1 µg/mL doxorubicin (+Doxo) alone or with D,L-methadone (3, 1 µg/mL) in addition to 0.1 µg/mL doxorubicin (+Doxo). After 144 h, western blot analyses were performed. Downregulation of caspase-10 was detected at ~58 kDa and of caspase-2 at ~48 kDa. The active fragment of caspase-9 was detected at ~37 kDa, the active fragment of caspase-3 at ~17 kDa and PARP cleavage at ~85 kDa. Equal protein loading was controlled by anti-β-actin antibody. (B) Inhibition of caspases activation blocks opioid receptor activation-sensitized glioblastoma cells for doxorubicin-induced apoptosis. Glioblastoma cells A172 were treated with different concentrations of D,L-methadone (10, 3, 1 µg/mL) in combination with 0.1 µg/mL doxorubicin (+0.1 µg/mL Doxo) without (−zVAD, black columns) or with addition of 50 µmol/L zVAD.fmk (+zVAD, white columns). After 120 h and 144 h, the percentages of apoptotic cells were measured by hypodiploid DNA analysis. The percentage of specific apoptosis was calculated as described in Figure 1B. (C) Opioid receptor activation using D,L-methadone sensitizes glioblastoma cell by downregulation of XIAP and Bcl-xL. Glioblastoma cells A172 were treated with different concentrations of D,L-methadone (3, 1 µg/mL, -Doxo) alone, with 0.1 µg/mL doxorubicin (+Doxo) alone, or with D,L-methadone (3, 1 µg/mL) in addition to 0.1 µg/mL doxorubicin (+Doxo) for 144 h. Western blot analyses for XIAP and Bcl-xL were performed. XIAP was detected at ~58 kDa and Bcl-xL at ~30 kDa. Equal protein loading was controlled by anti-β-actin antibody.
Opioid receptor activation reverses deficient downregulation of XIAP and Bcl-xL by doxorubicin in glioblastoma cells
Inhibition of XIAP by XIAP inhibitors or Bcl-xL by using Bcl-xL-antisense oligonucleotides sensitizes glioblastoma cells for chemotherapeutic drugs or radiation.20,21 Therefore, we examined if opioid receptor activation using D,L-methadone can also downregulate XIAP or Bcl-xL in doxorubicin-treated A172 glioblastoma cells. Strong downregulation of XIAP and Bcl-xL was found in glioblastoma cells treated with D,L-methadone (3, 1 µg/mL) in addition to doxorubicin (0.1 µg/mL) (Fig. 3C), indicating that opioid receptor activation using D,L-methadone in addition to doxorubicin sensitizes glioblastoma cells via downregulation of XIAP and Bcl-xL.
Opioid receptor activation reverses deficient activation of apoptosis pathways by doxorubicin in chemo- and radioresistant glioblastoma stem cells
Glioblastoma stem cells were reported to possess enhanced chemoresistance and radioresistance, resulting in the regulation of tumor progression and recurrence.5 We next analyzed, if caspases were activated in glioblastoma stem cell-induced apoptosis after co-treatment with D,L-methadone and doxorubicin. We treated glioblastoma stem cells with D,L-methadone (3 µg/mL) or doxorubicin (0.1 µg/mL) alone or in combination. 144 h after treatment with D,L-methadone in addition to doxorubicin, we observed a strong caspase activation in glioblastoma-intitiating-stem cells by activation of caspase-3, -9, -2, and -10 and cleavage of Poly-(ADP-ribose)-polymerase (PARP) (Fig. 4A). This demonstrates that opioid receptor activation using D,L-methadone reversed deficient caspases activation by doxorubicin in glioblastoma stem cells.
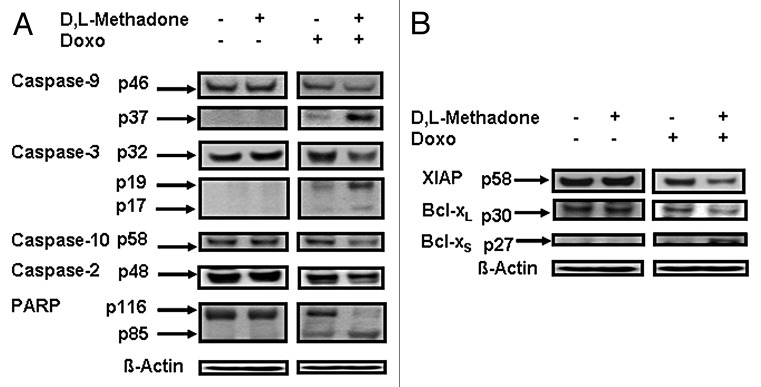
Figure 4. Opioid receptor activation using D,L-methadone reversed deficient activation of apoptotic pathways by doxorubicin in glioblastoma stem cells. (A) Glioblastoma stem cells were treated with D,L-methadone (3 µg/mL) alone, with 0.1 µg/mL doxorubicin (0.1 µg/mL Doxo) alone or with D,L-methadone (3 µg/mL) in addition to doxorubicin (0.1 µg/mL Doxo). After 144 h, western blot analyses for caspase-10, -2, -9, -3, and PARP were performed. Downregulation of caspase-10 was detected at ~58 kDa and of caspase-2 at ~48 kDa. The active fragment of caspase-9 was detected at ~37 kDa, the active fragment of caspase-3 at ~19 kDa and ~17 kDa and PARP cleavage at ~85 kDa. Equal protein loading was controlled by anti-β-actin antibody. (B) Glioblastoma stem cells were treated with D,L-methadone (3 µg/mL) alone, with 0.1 µg/mL doxorubicin (0.1 µg/mL Doxo) alone or with D,L-methadone (3 µg/mL) in addition to doxorubicin (0.1 µg/mL Doxo) for 144h. Western blot analyses for XIAP, Bcl-xL, and Bcl-xS were performed. XIAP was detected at ~58 kDa, Bcl-xL was detected at ~30 kDa, and Bcl-xS was detected at ~27 kDa. Equal protein loading was controlled by anti-β-actin antibody.
To determine the role of XIAP and Bcl-xL in chemoresistance of glioblastoma stem cells, we treated glioblastoma stem cells with D,L-methadone in addition to doxorubicin. After 144 h, we observed a strong downregulation of XIAP and Bcl-xL as well a strong upregulation of the pro-apoptotic protein Bcl-xS in glioblastoma stem cells treated with D,L-methadone (3 µg/mL) in combination with doxorubicin (0.1 µg/mL) (Fig. 4B). This suggests that opioid receptor activation using D,L-methadone sensitizes glioblastoma stem cells for doxorubicin-induced apoptosis by downregulation of XIAP, Bcl-xL and upregulation of Bcl-xS.
Opioid receptor activation using D,L-methadone enhances doxorubicin uptake and inhibits its efflux in glioblastoma cells
Doxorubicin resistance is associated with the overexpression of P-glycoprotein (p-gp).26-28 D,L-methadone increases doxorubicin uptake and inhibits doxorubicin efflux in leukemia cells.13 Therefore, we tested the effect of D,L-methadone in doxorubicin uptake and efflux in glioblastoma cells incubated with 0.3 µg/mL doxorubicin alone or in combination with 3 µg/mL D,L-methadone using flow cytometry. Four, 8, and 24 h after treatment, the amount of doxorubicin was enhanced in cells treated with the combination of doxorubicin and D,L-methadone (Fig. 5A). These findings suggest that D,L-methadone improves doxorubicin-uptake in glioblastoma cells.
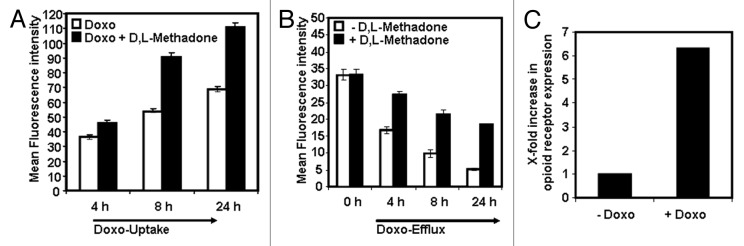
Figure 5. D,L-methadone enhances doxorubicin uptake and inhibits its efflux whereas doxorubicin enhances opioid receptor expression. (A) D,L-Methadone enhances doxorubicin-uptake in glioblastoma cells A172. A172 were incubated with 0.3 µg/mL doxorubicin alone (Doxo, white columns) or in combination with 3 µg/mL D,L-methadone (Doxo + D,L-Methadon, black columns). After 4, 8, and 24 h incubation, the fluorescence intensity of doxorubicin was determined using flow cytometry analysis. The graphic depicts the relative doxorubicin uptake. Columns, mean of triplicates; bars, SD < 10%. Similar results were obtained in 3 independent experiments. (B) D,L-methadone inhibits doxorubicin-efflux out of glioblastoma cells A172. A172 were incubated with 0.3 µg/mL doxorubicin for 4 h. After washing doxorubicin-treated A172 cells, A172 cells were either left untreated (−D,L-methadone) or treated with 3 µg/mL D,L-methadone (+D,L-Methadone) and incubated for different points in time (4 h, 8 h, 24 h). Doxorubicin efflux was analyzed via doxorubicin fluorescence in cells using flow cytometry. Columns, mean of triplicates; bars, SD < 10%. Similar results were obtained in 3 independent experiments. (C) Doxorubicin enhances opioid receptor expression. The glioblastama cell line A172 was treated for 120 h with 0.1 µg/mL doxorubicin. After staining of doxorubicin-treated (+Doxo) and untreated cells (−Doxo) with naloxone-fluoresceine. Relative fluorescence intensities were determined flow cytometrically. X-fold increase in opioid receptor expression is shown after subtracting the cells autofluorescence (−Doxo) and doxorubicin fluorescence (+Doxo).
To test whether D,L-methadone inhibits doxorubicin-efflux out of glioblastoma cells, we incubated the glioblastoma cell line A172 for 4 h with 0.3 µg/mL doxorubicin alone. After removing doxorubicin from supernatant, fresh medium was added without doxorubicin, and cells were treated with or without D,L-methadone. Cells treated with D,L-methadone had a delayed doxorubicin-efflux after 4, 8, and 24 h (Fig. 5B), suggesting that D,L-methadone inhibits doxorubicin efflux.
Doxorubicin increases opioid receptor expression in glioblastoma cells
In leukemia cells, doxorubicin increases opioid receptor expression.13 We analyzed the effect of doxorubicin on expression of opioid receptors in glioblastoma cells. Glioblastoma cells (A172) were incubated with 0.1 µg/mL doxorubicin or left untreated. After treatment, the relative amount of opioid receptors on the cell surface of untreated and doxorubicin-treated cells was measured flow cytometrically. Doxorubicin enhanced strongly the expression of opioid receptors in glioblastoma cells (Fig. 5C), indicating that more D,L-methadone molecules can bind to glioblastoma cells co-treated with doxorubicin.
Apoptosis induction and caspase activation depend on opioid receptor activation inducing cAMP downregulation
cAMP-related signaling can control apoptosis induction and cell growth.7,8 Opioid receptor activation induces cAMP downregulation and plays a critical role in D,L-methadone-sensitized leukemia cells for doxorubicin treatment.13 To analyze the role of opioid receptor activation in apoptosis induction and caspase activation in glioblastoma cells, glioblastoma cells A172 were treated with the opioid receptor agonist D,L-methadone, doxorubicin, or with the opioid receptor antagonist naloxone alone or in different combinations (Fig. 6A–C). Blocking opioid receptors by naloxone strongly reduced apoptosis (Fig. 6A and B) and activation of caspase-9, caspase-3, and cleavage of PARP (Fig. 6C) induced by combination treatment with D,L-methadone and doxorubicin. This indicates that opioid receptor activation plays a critical role in apoptosis induction and caspase activation. Opioid receptor stimulation activates inhibitory Gi-proteins, which, in turn, block adenylyl cyclase activity, reducing cAMP.29 IBMX, however, increases cAMP levels due to phosphodiesterase inhibition. To analyze the critical role of cAMP in opioid receptor activation-induced apoptosis, A172 cells were treated with D,L-methadone, doxorubicin, and IBMX, either alone or in different combinations (Fig. 6D). Upregulation of cAMP by IBMX strongly reduced apoptosis induction by combination treatment with D,L-methadone and doxorubicin, indicating that opioid receptor activation via cAMP downregulation sensitizes glioblastoma cells for doxorubicin-induced apoptosis and caspase activation.

Figure 6. Opioid receptor activation triggering downregulation of cAMP plays a critical role in sensitizing glioblastoma cells for doxorubicin treatment. (A and B) Blocking opioid receptor activation inhibits apoptosis induction. Glioblastoma cell line A172 was incubated with 100 µg/mL naloxone (Naloxone), 3 µg/mL D,L-methadone (D,L-Methadone) and 0.1 µg/mL doxorubicin (Doxo) alone or in different combinations as indicated. After (A) 120 h and (B) 144 h, the percentages of apoptotic cells were measured by hypodiploid DNA analysis. (C) Inhibition of opioid receptor activation inhibits caspase activation. A172 cells were incubated with 100 µg/mL naloxone (Naloxone), 3 µg/mL D,L-methadone (D,L-Methadone), and 0.1 µg/mL doxorubicin (Doxo) alone or in different combinations as indicated. Western blot analyses for caspase-9, caspase-3, and PARP were performed after 120 h of incubation. The active fragment of caspase-9 was detected at ~37 kDa, of caspase-3 at ~17 kDa, and PARP cleavage at ~85 kDa. Equal protein loading was controlled by anti-β-actin antibody. (D) Increasing cAMP levels via repression of phosphodiesterase activity inhibits apoptosis. A172 cells were incubated for 120 h with 25 µM 3-Isobutyl-1-methylxanthine (IBMX), 3 µg/mL D,L-methadone (D,L-Methadone), and 0.1 µg/mL doxorubicin (Doxo) alone or in different combinations as indicated. After 120 h, the percentages of apoptotic cells were measured by hypodiploid DNA analysis. The percentages of specific apoptosis was calculated as described in Figure 1B.
Opioid receptor activation using D,L-methadone inhibits tumor growth in vivo
We extended our in vitro studies using a nude mouse model to test the clinical relevance of the anti-cancer potential of the µ-opioid receptor agonist D,L-methadone in vivo. Therefore, 107 U87MG glioblastoma cells were subcutaneously inoculated per nude mouse. After randomization of 16 mice, D,L-methadone was daily orally administered in 8 mice starting at day 1 until the end of experiment, (Fig. 7). D,L-methadone dosage was increased weekly from 60 to 120 to 240 mg/kg/d bid. At day 33, 24 h after the last treatment with D,L-methadone, the mice were sacrificed. For analyzing serum concentrations of D,L-methadone in mice 0.5, 1, and 4 h after last D,L-methadone-application, serum was taken, and D,L-methadone quantified by mass spectrometry. In comparison to vehicle-treated mice of the control group, the D,L-methadone-treated mice had a significantly reduced tumor size at day days 19 to 33 with an optimum T/C value of 49% (Fig. 7). The D,L-methadone treatment was well tolerated in the dose used and induced only a minor body weight loss of 9%. Serum concentrations were found between 136 ng/mL and 1608 ng/mL of methadone in the time course of 0.5 to 4 h after D,L-methadone application. These findings demonstrate that opioid receptor activation using D,L-methadone inhibits tumor growth of glioblastomas in vivo.
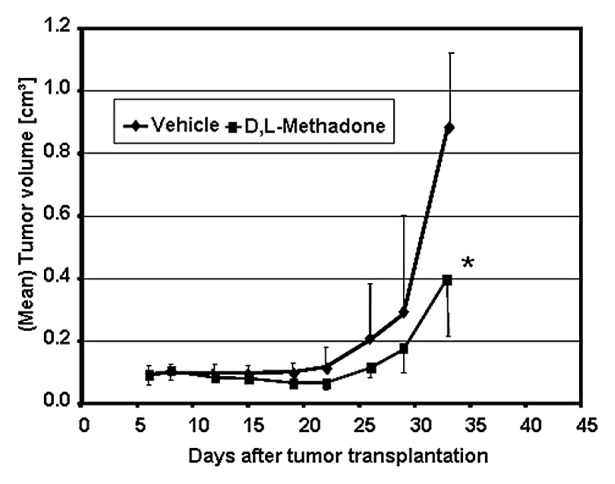
Figure 7. Opioid receptor activation using D,L-methadone inhibits tumor growth of glioblastoma. The glioblastoma cell linie U87MG (U87) was transplanted into nude-mice. Mice were daily treated with D,L-methadone (n = 8, black quadrate) or vehicle 10%Tween 80 in saline (n = 8, black diamond). After transplantation mice were observed for 33 d. At distinct points in time (as indicated), tumor growth and tumor volume were measured. *Significant to control (P < 0.05, Mann–Whitney U test).
Discussion
Resistance to current treatment strategies, such as chemotherapy and radiotherapy is still a major problem in oncology and may be caused by defects in apoptosis programs.30-32 Current attempts to improve therapeutic success and survival of cancer patients will likely depend on the successful development of novel strategies to increase the therapeutic efficacy of conventional therapies and to overcome chemo- and radioresistance in cancer.17,32,33 Induction of apoptosis is a key cytotoxic mechanism of most antitumor therapies.17,33 This complex pathway of programmed cell death is blocked in chemo- and radioresistant tumor cells.30,31,34 Glioblastoma are hardly treatable with anti-cancer drugs. The prognosis of glioma patients remains very poor.1 Glioblastoma patients have a median overall survival expectancy of 14 mo on the standard treatment of surgical resection followed by radio- and chemotherapy.1 One of the major problems for glioblastoma treatment is the low efficacy of blood–brain penetration of cytotoxic agents.22 Temozolomide, carmustine (BCNU), or lomustine (CCNU) are used in conventional therapies alone and/or in combination to γ-radiation.35,36 In vitro and in vivo studies showed that doxorubicin is an effective substance in treatment of malignant gliomas.22 To enhance penetration of doxorubicin, doxorubicin is used as liposomal encapsulated formulation (Caelyx) in vivo.22,23
The second messenger cAMP controls cell proliferation, differentiation, and apoptosis.7,8 For various tumor cells, like pancreatic or leukemia cells, stimulation with different agents could demonstrate that cAMP elevation is associated with impaired cell death.37-39 This is due to the synthesis of anti-apoptotic proteins, inactivation of pro-apoptotic proteins, and activation of PI3K-dependent Akt. Activation of cAMP plays also a critical role in inhibiting DNA damage- and doxorubicin-induced apoptosis via p53 dephosphorylation and, furthermore, by NF-κB activation.9,10 Opioid receptor agonists, exogenous opioids such as mophine, methadone, as well as endogenous opioids like endorphins, can activate opioid receptors. Opioid receptor stimulation activates inhibitory Gi-proteins, which, in turn, block adenylyl cyclase activity, reducing cAMP.29 The µ-opioid receptor agonist D,L-methadone is an attractive opioid analgesic, especially for patients with pain unresponsive to other analgesics40 and opioid withdrawal treatment.41,42 Opioid receptor activation using the µ-opioid receptor agonist D,L-methadone sensitizes leukemia cells for anti-cancer treatment and breaks chemo- and radioresistance in leukemia cells expressing opioid receptors.12,13 Glioblastoma cells express opioid receptors on their surface. We found that the µ-opioid receptor agonist D,L-methadone sensitizes glioblastoma cells for doxorubicin-induced apoptosis. For glioblastoma stem cells, it was shown that their presence correlates with clinical outcome.43 Human glioblastoma stem cells are known for their resistance toward radiation and chemotherapeutic drugs, which are in charge of failure of conventional therapies.5,24 Thus, finding strategies that also target the tumorigenic population of brain tumor stem cells is of importance in glioblastoma therapy to enhance progression-free survival and patients’ outcome.43 Glioblastoma stem cells express high levels of opioid receptors. Treatment of resistant glioblastoma stem cells with D,L-methadone in addition to doxorubicin strongly induced apoptosis, demonstrating that the µ-opioid receptor agonist D,L-methadone sensitizes glioblastoma stem cells for doxorubicin and overcomes chemoresistance of glioblastoma stem cells, suggesting that this combination treatment may provide a therapeutic advantage to reduce brain tumor recurrence. In addition, we found that opioid receptor activation using D,L-methadone inhibited tumor growth significantly in a nude mouse model in vivo. Opioid receptor antagonists like naloxone bind to opioid receptors with higher affinity than agonists but do not activate opioid receptors.13 This effectively blocks opioid receptors, preventing the response to opioids, opiates, and endorphins. We found that blocking opioid receptor signaling with the opioid receptor antagonist naloxone inhibited D,L-methadone-sensitized glioblastoma cells for doxorubicin-induced apoptosis, indicating that activation of opioid receptor signaling pathway is involved in apoptosis induction as well as in sensitization for doxorubicin treatment. In addition, upregulation of cAMP by inhibition of cAMP phosphodiesterases using IBMX reduced the cytotoxicity of combination treatment using D,L-methadone and doxorubicin in glioblastoma cells, indicating that the opioid receptor agonist D,L-methadone sensitizes glioblastoma cells for doxorubicin-induced apoptosis through triggering opioid receptors via downregulation of cAMP.
Deficient activation of caspases was found in chemo- and radioresistant cancer cells after anti-cancer drug treatment.44-46 The µ-opioid receptor agonist D,L-methadone activated caspases in chemo- and radioresistant leukemia cells.12 In glioblastoma cells and in the highly resistant glioblastoma stem cells D,L-methadone reversed deficient activation of caspases by doxorubicin. In addition, sensitization by D,L-methadone for doxorubicin treatment was almost completely inhibited by the broad-spectrum inhibitor of caspases zVAD.fmk, suggesting that caspases play a central role. Blocking opioid receptor signaling with the opioid receptor antagonist naloxone strongly reduced D,L-methadone-reversed deficient caspases activation of doxorubicin, suggesting that activation of opioid receptors play a critical role in caspase activation in glioblastoma.
Inhibitor of apoptosis proteins (IAPs), which are found at high expression levels in many tumors, suppress apoptosis by inhibiting caspases.47 Therapeutic modulation of IAPs could target an important checkpoint in resistance.47,48 Among the IAP family of proteins, XIAP displays the strongest anti-apoptotic properties. XIAP plays a crucial role in deficient activation of caspases-3, -7, and caspase-9.48 In glioblastoma cells as well as in gliobliastoma-initiating stem cells a strong expression of XIAP was found, suggesting that glioblastoma cells are hard to kill.21,47 Inactivation of XIAP with XIAP inhibitors sensitizes glioblastomas for anti-cancer drug treatment and γ-irradiation.47 The µ-opioid receptor agonist D,L-methadone downregulates XIAP in chemo- and radioresistant leukemia cells.12 We demonstrate that D,L-methadone in co-treatment with doxorubicin strongly downregulated XIAP in glioblastomas and glioblastoma stem cells. Blocking opioid receptor signaling with the opioid receptor antagonist naloxone strongly reduced downregulation of XIAP, suggesting that activation of opioid receptor signaling pathway using D,L-methadone could sensitize glioblastoma cells and the highly resistant glioblastoma stem cells for doxorubicin treatment by downregulation of XIAP.
Apoptotic signaling can be blocked by an increase in anti-apoptotic molecules and/or by a decrease or defective function of pro-apoptotic proteins.30 The Bcl-2 family of proteins consists of both anti-apoptotic proteins, for example Bcl-2 and Bcl-xL, as well as pro-apoptotic molecules, like Bcl-xS, Bax.49 Bcl-xL is highly expressed in glioblastoma cells and involved in tumor angiogenesis and resistance to chemotherapy.50 Glioblastoma demonstrating a recurrent or progressive disease have an upregulation of Bcl-xL. Inhibition of Bcl-xL overexpression by antisense oligonucleotides leads to a decrease of viability and an increase of chemosensitivity in glioblastoma cells.20 We found that therapeutic concentrations of the µ-opioid receptor agonist D,L-methadone in addition to therapeutic concentrations of doxorubicin strongly downregulate Bcl-xL. Blocking opioid receptor signaling with the opioid receptor antagonist naloxone strongly reduced downregulation of Bcl-xL, demonstrating that activation of opioid receptor signaling pathway using D,L-methadone could sensitize glioblastoma cells and glioblastoma stem cells for doxorubicin treatment by downregulation of Bcl-xL.
Overexpression of p-glycoproteins (p-gp), e.g., MDR-1, is involved in the resistance toward chemotherapeutic drugs such as doxorubicin and correlates with an enhanced rate of drug efflux.27 In glioblastoma cells p-gp is overexpressed. Next to doxorubicin, D,L-methadone is a substrate of p-gp.28 D,L-methadone increases doxorubicin influx and decreases doxorubicin efflux in leukemia cells.13 We found that D,L-methadone treatment led to an increase of doxorubicin also in glioblastoma cells by enforcing its uptake and inhibiting its efflux, suggesting that increased doxorubicin content in glioblastoma cells induced by D,L-methadone may be involved in overcoming chemo- and radioresistance. Doxorubicin stimulates expression of opioid receptors in leukemia cells.13 We found that, doxorubicin also enhances stongly expression of opioid receptors in glioblastoma cells, indicating that higher amounts of D,L-methadone can bind to glioblastoma cells that are co-treated with doxorubicin. These results indicate that the enhanced toxicity in combination treatment with D,L-methadone and doxorubicin is associated with the upregulation of opioid receptor expression mediated by doxorubicin and furthermore with an increased uptake and decreased efflux of doxorubicin mediated by D,L-methadone.
Taken together, activation of opioid receptor pathways using the µ-opioid receptor agonist D,L-methadone sensitizes chemo- and radioresistant glioblastoma cells and glioblastoma stem cells, which are in charge of failure of conventional therapies for doxorubicin treatment. In addition, activation of opioid receptors inducing downregulation of cAMP reversed deficient caspase activation and downregulation of XIAP and Bcl-xL by doxorubicin. Futhermore, opioid receptor activation using D,L-methadone inhibited tumor growth significantly in vivo. Our findings provide the foundation for new strategies establishing opioid receptor agonists such as D,L-methadone triggering opioid receptor activation as an additional therapeutic anti-cancer drug to improve therapeutic success and outcome.
Materials and Methods
Drugs and reagents
D,L-methadone hydrochloride (Methadone, Sigma), doxorubicin (Sigma), and naloxone (Fagron GmbH&Co) were freshly dissolved in sterile distilled water prior to each experiment to ensure constant quality of the preparations. 3-Isobutyl-1-methylxanthine (IBMX, Sigma) was freshly dissolved in 0.01N NaOH.
For in vivo application, we used D,L-methadone (Methaddict, Hexal) as 5 mg tablets purchased from the local pharmacy. The tablets were pulverized and solubilized freshly before use in 10% Tween 80 in saline.
Cell culture
Glioblastoma cell lines A172 and U118MG were obtained from the American Type Culture Collection and cultured in DMEM supplemented with 1 mmol/L glutamine, 1% penicillin/streptomycin (all of them Invitrogen), 25 mmol/L HEPES (Biochrom AG), and with 10% FCS (Lonza) at 37 °C, 95% air/5% CO2. For experiments, cells were seeded at 7000 cells/cm2 and allowed to settle for 24 h. From human specimen isolated primary cultured glioblastoma cells51,52 and glioblastoma stem cells52,53 were isolated, cultured, and characterized as described.51-53 The study was approved by the Ethics Committee, Medical Faculty, University of Ulm.
Serum concentrations of methadone
Determination of methadone in serum samples was performed after liquid/liquid extraction using a mass spectrometer equipped with a gas chromatograph (GC/MS). As internal standard, d9-methadone was added. The mass selective detector was operated in electron impact mode. Data were acquired in the selected-ion monitoring mode. The analytes were identified with the following masses m/z 294, 223, 72 (target ion) for methadone and m/z 303, 226, and 78 for d9-methadone with a limit of detection of 0.8 ng/ml and a limit of quantification of 1.2 ng/mL.
Induction of apoptosis
Glioblastoma cells (7000 cells/cm2) were treated with 10, 3, 1 µg/mL D,L-methadone (≤3 µg/mL therapeutic plasma concentration) alone or in addition doxorubicin in 75 cm2 flasks. Further experiments were performed simultaneously after addition of 100 µg/mL naloxone, 25 µM IBMX. Quantification of apoptosis was measured by flow cytometry as described.54 In brief, to determine apoptosis, cells were lysed with Nicoletti buffer containing 0.1% sodium citrate plus 0.1% Triton X-100 and propidium iodide 50 µg/mL as described by Nicoletti et al.54 Propidium iodide (PI) stained nuclei were analyzed by flow cytometry (FACSCalibur).
Inhibition of D,L-methadone in combination with doxorubicin induced-caspases activation by zVAD.fmk
The broad spectrum tripeptide inhibitor of caspases zVAD.fmk (benzoylcarbonyl-Val-Ala-Asp-fluoromethyl ketone, Enzyme Systems Products) was used in a concentration of 50 µmol/L. Glioblastoma cells were preincubated with zVAD.fmk 1 h before D,L-methadone and doxorubicin treatment. After 120 h and 144 h the percentage of apoptotic cells was measured as described above.54
Western blot analysis
Western blot analyses were done as described.55,56 Immunodetection of PARP, caspase-3, caspase-9, caspase-2, caspase-10, XIAP, Bcl-xL, and β-actin was done using rabbit-anti-PARP polyclonal antibody (1:5000, Roche), mouse-anti-caspase-10 (1:1000, MoBiTec), mouse-anti-caspase-2 (1:1000), mouse-anti-XIAP monoclonal antibody (1:1000, BD Transduction-Laboratories), rabbit-anti-caspase-3 monoclonal antibody (1:1000), rabbit-anti-caspase-9 (1:1000, Cell-Signaling), rabbit-anti-active-caspase-3 polyclonal antibody (1:200, Millipore Bioscience Research Reagents), rabbit-anti-Bcl-XS/L polyclonal antibody (1:1000, Santa-Cruz), and mouse-anti-β-actin monoclonal antibody (1:5000, Sigma). Peroxidase-conjugated goat-anti-mouse IgG or peroxidase-conjugated goat-anti-rabbit IgG (1:5000, Santa-Cruz) as secondary antibody were used for the enhanced chemoluminescence system (ECL, Amersham-Pharmacia). Equal protein loading was controlled by β-actin detection.
Analysis of doxorubicin uptake and efflux
For analysis of doxorubicin uptake, the glioblastoma cell line A172 was seeded in a density of 7000 cells/cm2 in 12-well plates and was either left untreated or incubated with 0.3 µg/mL doxorubicin or a combination of 0.3 µg/mL doxorubicin and 3 µg/mL D,L-methadone. After 4, 8, and 24 h, cells were washed and harvested with cell scrapers without trypsinizing. Afterwards the relative doxorubicin uptake in cells was analyzed using flow cytometry as described.57,58 After 4, 8, and 24 h treatment, apoptosis induction could not be observed in glioblastoma cells.
For analysis of doxorubicin efflux, cells were incubated with 0.3 µg/mL doxorubicin alone for 4 h. After 4 h doxorubicin treatment, cells were washed to remove doxorubicin from medium. Next, cells were incubated with fresh medium without doxorubicin or fresh medium containing 3 µg/mL D,L-methadone at 37 °C/5%CO2 without doxorubicin to measure doxorubicin efflux. After different time points, cells were harvested, washed, and relative doxorubicin content in glioblastoma cells was analyzed using flow cytometry as described.57,58
Flow cytometric assay for determination of cell surface opioid receptors
Cells were washed in PBS supplemented with 1% FCS, centrifuged, and resuspended in PBS/1% FCS containing naloxone-fluoresceine (0.05 mM, Invitrogen). After 30 min of incubation at RT, the cells were washed, centrifuged, and resuspended in ice-cold PBS/1% FCS. Flow cytometry analysis was performed using FACSCalibur (BD).
Nude mice studies
For in vivo use the glioblastoma cell line U87MG (U87)59 was chosen. 107 U87 cells were transplanted at day 0 subcutaneously into 16 female nude-mice. After randomization oral treatment (by gavage) with D,L-methadone was initiated at day 1 and performed daily until the end of the experiment with increasing doses: first week 60 mg/kg/d, second week 120 mg/kg/d, third week 2 × 120 mg/kg/d. Tumor size was measured twice weekly at 2 dimensions, and tumor volumes were calculated according to the formula (length × width2)/2. Mean tumor volumes and standard deviations were calculated per group. Treated to control values (T/C) in percent were calculated by relating mean tumor volumes of each group at each measurement day to the controls. Individual body weight was measured twice per week as parameter for tolerability and body weight changes in percent were calculated by relating the mean values of each group to the first measurement day. Serum from D,L-methadone treated mice was taken 0.5, 1, 4, and 24 h after last D,L-methadone treatment at day 33, respectively. D,L-methadone was quantified by mass spectrometry. The serum concentrations of methadone were found between 136 ng/mL and 1608 ng/mL in the time course of 0.5 h until 4 h after D,L-methadone application. Mice were sacrificed at day 33 for ethical reasons. All animal experiments were approved by the local responsible authorities (LaGeSo) and performed according to the guidelines for animal welfare in oncological experiments.60
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank L Ricci-Vitiani, R Pallini, A Eramo, Sri Hari Krishna Vellanki, Th Unterkircher, S Fulda, as well as KM Debatin for providing human primary glioblastoma cells and glioblastoma stem cells. Furthermore, we want to thank G Aggeler for technical assistance. Grant support: This work was supported by Deutsche Krebshilfe (http://www.krebshilfe.de), Bonn, Germany (grant 109035).
Glossary
Abbreviations:
- IAPs
inhibitor of apoptosis proteins
- cAMP
cyclic AMP
- Doxo
doxorubicin
- IBMX
3-isobutyl-1-methylxanthine
- XIAP
X-linked IAP
- p-gp
p-glycoproteins
- Caelyx
liposomal encapsulated formulation of doxorubicin
References
- 1.Lefranc F, Kiss R. The sodium pump alpha1 subunit as a potential target to combat apoptosis-resistant glioblastomas. Neoplasia. 2008;10:198–206. doi: 10.1593/neo.07928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kast RE, Boockvar JA, Brüning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas-Gonzalez A, Efferth T, Focosi D, et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget. 2013;4:502–30. doi: 10.18632/oncotarget.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 4.Würth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, Sirito R, Massollo M, Marini C, Zona G, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle. 2013;12:145–56. doi: 10.4161/cc.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabu K, Sasai K, Kimura T, Wang L, Aoyanagi E, Kohsaka S, Tanino M, Nishihara H, Tanaka S. Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res. 2008;18:1037–46. doi: 10.1038/cr.2008.270. [DOI] [PubMed] [Google Scholar]
- 6.Iwanami A, Cloughesy TF, Cavenee WK, Mischel PS. Arsenic reverses glioblastoma resistance to mTOR-targeted therapies. Cell Cycle. 2013;12:1473–4. doi: 10.4161/cc.24747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Insel PA, Zhang L, Murray F, Yokouchi H, Zambon AC. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol (Oxf) 2012;204:277–87. doi: 10.1111/j.1748-1716.2011.02273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frisch SM. cAMP takes control. Nat Cell Biol. 2000;2:E167–8. doi: 10.1038/35023650. [DOI] [PubMed] [Google Scholar]
- 9.Safa M, Kazemi A, Zand H, Azarkeivan A, Zaker F, Hayat P. Inhibitory role of cAMP on doxorubicin-induced apoptosis in pre-B ALL cells through dephosphorylation of p53 serine residues. Apoptosis. 2010;15:196–203. doi: 10.1007/s10495-009-0417-8. [DOI] [PubMed] [Google Scholar]
- 10.Naderi EH, Findley HW, Ruud E, Blomhoff HK, Naderi S. Activation of cAMP signaling inhibits DNA damage-induced apoptosis in BCP-ALL cells through abrogation of p53 accumulation. Blood. 2009;114:608–18. doi: 10.1182/blood-2009-02-204883. [DOI] [PubMed] [Google Scholar]
- 11.Standifer KM, Pasternak GW. G proteins and opioid receptor-mediated signalling. Cell Signal. 1997;9:237–48. doi: 10.1016/S0898-6568(96)00174-X. [DOI] [PubMed] [Google Scholar]
- 12.Friesen C, Roscher M, Alt A, Miltner E. Methadone, commonly used as maintenance medication for outpatient treatment of opioid dependence, kills leukemia cells and overcomes chemoresistance. Cancer Res. 2008;68:6059–64. doi: 10.1158/0008-5472.CAN-08-1227. [DOI] [PubMed] [Google Scholar]
- 13.Friesen C, Roscher M, Hormann I, Fichtner I, Alt A, Hilger RA, Debatin KM, Miltner E. Cell death sensitization of leukemia cells by opioid receptor activation. Oncotarget. 2013;4:677–90. doi: 10.18632/oncotarget.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh A, Jayanthan A, Farran A, Elwi AN, Kim SW, Farran P, Narendran A. Induction of apoptosis in pediatric acute lymphoblastic leukemia (ALL) cells by the therapeutic opioid methadone and effective synergy with Bcl-2 inhibition. Leuk Res. 2011;35:1649–57. doi: 10.1016/j.leukres.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 15.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 16.Friesen C, Herr I, Krammer PH, Debatin KM. Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat Med. 1996;2:574–7. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- 17.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 18.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–67. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 19.LaCasse EC, Baird S, Korneluk RGK, MacKenzie AE. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998;17:3247–59. doi: 10.1038/sj.onc.1202569. [DOI] [PubMed] [Google Scholar]
- 20.Guensberg P, Wacheck V, Lucas T, Monia B, Pehamberger H, Eichler HG, Jansen B. Bcl-xL antisense oligonucleotides chemosensitize human glioblastoma cells. Chemotherapy. 2002;48:189–95. doi: 10.1159/000063873. [DOI] [PubMed] [Google Scholar]
- 21.Vellanki SH, Grabrucker A, Liebau S, Proepper C, Eramo A, Braun V, Boeckers T, Debatin KM, Fulda S. Small-molecule XIAP inhibitors enhance gamma-irradiation-induced apoptosis in glioblastoma. Neoplasia. 2009;11:743–52. doi: 10.1593/neo.09436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fabel K, Dietrich J, Hau P, Wismeth C, Winner B, Przywara S, Steinbrecher A, Ullrich W, Bogdahn U. Long-term stabilization in patients with malignant glioma after treatment with liposomal doxorubicin. Cancer. 2001;92:1936–42. doi: 10.1002/1097-0142(20011001)92:7<1936::AID-CNCR1712>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.Safra T, Muggia F, Jeffers S, Tsao-Wei DD, Groshen S, Lyass O, Henderson R, Berry G, Gabizon A. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11:1029–33. doi: 10.1023/A:1008365716693. [DOI] [PubMed] [Google Scholar]
- 24.Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, Hjelmeland AB, Rich JN. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–8. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussaini IM, Carpenter JE, Redpath GT, Sando JJ, Shaffrey ME, Vandenberg SR. Protein kinase C-eta regulates resistance to UV- and gamma-irradiation-induced apoptosis in glioblastoma cells by preventing caspase-9 activation. Neuro Oncol. 2002;4:9–21. doi: 10.1093/neuonc/4.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouër R, Barthe L, Philibert C, Tournaire C, Woodley J, Houin G. The roles of P-glycoprotein and intracellular metabolism in the intestinal absorption of methadone: in vitro studies using the rat everted intestinal sac. Fundam Clin Pharmacol. 1999;13:494–500. doi: 10.1111/j.1472-8206.1999.tb00009.x. [DOI] [PubMed] [Google Scholar]
- 27.Callaghan R, Riordan JR. Synthetic and natural opiates interact with P-glycoprotein in multidrug-resistant cells. J Biol Chem. 1993;268:16059–64. [PubMed] [Google Scholar]
- 28.Crettol S, Digon P, Golay KP, Brawand M, Eap CB. In vitro P-glycoprotein-mediated transport of (R)-, (S)-, (R,S)-methadone, LAAM and their main metabolites. Pharmacology. 2007;80:304–11. doi: 10.1159/000107104. [DOI] [PubMed] [Google Scholar]
- 29.Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- 30.Fulda S. Tumor resistance to apoptosis. Int J Cancer. 2009;124:511–5. doi: 10.1002/ijc.24064. [DOI] [PubMed] [Google Scholar]
- 31.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–64. doi: 10.1016/S0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 32.Blagosklonny MV. Common drugs and treatments for cancer and age-related diseases: revitalizing answers to NCI’s provocative questions. Oncotarget. 2012;3:1711–24. doi: 10.18632/oncotarget.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golstein P, Ojcius DM, Young JD. Cell death mechanisms and the immune system. Immunol Rev. 1991;121:29–65. doi: 10.1111/j.1600-065X.1991.tb00822.x. [DOI] [PubMed] [Google Scholar]
- 34.Bergman PJ, Harris D. Radioresistance, chemoresistance, and apoptosis resistance. The past, present, and future. Vet Clin North Am Small Anim Pract. 1997;27:47–57. doi: 10.1016/s0195-5616(97)50005-2. [DOI] [PubMed] [Google Scholar]
- 35.Clarke J, Butowski N, Chang S. Recent advances in therapy for glioblastoma. Arch Neurol. 2010;67:279–83. doi: 10.1001/archneurol.2010.5. [DOI] [PubMed] [Google Scholar]
- 36.Triscott J, Lee C, Hu K, Fotovati A, Berns R, Pambid M, Luk M, Kast RE, Kong E, Toyota E, et al. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget. 2012;3:1112–23. doi: 10.18632/oncotarget.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boucher MJ, Duchesne C, Lainé J, Morisset J, Rivard N. cAMP protection of pancreatic cancer cells against apoptosis induced by ERK inhibition. Biochem Biophys Res Commun. 2001;285:207–16. doi: 10.1006/bbrc.2001.5147. [DOI] [PubMed] [Google Scholar]
- 38.García-Bermejo L, Pérez C, Vilaboa NE, de Blas E, Aller P. cAMP increasing agents attenuate the generation of apoptosis by etoposide in promonocytic leukemia cells. J Cell Sci. 1998;111:637–44. doi: 10.1242/jcs.111.5.637. [DOI] [PubMed] [Google Scholar]
- 39.Sastry KS, Karpova Y, Prokopovich S, Smith AJ, Essau B, Gersappe A, Carson JP, Weber MJ, Register TC, Chen YQ, et al. Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J Biol Chem. 2007;282:14094–100. doi: 10.1074/jbc.M611370200. [DOI] [PubMed] [Google Scholar]
- 40.Weschules DJ, Bain KT. A systematic review of opioid conversion ratios used with methadone for the treatment of pain. Pain Med. 2008;9:595–612. doi: 10.1111/j.1526-4637.2008.00461.x. [DOI] [PubMed] [Google Scholar]
- 41.Mercadante S. Opioid titration in cancer pain: a critical review. Eur J Pain. 2007;11:823–30. doi: 10.1016/j.ejpain.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Krantz MJ, Mehler PS. Treating opioid dependence. Growing implications for primary care. Arch Intern Med. 2004;164:277–88. doi: 10.1001/archinte.164.3.277. [DOI] [PubMed] [Google Scholar]
- 43.Pallini R, Ricci-Vitiani L, Banna GL, Signore M, Lombardi D, Todaro M, Stassi G, Martini M, Maira G, Larocca LM, et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin Cancer Res. 2008;14:8205–12. doi: 10.1158/1078-0432.CCR-08-0644. [DOI] [PubMed] [Google Scholar]
- 44.Friesen C, Uhl M, Pannicke U, Schwarz K, Miltner E, Debatin K-M. DNA-ligase IV and DNA-protein kinase play a critical role in deficient caspases activation in apoptosis-resistant cancer cells by using doxorubicin. Mol Biol Cell. 2008;19:3283–9. doi: 10.1091/mbc.E08-03-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–9. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 46.Los M, Herr I, Friesen C, Fulda S, Schulze-Osthoff K, Debatin K-M. Cross-resistance of CD95- and drug-induced apoptosis as a consequence of deficient activation of caspases (ICE/Ced-3 proteases) Blood. 1997;90:3118–29. [PubMed] [Google Scholar]
- 47.Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med. 2002;8:808–15. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 48.Schimmer AD, Dalili S. Targeting the IAP family of caspase inhibitors as an emerging therapeutic strategy. Hematology Am Soc Hematol Educ Program. 2005;•••:215–9. doi: 10.1182/asheducation-2005.1.215. [DOI] [PubMed] [Google Scholar]
- 49.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gabellini C, Castellini L, Trisciuoglio D, Kracht M, Zupi G, Del Bufalo D. Involvement of nuclear factor-kappa B in bcl-xL-induced interleukin 8 expression in glioblastoma. J Neurochem. 2008;107:871–82. doi: 10.1111/j.1471-4159.2008.05661.x. [DOI] [PubMed] [Google Scholar]
- 51.Opel D, Westhoff MA, Bender A, Braun V, Debatin KM, Fulda S. Phosphatidylinositol 3-kinase inhibition broadly sensitizes glioblastoma cells to death receptor- and drug-induced apoptosis. Cancer Res. 2008;68:6271–80. doi: 10.1158/0008-5472.CAN-07-6769. [DOI] [PubMed] [Google Scholar]
- 52.Unterkircher T, Cristofanon S, Vellanki SH, Nonnenmacher L, Karpel-Massler G, Wirtz CR, Debatin KM, Fulda S. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin Cancer Res. 2011;17:4019–30. doi: 10.1158/1078-0432.CCR-11-0075. [DOI] [PubMed] [Google Scholar]
- 53.Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–41. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- 54.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–9. doi: 10.1016/0022-1759(91)90198-O. [DOI] [PubMed] [Google Scholar]
- 55.Friesen C, Kiess Y, Debatin K-M. A critical role of glutathione in determining apoptosis sensitivity and resistance in leukemia cells. Cell Death Differ. 2004;11(Suppl 1):S73–85. doi: 10.1038/sj.cdd.4401431. [DOI] [PubMed] [Google Scholar]
- 56.Roscher M, Hormann I, Leib O, Marx S, Moreno J, Miltner E, Friesen C. Targeted alpha-therapy using [Bi-213]anti-CD20 as novel treatment option for radio- and chemoresistant non-Hodgkin lymphoma cells. Oncotarget. 2013;4:218–30. doi: 10.18632/oncotarget.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Durand RE. Flow cytometry studies of intracellular adriamycin in multicell spheroids in vitro. Cancer Res. 1981;41:3495–8. [PubMed] [Google Scholar]
- 58.Durand RE, Olive PL. Flow cytometry studies of intracellular adriamycin in single cells in vitro. Cancer Res. 1981;41:3489–94. [PubMed] [Google Scholar]
- 59.Jaszberenyi M, Schally AV, Block NL, Nadji M, Vidaurre I, Szalontay L, Rick FG. Inhibition of U-87 MG glioblastoma by AN-152 (AEZS-108), a targeted cytotoxic analog of luteinizing hormone-releasing hormone. Oncotarget. 2013;4:422–32. doi: 10.18632/oncotarget.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, et al. Committee of the National Cancer Research Institute Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–77. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]