

Abstract
Ischemia reperfusion processes induce damage in renal tubules and compromise the viability of kidney transplants. Understanding the molecular events responsible for tubule damage and recovery would help to develop new strategies for organ preservation. CDK5 has been traditionally considered a neuronal kinase with dual roles in cell death and survival. Here, we demonstrate that CDK5 and their regulators p35/p25 and cyclin I are also expressed in renal tubular cells. We show that treatment with CDK inhibitors promotes the formation of pro-survival CDK5/cyclin I complexes and enhances cell survival upon an ischemia reperfusion pro-apoptotic insult. These findings support the benefit of treating with CDK inhibitors for renal preservation, assisting renal tubule protection.
Keywords: apoptosis, CDK5, kidney injury, ischemia reperfusion, renal preservation
Introduction
Chronic kidney disease (CKD) is a progressive and irreversible loss of the kidney function characterized by a lower glomerular filtration rate below 50%.1 Two types of treatment are currently applied to patients with CKD: dialysis (hemodialysis or peritoneal dialysis) and renal transplantation. When transplantation is possible, patients’ half-life and quality of life increase.1 However, 25% of available kidneys are discarded for transplantation. It is known that short periods of ischemia are sufficient to induce pathophysiological events in renal tubules that could compromise transplant viability. Kidneys are susceptible to alterations in oxygen supply in procedures of organ transplantation protocols,2 particularly due to an increase in both reactive oxygen species and inflammatory response activation, and to the appearance of cell death, since susceptible, or early damaged cells, find the energy needed to carry out these processes at the time of reperfusion after transplantation.3 Research into new protocols to define preconditioning conditions that could render an organ resistant to ischemic insults is an area that receives increasing interest, from which new concepts emerge. We hypothesized that cell cycle arrest using CDK inhibitors may allow cells to initiate a process to repair ischemia-induced early damage in kidney tissues to later prevent reperfusion-induced damage. It has been previously proposed that cyclin-dependent kinases (CDK) participate in ischemia-induced neuronal death,4 and that CDK inhibitors provide protection.5 Here we report that the CDK inhibitors roscovitine6 and TAT-NBI17,8 provide protection against cell death in a well-established ischemia/reperfusion (I/R) model in porcine renal tubular cells (LLC-PK1).9 When analyzing the molecular mechanism, we found the protective effect required of CDK5. CDK5 is an atypical member of the CDK family10,11 that is highly conserved in mammals and is expressed in all tissues, although the highest expression levels are observed in the central nervous system.12 CDK5 is regulated by p35 and p39 proteins, and also by p25, a calpain-generated proteolytic fragment of p35.11,13 Furthermore, cyclin I has also been described as a regulatory protein of CDK5.13-15 Cyclin I is constitutively expressed in cells, including differentiated cells, and cyclin I-null mice present no developmental abnormalities or phenotypic abnormalities.16 However, the podocytes of these mice are more susceptible to apoptosis, while an increased expression of cyclin I prevents podocyte cell death both in vitro and in vivo.16 In these studies, we investigated the molecular mechanism by which CDK/cyclin inhibitors roscovitine and TAT-NBI1 protect renal tubular cells from I/R-induced death. We also show that TAT-NBI1 facilitates the formation of the complex CDK5/cyclin I to offer protection against I/R-induced death.
Results
CDK inhibitors increase cell viability in an ischemia/reperfusion-induced cell death model in renal tubular cells
To investigate the potential role of CDK/cyclin inhibitors in cell recovery of the kidney cells subjected to ischemia/reperfusion (I/R), we selected the general CDK/cyclin inhibitor roscovitine6 and the ATP non-competitive inhibitor TAT-NBI1.7,8 The cellular model we used was the I/R model developed from an epithelial cell line deriving from pig kidney proximal tubule cells (LLC-PK1).9 In this cell model, cells are subjected to hypoxia (1% O2) and hypercapnia (18% CO2) for 24 h, and are subsequently treated under normoxic conditions for 24 h (5% CO2) (Hx-Hp/Nx). Both roscovitine17 and TAT-NBI17,8 induced cell death in tumor cell lines. Then we first determined the sensitivity of the LLC–PK1 cell line to these compounds in order to define sublethal concentrations. From the dose response curves, we selected 5 μM as an appropriate compound concentration, because the extent of cell death induced in LLC-PK1 under normal (normoxic) conditions was always below 20% (Fig. S1). Then, LLC–PK1 cells were subjected to Hx–Hp/Nx conditions in the presence and absence of the CDK/cyclin inhibitors. In the vehicle control-treated cells, Hx–Hp/Nx conditions induced cell death (Fig. 1A), which was characterized by increased caspase-3/-7 activity (Fig. 1B). The presence of CDK/cyclin inhibitors enhanced cell viability under Hx–Hp/Nx conditions, together with diminished caspase-3/-7 activity (Fig. 1A and B). It should be mentioned that despite the differences found in in vitro activity, the obtained cell recovery and diminished caspase-3/7 activity were similar for roscovitine and TAT-NBI1 when cells were treated with identical concentrations of these compounds (Fig. 1).
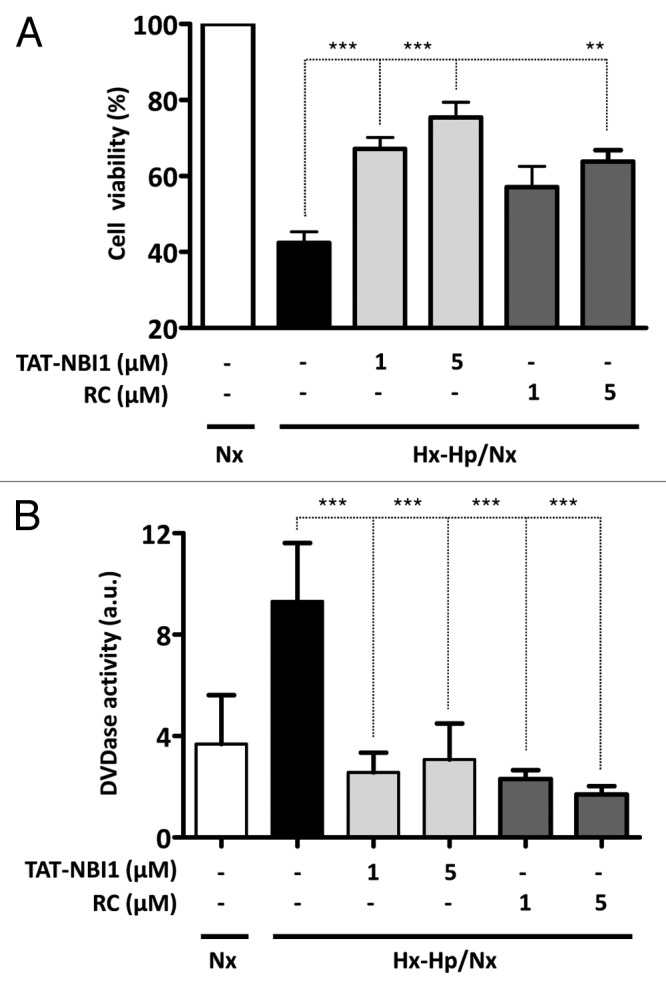
Figure 1. CDK inhibitors increase cell viability in a model of ischemia/reperfusion-induced cell death in renal tubular cells. LLC–PK1 cells were either maintained in Nx (white bars) or subjected to Hx–Hp conditions (1.5% O2; 18% CO2). After 24 h, cells were treated (gray bars) or not (black bars) with TAT-NBI1 (1 and 5 µM) or roscovitine (1 and 5 µM) and were maintained under Nx conditions (21% O2; 5% CO2) for 24 h. (A). Cell survival was measured by the trypan blue exclusion assay. (B) Caspase-3/7 activity was measured under the conditions described above. In all cases, data are expressed as the mean ± SE (n > 3). (***P < 0,0001; **P < 0,01).
Molecular mechanism of cell death protection provided by treatment with CDK/cyclin inhibitors
CDK/cyclin inhibitors have been proven to have a beneficial effect on ischemia-induced neuronal death through a CDK5-dependent mechanism. In these cellular models, CDK5 was implicated directly in the cell death mechanism, and the use of CDK5 inhibitors avoids neuronal damage.18,19 Nevertheless, the role of CDK5 in other cellular types, such as podocytes, is still an active research area.20 Both TAT-NBI1 and roscovitine in vitro inhibited the activity of the complex CDK5/p35 with the IC50 values of 7 μM (Table S1) and 0.2 μM,21 respectively. Thus, we evaluated the protein expression levels of CDK5 and its regulatory proteins, p35/p25 and cyclin I, in LLC–PK1 renal tubular cells in normoxia and Hx-Hp/Nx in the absence or presence of roscovitine and TAT-NBI1 (Fig. 2A and B). The CDK5 protein levels remained constant regardless of treatment type. In contrast, the levels of p35/p25 increased under Hx–Hp/Nx conditions in the absence of the inhibitors (Fig. 2A and B). However, the p35/25 protein levels were reversed to control levels in the presence of the CDK/cyclin inhibitors (Fig. 2A and B). In contrast, Hx–Hp/Nx conditions induced a decrease in the protein levels of cyclin I, which did not occur in the presence of the CDK/cyclin inhibitors (Fig. 2A and B). Recent reports suggest a possible role for CDK5 and cyclin I in cell survival through the modulation of the MEK–ERK pathway in podocytes.14,15,20 To determine whether the MEK–ERK pathway is involved in the CDK/cyclin inhibitors-mediated protection of LLC–PK1 cells under Hx–Hp/Nx conditions, we evaluated the phosphorylation levels of ERK1/2. Under normoxia conditions, the CDK/cyclin inhibitors did not influence the phosphorylation levels of ERK1/2 (Fig. 2C and D). In contrast, a number of modifications were noted when cells were subjected to Hx–Hp/Nx conditions. When CDK/cyclin inhibitors were absent, such conditions gave rise to a sharp drop in the phosphorylation levels, particularly of ERK1 (44 kDa), which would imply loss of cell survival signals. Interestingly, the ERK1 phosphorylation levels were similar to those observed under normoxic conditions, when cells were subjected to Hx-Hp/Nx in the presence of TAT-NBI1 or roscovitine (Fig. 2C and D). Loss of survival signals induced apoptosis, which, in LLC–PK1 cells under Hx–Hp/Nx conditions, was concurrent with an accumulation of damaged DNA, as demonstrated by the increased phosphorylation of histone 2A.x (H2A.x) at Ser139 and by the reduced phosphorylation of DNA damage effector checkpoint kinase 1 (Chk1) (Fig. 2C).22,23 These symptoms were prevented in the presence of the CDK/cyclin inhibitors (Fig. 2C and D).

Figure 2. TAT–NBI1 treatment restores the normoxic phenotype in the LLC–PK1 Hx–Hp/Nx model. LLC–PK1 cells were treated as indicated in Figure 1. The expression of CDK5, p35/p25, cyclin I in total cell extracts was analyzed by western blot in the presence of (A) TAT-NBI1 or (B) roscovitine. An analysis of the phosphorylation levels of ERK1/2, H2A.x ,and Chk1 detected by the western blotting of the cells treated with (C) TAT-NBI1 or (D) roscovitine, respectively. A densitrometric analysis of the western blots from 3 independent experiments are shown in the right-hand panels.
Taken together, these results suggest that CDK5 plays a key regulatory role in the resolution to cell death or to survival in the cellular pathways related to I/R-induced damage. We therefore examined whether CDK5 is in fact crucial for the process by silencing CDK5 in LLC–PK1 cells. Surprisingly, the silencing of CDK5 did not affect apoptosis in tubular renal cells, which occurs in other cellular damage models in neural or podocyte cells.13,14,24-26 Nevertheless, treatment of LLC–PK1 cells with CDK5 siRNA, but not with the control random siRNA, precluded the beneficial effect of CDK/cyclin inhibitors on Hx-Hp/Nx-induced caspase-3/7 activity (Fig. 3A). Interestingly, in the presence of CDK5 siRNA, the protein levels of p35/p25, which increased under Hx–Hp/Nx conditions, remained constant, even in the presence of CDK/cyclin inhibitors, while the Hx-Hp/Nx-induced low levels of cyclin I were not recovered by treatment with the CDK/cyclin inhibitors (Fig. 3B and C). These results indicate that upon CDK5 silencing, cells are susceptible to Hx-Hp/Nx-induced apoptosis, characterized by lower levels of ERK1/2 phosphorylation and increased γ-H2Ax phosphorylation (Fig. 3B and C), but they cannot engage in CDK/cyclin inhibitors-mediated recovery programs. To confirm these results, we turned our attention to using CDK5 dominant-negative constructions (DNcdk5).27 DNcdk5 competes with the endogenous CDK5 protein to bind to its activators and substrates. However, DNcdk5 has a point mutation at the catalytic site and is, thus, catalytically inactive.27 LLC–PK1 cells were transfected with either the DNcdk5 construct or a recombinant vector expressing wild-type CDK5 (WtCDK5) as a control and compared with non-transfected cells (Fig. 4). As observed in the CDK5 siRNA-treated cells (Fig. 3), DNcdk5 cells were also induced to apoptosis under Hx–Hp/Nx conditions, as demonstrated by the activation of caspase-3/7 activity, which was not inhibited in the presence of CDK/cyclin inhibitors (Fig. 4A). As expected, when cells were transfected with WtCDK5 and were subjected to Hx–Hp/Nx conditions, they start the apoptosis program, which was inhibited with CDK/cyclin inhibitors. However, unlike the control cells, higher concentrations of inhibitors were required for effective apoptosis inhibition (Fig. 4B). These data collectively suggest that CDK5 activity is required for the cell protection mechanism provided by CDK/cyclin inhibitors, although the presence of an excessive CDK5 concentration in the cell nullifies this effect, or at least makes it difficult. It is noteworthy that CDK5 does not appear to be involved in the cell death mechanism induced by Hx-Hp/Nx, since its silencing did not affect the apoptosis levels, although it was required for the cell recovery programs. These results suggest that CDK5 plays a dual regulatory role in exerting control on cell survival or cell death induced by I/R insults, depending on the molecular context. Such a role may relate to the regulatory subunit with which CDK5 is associated. Therefore, we decided to analyze the relevance of these subunits in the recovery mechanism mediated by CDK/cyclin inhibitors in LLC–PK1 cells.

Figure 3. CDK5 is not involved in apoptosis induction, but is required to engage the TAT-NBI1 treatment-induced recovery program. LLC–PK1 cells were untreated or transfected with a control (random) or a CDK5-specific siRNA. After 24 h of silencing, cells were treated as indicated in Figure 1. (A) The caspase-3/7 activity of the cytosolic extracts from treated cells. Error bars represent the mean of 3 experiments ± sd. The expression of CDK5, p35/p25, cyclin I, and the phosphorylation levels of ERK1/2 and H2A.x were analyzed by western blot in the cells treated either with TAT-NBI1 (B) or roscovitine (C). The densitrometric analysis of the western blots from 3 independent experiments are shown in the right-hand panels.

Figure 4. CDK5 activity is required to activate the TAT-NBI1 treatment-induced recovery program. LLC–PK1 cells were transfected with a mock vector or with dominant-negative Cdk5 (DNcdk5) (A) or WtCDK5 (B). After 24 h of transfection, cells were treated as indicated in Figure 1, and the caspase-3/7 activity of cytosolic cell extracts was measured under the conditions described above. The cellular extracts of the LLC–PK1 cells were analyzed by western blot for the overexpression of CDK5 (right-hand panels).
For this purpose, we downregulated the expression of cyclin I by siRNA and measured the protein levels of the related proteins (Fig. 5). In the knocked-down cyclin I cells subjected to Hx–Hp/Nx conditions, CDK inhibitors lost the beneficial effect, as in CDK5 silencing. In fact, caspase-3/7 activity was not modified when knocked-down cyclin I cells were subjected to Hx–Hp/Nx conditions in either the absence or presence of CDK/cyclin inhibitors (Fig. 5A). Moreover, treatment with the compounds increased ERK1/2 phosphorylation and diminished γ-H2A.x phosphorylation under normal basal conditions (Fig. 5B and C). Yet when the cyclin I levels were downregulated, neither TAT-NBI1 nor roscovitine modified the phosphorylation levels of these proteins. These data suggest a pronounced effect of cyclin I silencing on the CDK/cyclin inhibitors-based escape from Hx–Hp/Nx conditions-induced cell death, and demonstrate that the anti-apoptotic effect of CDK/cyclin inhibitors requires cyclin I to be present in the cell.

Figure 5. CDK5/cyclin I complex contributes to the cell recovery program. LLC–PK1 cells were untreated or transfected with control (random) or cyclin I-specific siRNA. After a 24 h post-transfection, cells were treated as described in Figure 1 (A). The caspase-3/7 activity of the cytosolic cell extracts was measured under the conditions described above. In all cases, bars represent the mean of 3 experiments ± sd (B). The western blot analysis for the expression of cyclin I, CDK5, p35/p25, and the phosphorylation levels of ERK1/2 and H2A.x in cellular extracts. The densitrometric analysis of the western blots from 3 independent experiments are shown in the right-hand panels.
Having shown that the CDK5/cyclin I complex contributes to the cell recovery program, we went on to evaluate the role of p35. To this end, the p35 expression was reduced by transfection with siRNA against p35. Reducing the p35 levels had a similar biological effect to that observed when the cells subjected to Hx-Hp/Nx were treated with TAT-NBI1 or roscovitine. Both the siRNA-induced decrease of p35 and TAT-NBI1/roscovitine treatment reduced the Hx-Hp/Nx-induced activation of caspase-3/-7 activity (Fig. 6A) and increased the cyclin I levels (Fig. 6B and C). Based on these data, we hypothesized that CDK inhibitors induce CDK5/cyclin I to probably compete for the binding of CDK5 to p35. In order to evaluate the influence of CDK inhibitors on the multiple equilibria between CDK5 and its regulatory subunits, we performed co-immunoprecipitation experiments. The cellular extracts from the LLC–PK1 control cells or from those cells subjected to Hx–Hp/Nx conditions in the presence or absence of TAT-NBI1 were prepared and used to immunoprecipitate CDK5. A western blotting analysis revealed that the immunoprecipitation of CDK5 was effective, and that the amount of precipitated protein was not influenced by Hx–Hp/Nx conditions (Fig. 7A and B). WB with anti-p35/p25 manifested the presence of p35/p25 in the precipitated material, suggesting the formation of CDK5/p35/p25 complexes (Fig. 7A). These complexes increased under Hx–Hp/Nx conditions, but reverted back to the control levels in the presence of TAT-NBI1 (Fig. 7A and B). Interestingly, when anti-cyclin I was employed to develop WB, we also obtained evidence for the presence of CDK5/cyclin I complexes, but showing the opposite behavior. The CDK5/cyclin I complexes decreased under Hx–Hp/Nx conditions, but increased in the presence of TAT-NBI1. Thus, TAT–NBI1 and roscovitine treatments favor CDK5/cyclin I over CDK5/p35 complexes as part of their mechanism of action to protect cells from Hx–Hp/Nx-induced apoptosis. To evaluate the relevance of our findings for in vivo studies, we performed immunohistochemistry assays in rat kidneys and showed the expression of CDK5 and its regulatory subunits, cyclin I and p35/p25, in renal tubules (Fig. 7C).

Figure 6. p35 siRNA had a similar biological effect to the treatment with CDK5 inhibitors. LLC–PK1 cells were untreated or transfected with control (random) or p35 specific siRNA. After 24 h of silencing, cells were treated as indicated in Figure 1. (A) The caspase-3/7 activity of the cytosolic cell extracts was measured. In all cases, bars represent the mean of 3 experiments ± sd. (B) The cellular extracts of the LLC–PK1 cells treated as described above were analyzed by western blot for the expression of p35/p25, CDK5 and cyclin I. A densitrometric analysis of the western blots from 3 independent experiments are shown in the right panels.

Figure 7. CDK5 inhibitors favored the formation of CDK5/cyclin I complexes to promote cellular recovery mechanisms. Cells were treated as described in Figure 1. (A) Immunoprecipitation (IP) of the CDK5 protein in the LLC–PK1 cell lysates probed with the CDK5, cyclin I and p35/p25 antibodies. S, supernatant. (B). A non-relevant immunoglobulin (IgG) was used as the control for non-specific immunoprecipitation. Control IP and total cell lysates (inputs) were immunoblotted with the CDK5, cyclin I, and p35/p25 antibodies. A densitrometric analysis of the western blots from 3 independent experiments are shown in the right panels. (C) Immunohistochemistry analysis of rat kidneys showing morphological hematoxylin and CDK5, cyclin I and p35/p25 staining of the tubular renal cells.
Discussion
CDK5 has been discovered to be a non-canonical CDK, given its association with non cyclin-like regulatory subunits p35/p25 and p39. As the expression of p35/p25 is restricted mainly to neurons, CDK5/p35 activity has been studied chiefly in neuronal-specific functions. In neurons under different stress stimuli, such as ischemia/reperfusion,4,28 given its association with p35, CDK5 plays an active role in the cell death induction process. In these models, CDK5 inhibition by classical CDK inhibitors, such as roscovitine, induces cell recovery.19,29-31
Nonetheless, the wide tissue distribution of CDK5 across cells and tissues suggests that the function of CDK5 is not restricted to neurons. Indeed, in recent years, novel regulatory proteins such as cyclins I or G have been described, and the activation of CDK5 has been involved in multiple cellular processes.32,33 Regarding kidney, in podocytes, which are the postmitotic kidney cells responsible for glomerular filtration processes, the involvement of CDK5, p35, and cyclin I in cell survival processes has been demonstrated.13,16 Double KO mice p35 and cyclin I exhibit a normal kidney phenotype, although glomerular disease induction triggers exacerbated apoptosis activation. These results highlight the involvement of CDK5 and its regulatory proteins in the cell survival processes linked to stress situations.15 Therefore, the balance between different CDK5 regulators, their cellular localization, and cell type seem to be key factors in determining the role of CDK5 in the equilibrium between cell survival and cell death.
Renal transplants in patients who present proximal tubular dysfunction produce functional alterations associated with a progressive decrease in the renal function.34 Therefore, protection of renal tubular cells would notably improve transplantation quality. In this report, we have studied the effect of CDK inhibitors on the prevention of Hx–Hp/Nx damage in renal tubular cells. Collectively, our data indicate that treatment with CDK5 inhibitors prevents Hx–Hp/Nx damage in renal tubular cells by disrupting the equilibrium state of CDK5 and by triggering the implication of the CDK5/cyclin I complex in the cell recovery process (Fig. 8). We demonstrate that the presence of CDK5 is necessary for Hx–Hp/Nx renal tubular cell recovery, and that CDK5 activity is required for roscovitine- and TAT-NBI1-mediated apoptosis protection, because the overexpression of DNcdk5 avoids cell recovery. In fact, silencing cyclin I hinders the protective action of compounds, but, in contrast, p35 silencing has a protective effect itself. Overall, the results obtained fit a model in which roscovitine and TAT-NBI1 preferentially inhibit the CDK5/p35/25 complex. Cyclin and cyclin-like proteins are proteolytically unstable unless associated with their CDK partners.35,36 Then, once the complex has been inhibited, the p35 protein would be degraded, and the CDK5 protein would be free to interact with cyclin I to stabilize the protein. This model is supported by the data reported in the bibliography, where the IC50 determined for CDK5/p35 with roscovitine is 0.2 µM,21 while 50 µM concentrations were needed to partially inhibit the CDK5/cyclin I complex.14 In the case of the TAT–NBI1 peptide, the IC50 determined for CDK5/p35 is 7 µM (Table S1). The IC50 value for the CDK5/cyclin I complex has not been in vitro determined due to cyclin I purification problems. Nevertheless, the mechanism of action of TAT-NBI1 is based on TAT-NBI1 binding to a cyclin groove present in cyclin A that is non-existent in cyclin E.7 TAT-NBI1 specificity is reflected in the IC50s obtained for both complexes (Table S1). The molecular modeling studies of cyclin I described in the bibliography show that cyclin I is a homolog to cyclin E,37,38 thereby supporting the differential inhibition proposed for TAT-NBI1 of both complexes.

Figure 8. Proposed model for the CDK5-mediated cell recovery of kidney tubular cells from Hx–Hp/Nx damage. (A) Under Nx conditions, CDK5 maintains a binding equilibrium between both partners p35/p25 and cyclin I signaling to cell survival. (B) Hx–Hp/Nx conditions would produce a decrease in the cyclin I protein levels to favor the formation of the CDK5/p35 complex. These changes would lead to the loss of pro-survival signaling induced by the CDK5/cyclin I complex. (C) Under Hx–Hp/Nx conditions, treatment with the CDK5 inhibitors would affect the CDK5/p35 complex without affecting the activity of the CDK5/cyclin I complex. This differential inhibition would promote the degradation of p35, the stabilization of the CDK5/cyclin I complex, and the engagement of a cell survival program.
Therefore in our model, Hx–Hp/Nx conditions would bring about a decrease in the cyclin I protein levels to favor the formation of the CDK5/p35 complex. These changes would lead to the loss of pro-survival signaling induced by the CDK5/cyclin I complex. We propose that treatment with CDK5 inhibitors at low concentrations can affect mainly the CDK5/p35 complex without affecting the activity of the CDK5/cyclin I complex. This differential inhibition would promote the degradation of p35, the stabilization of the CDK5/cyclin I complex, and the engagement of a cell survival program that would protect cells from Hx–Hp/Nx-induced damage (Fig. 8).
Hence, treatment of kidneys with CDK5 inhibitors has the dual benefit of increasing survival of not only podocytes, but also of the renal tubular cells subjected to ischemic damage. These results further highlight the need to in vivo evaluate CDK inhibitors as potential drugs for use in different stages of the kidney transplant process.
Materials and Methods
Antibodies, chemicals, and cell lines
Antibodies for phospho-Chk1 (#23425), γ-H2A.x (Ser 139) (#9718S), CDK5 (#2506) were obtained from Cell Signaling. Cyclin I (sc-5547), p35/p35 (sc-820), and phospho-ERK1/2 (sc-7383) were acquired from Santa Cruz Biotechnology. Tubulin (T8203) was purchased from Sigma-Aldrich. TAT-NBI1 was synthesized as previously described7 and is also available from Calbiochem (238808), while roscovitine (R7772) was obtained from Sigma-Aldrich. pCMV-HA-tagged Cdk5 (#1872) and HA-tagged dominant-negative (D145N) Cdk5 (#1873) (both) were purchased from Addgene.
Cell lines
Proximal tubular porcine LLC-PK-1 cells were obtained from ATCC. Cells were grown in M199 supplemented with 3% FBS and were maintained at 37 °C in a 5% carbon dioxide atmosphere.
Model renal ischemia–reperfusion (I/R) injury in LLC-PK1
LLC–PK1 proximal tubule cells were grown to form a confluent monolayer in a 6-well plate format at a cellular density of 4 × 105 cells/well. Then cells were subjected to hypoxia–hypercapnia conditions (Hx-Hp) (1.5% O2; 18% CO2). After 24 h, cells were treated with the compounds at the indicated concentrations and were maintained under normoxia conditions (Nx) (21% O2; 5% CO2) for an additional 24-h period.9
Cell viability assays
For the trypan blue cell viability assays, cell suspensions were stained with trypan blue 0.004% (1:1), and cells were counted using a hemocytometer. Viability values were calculated in relation to the control cells maintained under normoxia conditions.
Caspase 3/7 activity measurements
LLC–PK1 cells were lysed in extraction buffer (50 mM PIPES, 50 mM KCl, 5 mM EDTA, 2 mM MgCl2, 2 mM DTT, supplemented with protease inhibitors). After 3 freeze and thaw rounds, cell lysates were centrifuged at 14 000 rpm for 5 min, and supernatants were collected. Quantification of the total protein concentration was performed using the BCA protein assay (Thermo Scientific). Total protein (50 µg) was mixed with 200 μL of caspase assay buffer (PBS, 10% glycerol, 0.1 mM EDTA, 2 mM DTT) containing 20 μM of Ac-DEVD-afc (Enzo Life Sciences) of the caspase-3 substrate. DVDase activity was continuously monitored following the release of fluorescent afc at 37 °C with a Wallac 1420 Workstation (λexc = 400 nm; λem = 508 nm).
Kinase activity assays
TAT-NBI1 IC50 vs. different kinases (in vitro pharmacology kinase assays) were either previously described in references 7 and 8 or performed by Cerep.
Western blot analysis
Whole-cell extracts were obtained by lysing cells in a buffer containing 25 mM TRIS-HCl pH 7.4, 1 mM EDTA, 1 mM EGTA, 1% SDS, plus protease and phosphatase inhibitors. The protein concentration was determined by the BCA protein assay. Samples were separated by SDS-PAGE and blotted on a nitrocellulose membrane. The membrane was incubated in 5% nonfat milk powder in TBS solution (10 mM TRIS-HCl, pH 8.0, 150 mM NaCl), and the blot was incubated overnight at 4 °C with primary antibodies, followed by incubation with appropriate horseradish peroxidase secondary antibodies (GE Healthcare). Proteins were visualized using enhanced chemiluminescence technology (GE Healthcare).
Gene silencing with small interfering RNA
The siRNA for CDK5 mRNA (#6216) and the negative control siRNA (#6568) were purchased from Cell Signaling. The siRNAs for p35 and Cyclin I were acquired from Santa Cruz Biotechnology (sc-36153 and sc-35141, respectively). siRNA oligonucleotides were transfected in Optimem at the recommended concentrations using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. At 24-h post-transfection, cells were incubated under Hx–Hp conditions, treated with the compounds, and maintained under Nx conditions for 24 h.
DNcdk5, WtCDK5 transfection conditions
LLC–PK1 cells were seeded in 6-well plates at a cellular density of 4.5 × 105 cells/well and were transfected with 2 µg of DNA using Lipofectamine 2000 (Invitrogen), following the manufacturer’s instructions.
Immunoprecipitation studies
LLC-PK-1 cells were treated as described above, and 1 mg of cell extract was used for the immunoprecipitation analysis. The Co-IP kit (Pierce Classic IP Kit #26146) was used for the CDK5 immunoprecipitation studies to reduce IgG light- and heavy-chain contamination according to the manufacturer’s instructions.
Immunohistochemistry analysis
Indirect immunoperoxidase immunostaining was performed on formalin-fixed paraffin-embedded kidney specimens from normal rat. Briefly, 5-µm cut tissue sections were deparaffinized in xylol and rehydrated in graded ethanol. Antigen retrieval was performed by heating tissue sections in 0.1 M citrate solution (pH 6.0) using autoclave, and endogenous peroxidases were blocked with 3% hydrogen peroxide. Sections were blocked in PBS-T (PBS with 0.3% Triton X-100) with 5% goat serum for 30 min and then incubated for 1 h at room temperature with primary antibody. The sections were washed repeatedly in PBS before incubation with anti-rabbit HRP conjugated secondary antibody (Sigma) for 1 h at room temperature. 3,3-diaminobenzidine (Dako) was used as chromogen. Slides were counterstained with hematoxylin, dehydrated, and coverslipped.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This paper is dedicated to the memory of Enrique Pérez-Payá. We thank Ana Giménez and Rebeca Montava for technical assistance. We would like also to thank Zahara Garzón for valuable help in immunohistochemistry experiments. This work has been supported by grants from the Spanish Ministry of Science and Innovation (MICINN-BIO2007-60066, -SAF2010-15512), and from the Generalitat Valenciana (GV) Prometeo 2010/005 (funded in part with ERDF) to E.P.-P. T.G. was supported by a JAE-pre fellowship from the Consejo Superior de Investigaciones Científicas (CSIC).
REFERENCES
- 1.Bagshaw SM, Mortis G, Godinez-Luna T, Doig CJ, Laupland KB. Renal recovery after severe acute renal failure. Int J Artif Organs. 2006;29:1023–30. doi: 10.1177/039139880602901102. [DOI] [PubMed] [Google Scholar]
- 2.Legrand M, Mik EG, Johannes T, Payen D, Ince C. Renal hypoxia and dysoxia after reperfusion of the ischemic kidney. Mol Med. 2008;14:502–16. doi: 10.2119/2008-00006.Legrand. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kosieradzki M, Rowiński W. Ischemia/reperfusion injury in kidney transplantation: mechanisms and prevention. Transplant Proc. 2008;40:3279–88. doi: 10.1016/j.transproceed.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Rashidian J, Iyirhiaro G, Aleyasin H, Rios M, Vincent I, Callaghan S, Bland RJ, Slack RS, During MJ, Park DS. Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:14080–5. doi: 10.1073/pnas.0500099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyirhiaro GO, Brust TB, Rashidian J, Galehdar Z, Osman A, Phillips M, Slack RS, Macvicar BA, Park DS. Delayed combinatorial treatment with flavopiridol and minocycline provides longer term protection for neuronal soma but not dendrites following global ischemia. J Neurochem. 2008;105:703–13. doi: 10.1111/j.1471-4159.2007.05166.x. [DOI] [PubMed] [Google Scholar]
- 6.Hardcastle IR, Golding BT, Griffin RJ. Designing inhibitors of cyclin-dependent kinases. Annu Rev Pharmacol Toxicol. 2002;42:325–48. doi: 10.1146/annurev.pharmtox.42.090601.125940. [DOI] [PubMed] [Google Scholar]
- 7.Canela N, Orzáez M, Fucho R, Mateo F, Gutierrez R, Pineda-Lucena A, Bachs O, Pérez-Payá E. Identification of an hexapeptide that binds to a surface pocket in cyclin A and inhibits the catalytic activity of the complex cyclin-dependent kinase 2-cyclin A. J Biol Chem. 2006;281:35942–53. doi: 10.1074/jbc.M603511200. [DOI] [PubMed] [Google Scholar]
- 8.Orzáez M, Guevara T, Sancho M, Pérez-Payá E. Intrinsic caspase-8 activation mediates sensitization of erlotinib-resistant tumor cells to erlotinib/cell-cycle inhibitors combination treatment. Cell Death Dis. 2012;3:e415. doi: 10.1038/cddis.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hotter G, Palacios L, Sola A. Low O2 and high CO2 in LLC–PK1 cells culture mimics renal ischemia-induced apoptosis. Lab Invest. 2004;84:213–20. doi: 10.1038/labinvest.3700026. [DOI] [PubMed] [Google Scholar]
- 10.Lalioti V, Muruais G, Dinarina A, van Damme J, Vandekerckhove J, Sandoval IV. The atypical kinase Cdk5 is activated by insulin, regulates the association between GLUT4 and E-Syt1, and modulates glucose transport in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 2009;106:4249–53. doi: 10.1073/pnas.0900218106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hisanaga S, Endo R. Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J Neurochem. 2010;115:1309–21. doi: 10.1111/j.1471-4159.2010.07050.x. [DOI] [PubMed] [Google Scholar]
- 12.Dhariwala FA, Rajadhyaksha MS. An unusual member of the Cdk family: Cdk5. Cell Mol Neurobiol. 2008;28:351–69. doi: 10.1007/s10571-007-9242-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brinkkoetter PT, Pippin JW, Shankland SJ. Cyclin I-Cdk5 governs survival in post-mitotic cells. Cell Cycle. 2010;9:1729–31. doi: 10.4161/cc.9.9.11471. [DOI] [PubMed] [Google Scholar]
- 14.Brinkkoetter PT, Olivier P, Wu JS, Henderson S, Krofft RD, Pippin JW, Hockenbery D, Roberts JM, Shankland SJ. Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells. J Clin Invest. 2009;119:3089–101. doi: 10.1172/JCI37978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taniguchi Y, Pippin JW, Hagmann H, Krofft RD, Chang AM, Zhang J, Terada Y, Brinkkoetter P, Shankland SJ. Both cyclin I and p35 are required for maximal survival benefit of cyclin-dependent kinase 5 in kidney podocytes. Am J Physiol Renal Physiol. 2012;302:F1161–71. doi: 10.1152/ajprenal.00614.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffin SV, Olivier JP, Pippin JW, Roberts JM, Shankland SJ. Cyclin I protects podocytes from apoptosis. J Biol Chem. 2006;281:28048–57. doi: 10.1074/jbc.M513336200. [DOI] [PubMed] [Google Scholar]
- 17.MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65:5399–407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- 18.Hilton GD, Stoica BA, Byrnes KR, Faden AI. Roscovitine reduces neuronal loss, glial activation, and neurologic deficits after brain trauma. J Cereb Blood Flow Metab. 2008;28:1845–59. doi: 10.1038/jcbfm.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menn B, Bach S, Blevins TL, Campbell M, Meijer L, Timsit S. Delayed treatment with systemic (S)-roscovitine provides neuroprotection and inhibits in vivo CDK5 activity increase in animal stroke models. PLoS One. 2010;5:e12117. doi: 10.1371/journal.pone.0012117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brinkkoetter PT, Wu JS, Ohse T, Krofft RD, Schermer B, Benzing T, Pippin JW, Shankland SJ. p35, the non-cyclin activator of Cdk5, protects podocytes against apoptosis in vitro and in vivo. Kidney Int. 2010;77:690–9. doi: 10.1038/ki.2009.548. [DOI] [PubMed] [Google Scholar]
- 21.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–36. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 22.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 23.Capasso H, Palermo C, Wan S, Rao H, John UP, O’Connell MJ, Walworth NC. Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J Cell Sci. 2002;115:4555–64. doi: 10.1242/jcs.00133. [DOI] [PubMed] [Google Scholar]
- 24.Wang CX, Song JH, Song DK, Yong VW, Shuaib A, Hao C. Cyclin-dependent kinase-5 prevents neuronal apoptosis through ERK-mediated upregulation of Bcl-2. Cell Death Differ. 2006;13:1203–12. doi: 10.1038/sj.cdd.4401804. [DOI] [PubMed] [Google Scholar]
- 25.Piedrahita D, Hernández I, López-Tobón A, Fedorov D, Obara B, Manjunath BS, Boudreau RL, Davidson B, Laferla F, Gallego-Gómez JC, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J Neurosci. 2010;30:13966–76. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaminosono S, Saito T, Oyama F, Ohshima T, Asada A, Nagai Y, Nukina N, Hisanaga S. Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J Neurosci. 2008;28:8747–55. doi: 10.1523/JNEUROSCI.0973-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strock CJ, Park JI, Nakakura EK, Bova GS, Isaacs JT, Ball DW, Nelkin BD. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res. 2006;66:7509–15. doi: 10.1158/0008-5472.CAN-05-3048. [DOI] [PubMed] [Google Scholar]
- 28.Paoletti P, Vila I, Rifé M, Lizcano JM, Alberch J, Ginés S. Dopaminergic and glutamatergic signaling crosstalk in Huntington’s disease neurodegeneration: the role of p25/cyclin-dependent kinase 5. J Neurosci. 2008;28:10090–101. doi: 10.1523/JNEUROSCI.3237-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aydemir A, Abbasoglu O, Topaloglu S, Ertoy D, Ayhan A, Kilinç K, Karabulut E, Sayek I. Protective effect of roscovitine on renal ischemia reperfusion injury. Transplant Proc. 2002;34:2027–8. doi: 10.1016/S0041-1345(02)02838-5. [DOI] [PubMed] [Google Scholar]
- 30.Timsit S, Menn B. Cerebral ischemia, cell cycle elements and Cdk5. Biotechnol J. 2007;2:958–66. doi: 10.1002/biot.200700072. [DOI] [PubMed] [Google Scholar]
- 31.Topaloglu S, Abbasoglu O, Ayhan A, Sokmensuer C, Kilinc K. Antiapoptotic and protective effects of roscovitine on ischemia reperfusion injury of the rat liver. Liver Int. 2003;23:300–7. doi: 10.1034/j.1600-0676.2003.00842.x. [DOI] [PubMed] [Google Scholar]
- 32.Lew J. CDK5: A new lead to survival. Cell Cycle. 2013;12:1981–2. doi: 10.4161/cc.25304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Contreras-Vallejos E, Utreras E, Gonzalez-Billault C. Going out of the brain: non-nervous system physiological and pathological functions of Cdk5. Cell Signal. 2012;24:44–52. doi: 10.1016/j.cellsig.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 34.de Matos AC, Camara NO, de Oliveira AF, Franco MF, Moura LA, Nishida S, Pereira AB, Pacheco-Silva A. Functional and morphologic evaluation of kidney proximal tubuli and correlation with renal allograft prognosis. Transplant international: official journal of the European Society for Organ Transplantation. 2010;23:493–9. doi: 10.1111/j.1432-2277.2009.01005.x. [DOI] [PubMed] [Google Scholar]
- 35.Chibazakura T. Cyclin proteolysis and CDK inhibitors: two redundant pathways to maintain genome stability in mammalian cells. Cell Cycle. 2004;3:1243–5. doi: 10.4161/cc.3.10.1199. [DOI] [PubMed] [Google Scholar]
- 36.King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–9. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 37.Landberg G, Nilsson K, Jirström K, Rydén L, Kitching R, Burger AM, Seth A. Cyclin I is expressed in human breast cancer and closely associated with VEGF and KDR expression. Breast Cancer Res Treat. 2005;89:313–6. doi: 10.1007/s10549-004-2230-y. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura T, Sanokawa R, Sasaki YF, Ayusawa D, Oishi M, Mori N. Cyclin I: a new cyclin encoded by a gene isolated from human brain. Exp Cell Res. 1995;221:534–42. doi: 10.1006/excr.1995.1406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.