

Autosomal recessive primary microcephaly (MCPH) delineates a genetically heterogeneous and rare subgroup of congenital microcephalies characterized by a pronounced reduction of brain volume at birth and intellectual disability.1,2 Genetic causes of MCPH subtypes 1–12 include mutations in genes encoding microcephalin (MCPH1; MIM*607117), WD-repeat-containing protein 62 (WDR62, MCPH2; MIM*613583), cyclin-dependent kinase 5 regulatory subunit-associated protein 2 (CDK5RAP2, MCPH3; MIM*608201), cancer susceptibility candidate 5 (CASC5, MCPH4; MIM*609173), abnormal spindle-like, microcephaly-associated protein (ASPM, MCPH5; MIM*605481), centromeric protein J (CENPJ, MCPH6; MIM*609279), SCL/TAL1-interrupting locus (STIL, MCPH7; MIM*181590), centrosomal protein 135 kD (CEP135, MCPH8; MIM*611423), centrosomal protein 152 kD (CEP152, MCPH9; MIM*613529), zinc finger protein 335 (ZNF335, MCPH10; MIM*610827), polyhomeotic-like 1 (PHC1, MCPH11; MIM*602978), and cyclin-dependent kinase 6 (CDK6; MCPH12, MIM*603368). A respectable percentage of families with an MCPH phenotype do not carry mutations in the known MCPH genes, indicating further genetic heterogeneity. In 2011, Marchal et al. reported a consanguineous family with a classic MCPH phenotype and demonstrated linkage to chromosome 10q11.23–21.3; thereby they postulated a new and hitherto unrecognized MCPH locus.3 However, within this region, a new candidate gene could not be identified. We reinvestigated this family by a whole-exome sequencing (WES) approach, and identified compound heterozygous mutations in the ASPM gene as the causal gene defect.
We studied the index family previously reported by Marchal et al. 20113 with approval of the ethics committee of the Charité (EA1/212/08) and written informed consent of the parents (Fig. 1). The 3 affected children of healthy, consanguineous parents of Turkish descent were born at term without complications and with normal weight and height. Microcephaly was diagnosed through prenatal ultrasound in all patients by the 24th week of gestation, and one further pregnancy (II.4) was terminated through abortion following prenatal microcephaly diagnosis. The relative head circumference decreased further in the youngest and most affected child (from −3 to −7 SD). Motor milestones were delayed, and all 3 children presented with intellectual disability and an expressive speech delay despite normal hearing. First words were spoken between ages 2 and 4.5 years of age, but even by the age of 23 y, only single words were expressed in the eldest patient. In the youngest patient, II.5, a spastic quadriplegia with increased muscle tone and extensor plantar reflex, bilateral atypical transverse palmar creases, and a short left proximal fifth phalanx were apparent. Patient II.3 had fine motor problems and required surgery for strabismus. The 2 patients II.3 and II.5 developed focal and generalized epilepsy at ages 4 and 9 y, respectively, which were successfully treated with carbamazepine and valproic acid. In addition to the microencephaly, cranial magnetic resonance imaging (MRI) revealed a parieto–occipital and left temporo–occipital gyration defect in patient II.3 as well as reduced gyration, parietal polymicrogyria and a myelination defect in patient II.5. The results of abdominal ultrasound, echocardiography, routine blood tests, and metabolic work-up were normal. In all 3 affected children, we identified, by means of WES and Medical Resequencing Analysis Pipeline (MERAP; https://sourceforge.net/projects/merap/), a compound heterozygous mutation in the ASPM gene: (1) the nonsense mutation c.8227C > T (p.R2743X); and (2) the frameshift mutation c.7772_7775delAAAA (p.2591fs) (NM_018136). These mutations, which have not been reported so far in the dbSNP138 (http://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genome Project (http://www.1000genomes.org/), the NHLBI GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), and our in-house 417 exome data of the Middle East origin, were confirmed by Sanger sequencing, and show perfect cosegregation with biparental inheritance (Fig. 1). Phenotype and clinical presentation are in agreement with the phenotypic spectrum of ASPM mutations.4 However, compound heterozygosity is not to be expected with close consanguinity a priori, though not impossible and not contradictory in principle. Likewise, the occurrence of multiple independent mutations in an isolated inbred population is an unexpected finding. It was first reported on Reunion Island with limb girdle muscular dystrophy, and hence referred to as the “Reunion paradox”.5 Subsequently, several examples have been reported, and until recently, there was no satisfactory explanation.6 Standard population genetic theory has been extended recently, with the result that compound heterozygosity in inbred populations, and, hence, in consanguineous marriages, is severely underestimated (or “misunderestimated” according to George W Bush) by neglecting population sub-structuring and other phenomena.7 There is a lesson to be learned8 when autozygosity mapping is applied as a research strategy for elucidating the genetic causes of rare recessive diseases, and here we report another example where the correct molecular diagnosis was missed previously by applying incomplete and insufficient, though simplistic and standard, population genetic theory.
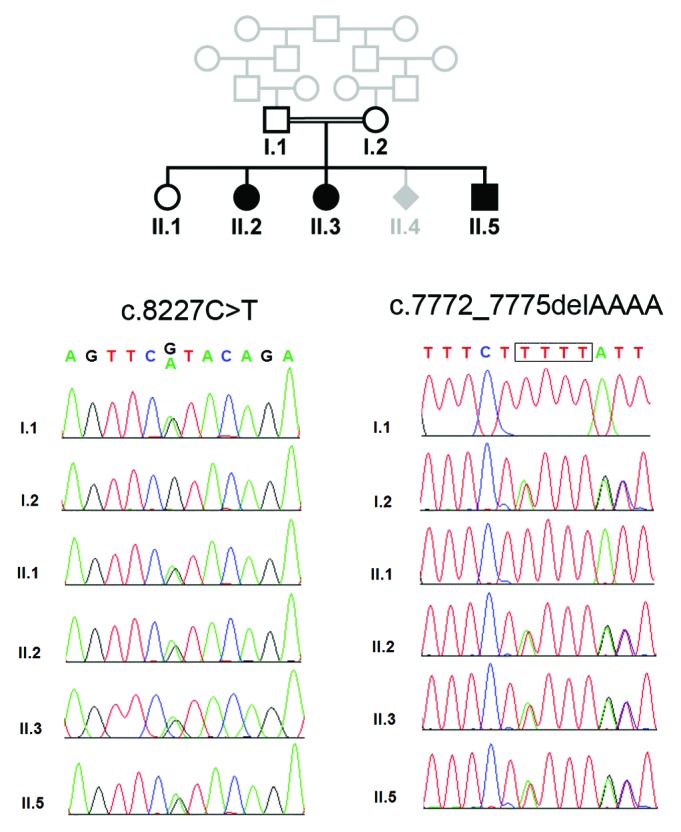
Figure 1. Compound heterozygous ASPM mutation in family with MCPH. Pedigree and results of Sanger sequencing.
Acknowledgments
The authors thank the family members who participated in this study. This work was supported by the German Research Foundation (SFB665), the Sonnenfeld Stiftung, the Berlin Institute of Health (BIH), the Max-Planck Society, the European Commission Framework Program 7 (FP7) project GENCODYS (grant no. 241995, coordinator H van Borkhoven, Nijmegen, NL), and the Charité. We acknowledge B Weschke, M Tountopoulou, L Graul-Neumann, and H Neitzel for their contribution of clinical data and discussions.
References
- 1.Thornton GK, et al. Trends Genet. 2009;25:501–10. doi: 10.1016/j.tig.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaindl AM, et al. Prog Neurobiol. 2010;90:363–83. doi: 10.1016/j.pneurobio.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Marchal JA, et al. Cell Cycle. 2011;10:2967–77. doi: 10.4161/cc.10.17.16871. [DOI] [PubMed] [Google Scholar]
- 4.Passemard S, et al. Neurology. 2009;73:962–9. doi: 10.1212/WNL.0b013e3181b8799a. [DOI] [PubMed] [Google Scholar]
- 5.Richard I, et al. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- 6.Zlotogora J, et al. Am J Hum Genet. 1996;58:241–3. [PMC free article] [PubMed] [Google Scholar]
- 7.Overall AD. Genetica. 2011;139:403–9. doi: 10.1007/s10709-011-9559-z. [DOI] [PubMed] [Google Scholar]
- 8.Petukhova L, et al. Hum Hered. 2009;68:117–30. doi: 10.1159/000212504. [DOI] [PMC free article] [PubMed] [Google Scholar]