

Highlights
-
•
Familial Alzheimer disease (FAD) mutations affect presenilin membrane integration.
-
•
The transmembrane domains around the catalytic site are vulnerable to changes.
-
•
All FAD mutations cause changes in the active site of the γ-secretase complex.
-
•
The FAD mutants lead to a complex processing pattern of the amyloid precursor protein.
Abbreviations: APP, amyloid precursor protein; Aβ, amyloid-β peptide; AICD, amyloid precursor protein intracellular domain; AD, Alzheimer disease; FAD, familial AD; TMD, transmembrane domains; PS, presenilin; NTF, N-terminal fragment; CTF, C-terminal fragment; ER, endoplasmic reticulum; Lep, leader peptidase; BD8, blastocyst-derived embryonic stem cells; GVP, Gal4VP16; GCB, γ-secretase inhibitor coupled to biotin; WT, wild type; FLIM/FRET, Fluorescence Lifetime Imaging/ Fluorescence Resonance Energy Transfer; CHAPSO, 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid; Bis-Tris, 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol; MSD, Meso Scale Discovery; RM, rough microsomes; CRM, column-washed dog pancreas rough microsomes; Endo H, endoglycosidase H; MGD, minimal glycosylation distance
Keywords: Alzheimer disease, γ-Secretase, Membrane integration, Amyloid β-peptide, Protein structure
Abstract
The enzyme complex γ-secretase generates amyloid β-peptide (Aβ), a 37–43-residue peptide associated with Alzheimer disease (AD). Mutations in presenilin 1 (PS1), the catalytical subunit of γ-secretase, result in familial AD (FAD). A unifying theme among FAD mutations is an alteration in the ratio Aβ species produced (the Aβ42/Aβ40 ratio), but the molecular mechanisms responsible remain elusive. In this report we have studied the impact of several different PS1 FAD mutations on the integration of selected PS1 transmembrane domains and on PS1 active site conformation, and whether any effects translate to a particular amyloid precursor protein (APP) processing phenotype. Most mutations studied caused an increase in the Aβ42/Aβ40 ratio, but via different mechanisms. The mutations that caused a particular large increase in the Aβ42/Aβ40 ratio did also display an impaired APP intracellular domain (AICD) formation and a lower total Aβ production. Interestingly, seven mutations close to the catalytic site caused a severely impaired integration of proximal transmembrane/hydrophobic sequences into the membrane. This structural defect did not correlate to a particular APP processing phenotype. Six selected FAD mutations, all of which exhibited different APP processing profiles and impact on PS1 transmembrane domain integration, were found to display an altered active site conformation. Combined, our data suggest that FAD mutations affect the PS1 structure and active site differently, resulting in several complex APP processing phenotypes, where the most aggressive mutations in terms of increased Aβ42/Aβ40 ratio are associated with a decrease in total γ-secretase activity.
1. Introduction
The presence of senile plaques and neurofibrillary tangles in cerebral cortex and hippocampus are the main neuropathological hallmarks of Alzheimer disease. Processes leading to plaque and tangle formation result in neuronal loss in the affected brain areas. The senile plaques are composed of extracellular aggregates of the small secreted amyloid β-peptide (Aβ) [1] that can range from 37–43 amino acids in length [2]. Aβ is derived from the amyloid precursor protein (APP), which is sequentially cleaved by the membrane bound β- and γ-secretases [3]. Simultaneously, the APP intracellular domain (AICD) is released into the cytosol. Aβ40 and Aβ42 are the most common forms of Aβ, where the Aβ42 peptide is more prone to aggregate and form soluble dimers, oligomers, protofibrills and eventually insoluble plaques [4–6]. The soluble forms are implied to be the toxic species that causes synaptic and neuronal damage as well as impaired memory and long-term potentiation in the rodent brain [7–9].
The multiprotein complex, γ-secretase is an unusual intramembrane-cleaving aspartyl protease composed of presenilin (PS), nicastrin, Pen-2 and Aph-1 [10–14]. PS is a highly conserved membrane protein with nine transmembrane domains (TMDs) and it harbours the catalytic site with the two conserved aspartate residues that are located in TMD6 and 7 [13,15–18]. Once all components are assembled, the PS molecule becomes activated by endoproteolysis, generating an N-terminal and a C-terminal fragment (NTF and CTF). γ-Secretase hydrolyzes its substrate within the hydrophobic TMDs, which require access to water molecules in the catalytic site [19].
Currently, more than 185 autosomal dominantly inherited familial AD (FAD) causing mutations have been identified in PS1, 13 in PS2 and 33 in APP, strongly suggesting that an altered Aβ metabolism plays a pivotal role in AD pathogenesis. Most PS FAD mutations are situated within or flanking the conserved hydrophobic TMDs and are, except for the Δexon9 mutation, missense mutations resulting in single amino acid changes or deletion of two amino acid residues. In general, FAD mutations in PS1 and 2 cause an increase in Aβ42/Aβ40 ratio, either by decreasing the production of Aβ40 or by increasing the Aβ42 generation [20–24]. Previously, Takami et al presented an Aβ product line-model where APP is sequentially cleaved after every third or fourth amino acid by γ-secretase, depending on the initial ε-cleavage site [25]. Recently, Cháves-Gutiérraz et al studied six FAD PS1 mutations’ impact on ε- and γ-cleavage sites and reported that they impaired the fourth cleavage site in both Aβ40 (AICD50–99 + Aβ49 > Aβ46 > Aβ43 > Aβ40) and Aβ42 (AICD49–99 + Aβ48 > Aβ45 > Aβ42 > Aβ38) product lines and thus gave increased Aβ42/Aβ40 ratio [26]. The altered Aβ42/Aβ40 ratio appears to be a very important determinant; both for the process for amyloidosis and for disease onset [21,27]. In this report, we have performed an even more extensive study and examined 13 different PS1 FAD mutations and their effect on the production of five APP processing products: Aβ38, Aβ40, Aβ42, Aβ43 and AICD.
The topology of a membrane protein is largely dependent on the hydrophobicity of the TMDs and the charged residues flanking the TMDs play an important role during insertion into the membrane [28]. Most eukaryotic membrane proteins, including the human PS1, are co-translationally integrated into the endoplasmic reticulum (ER) membrane by the Sec61 translocon [29,30]. The translocon recognizes the hydrophobic TM sequences and allows them to partition into the membrane while hydrophilic loops are translocated into the ER lumen or retained in the cytosol [31]. However, in some cases the TM sequences of multi-spanning membrane proteins fail to be recognized (marginally hydrophobic TMs) and need assistance from other parts of the protein for efficient integration and folding [32–34]. Previously, by introducing glycosylation acceptor sites into the molecule and measuring the glycosylation status, it has been shown that PS1 contains nine TMDs [16]. Here we report how efficiently each PS1 wild type TM segment is recognized and inserted into the membrane by the translocon, by engineering them into the model protein leader peptidase (Lep) [35,36]. Moreover, we have selected 35 disease-related PS1 FAD mutations and studied whether the insertion process is affected in terms of efficiency in targeting and recognition in comparison to the wild type protein. Finally, we have analyzed how six out of 35 FAD mutants affected the conformation of the catalytic site of PS1, using an active site inhibitor pull-down method.
2. Results
2.1. PS FAD mutations differentially affect Aβ42/40 ratio, Aβ38, Aβ43 and AICD production
In order to get a deeper understanding of the mechanisms behind PS1 FAD mutations, we selected 13 different FAD mutations scattered throughout the protein, and transiently transfected them into cells deficient for PS1 and PS2 (BD8), to nullify the influence of endogenous PS. It has previously been reported that generally all PS FAD mutations have the same phenotype regarding elevation of the Aβ42/Aβ40 ratio [20–24,26]. Here we extend these studies by looking at more Aβ products and AICD generation in order to get a clearer understanding of the PS1 FAD mutations role in APP processing. The selected mutations were located in TM2, TM3, TM6, H7 and TM7; some have been studied previously by other research groups (L166P, A246E, Δexon9, G384A and F386S) and others have not been examined in this context before. All PS1 protein variants were expressed and endoproteolysed with the exception for the Δexon9 mutant which lacks the endoproteolytic site [37] (Fig. 1A). When measuring Aβ38, Aβ40 and Aβ42 generation by MSD technology, we observed that all mutations increased the Aβ42/Aβ40 ratio to different extent, except for the A246E mutant that did not affect the ratio compared to PS1 wild type (Fig. 1B). This is consistent with previously published results [20–24,26]. The highest ratios were generated by the L166P and G384A mutants that gave rise to substantially elevated levels compare to PS1 wild type, 7.3 and 6.0 times, respectively. In addition, I143T, L392P and Δexon9 increased the Aβ42/Aβ40 ratio, with 4.4, 3.6 and 3.2 times, respectively. Interestingly, the L166P, I143T, L392P and G384A mutations, also had a pronounced decrease in Aβ40, with a minimum of 50% reduction compared to wild type. Moreover, these mutations, except L166P, had elevated Aβ42 levels and especially G384A increased Aβ42 (four times compared to wild type) (Fig. 1C). The rest of the mutations were more homogeneous, displaying an increased Aβ42/Aβ40 ratio ranging from 1.2-2.6 in relation to wild type and had a similar pattern in the Aβ40 and Aβ42 distribution (Fig. 1B and C). However, the Aβ38 values were more similar between the FAD mutations. All, except A246E and L250S, showed a reduction in Aβ38 levels (39–79% of wild type) (Fig. 1C). Only six of all FAD mutations showed Aβ43 values, above the detection limit of 0.62pM (WT, E280A, P284L, Δexon9, G384A, F386S and S390I). All, except E280A and F386S, showed increased Aβ43/Aβ40 ratio compared to wild type. The most pronounced increase was for P284L and G384A (6.1 and 14 times increase compared to wild type, respectively) (Fig. 1D).
Fig. 1.
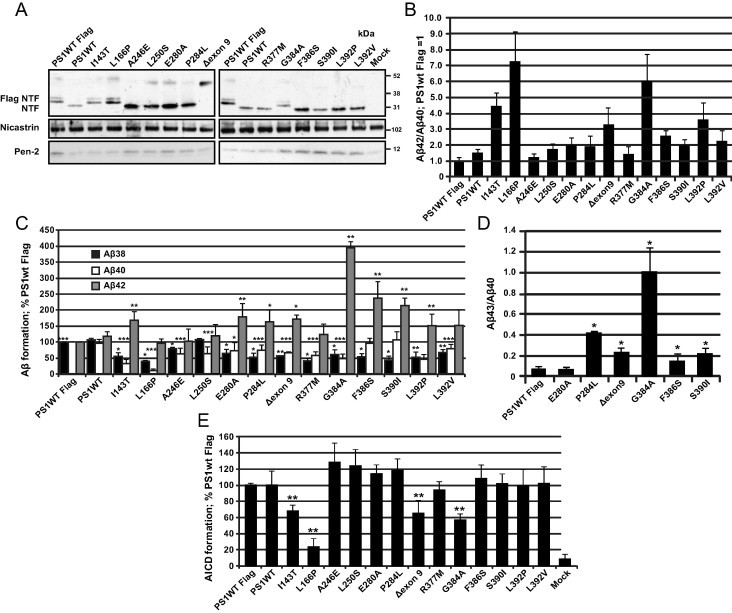
PS1 FAD mutations differentially affect Aβ42/Aβ40 ratio and the AICD processing. (A) Expression levels of PS1 NTF, CTF, Nicastrin, Pen-2 in PS−/− BD8 cells expressing PS1 WT, I143T, L166P, A246E, L250S, E280A, P284L, Δexon9, R377M, G384A, F386S, S390I, L392P or L392V. All PS1 FAD mutants are expressed to a similar extent and subjected to endoproteolysis with the exception for the Δexon9 mutant that lacks the endoproteolytic site. (B–C) Conditioned media from BD8 cells expressing PS1 WT or the mutant forms were analyzed for levels of secreted Aβ38, Aβ40 and Aβ42 species using MSD technology. (B) The Aβ42/Aβ40 ratio and (C) the levels of Aβ38, Aβ40 and Aβ42 are plotted in relative to PS1 WT Flag. (D) Conditioned media from BD8 cells expressing PS1 WT and six FAD mutations (E280A, P284L, Δexon9, G384A, F386S and S390I) were used to determine the Aβ43/Aβ40 ratio using ELISA. The other mutations were below detection limit. (E) Lysates from BD8 cells expressing PS1 WT and the 13 FAD mutations were monitored with a Luciferase-based reporter gene assay for AICD production using transfected C99-GVP, MH100, and CMV-β-gal constructs. The transfection efficiency was determined by β-gal activity and the mean value for PS1 wt was set to normalization factor 1, and the FAD mutants in relation to this factor (>1 or <1). The Aβ values from each mutant from the standard curve were then divided with its normalization factor to get the normalized Aβ value. The relative Aβ value (%) for each mutant to PS1 wt was calculated by dividing Aβ normalized from PS1 FAD mutant with normalized Aβ from PS1wt and multiply with 100. Bars represent mean of 3–7 experiments with error bars indicating S.E. Statistical significance is calculated by the non-parametric Mann–Whitney U test. ∗p < 0.05, ∗∗p < 0.01; ∗∗∗p < 0.001.
Next, we characterized the FAD mutations further by examining their overall effect on γ-secretase activity by monitoring AICD production from the ε-cleavage site by using a Luciferase-reporter gene assay [38]. In line with a recent report from Chávez-Gutiérrez et al. [26] and Bentahir et al. [22], we observed that not all FAD mutations affect AICD formation. Most mutations that we investigated showed no decrease in AICD formation, except I143T, L166P, Δexon9 and G384A, which all lowered the production (Fig. 1E). The most impaired production was observed for L166P that reduced AICD formation to 20% compared to wild type. In addition, I143T, Δexon9 and G384A decreased the AICD formation with 30–40% in comparison to PS1 wild type. Overall, the PS1 FAD mutations decreased the formation of the smaller Aβ peptides, Aβ38 and Aβ40, in favor to the production of the longer forms (Aβ42 and Aβ43), suggesting an impairment in the fourth cleavage sites of both Aβ product lines. However, the PS1 FAD mutations with a Aβ42/Aβ40 ratio above 3.2, (L392P, I143T, L166P, Δexon9, G384A, Fig. 1C) showed also an impairment in the first cleavage site (ε-site), indicating that this could contribute to the aggressive pathology and early onset of the disease in these FAD patients.
2.2. Efficient membrane integration of all TMDs in PS1, except H7 and TMD7
One proposed mechanism of PS FAD mutations is a conformational change of PS1. This could be a three dimensional structural change of the PS molecule in the catalytic site [39,40] due to altered membrane integration of the TMD segment that harbours the mutation. To explore how PS1 FAD-linked mutations affect the translocon mediated membrane insertion of PS1, we introduced each TM segment with their respective flanking residues into the well-characterized Escherichia coli inner membrane protein Lep [35,36,41,42]. Lep has two hydrophobic domains (H1 and H2) and a large C-terminal periplasmic domain (P2). When expressed in vitro in the presence of rough microsomes (rough ER membrane vesicles, RM) it adopts its natural topology, Nout–Cout, with its N- and C-terminus in the microsomal ER lumen [43]. In vitro translation supplemented with ER membranes is a well established system where the membrane proteins are efficiently targeted to the ER membrane with the help of the signal recognition particle (SRP) and its receptor. The proteins are integrated into the membrane and glycosylated during translation. The glycosylation status can be used to study the topology of the protein. The introduction of the PS1 TMDs was based on the nine-TM topology model of PS1 [15–18]. In order to study PS1 TMDs in their natural orientation, we used two versions of Lep. In the first version called LepH2, H2 (Nin–Cout orientation) was replaced by the PS1 TMD of interest and in the second version, LepH3, the selected TMD was introduced into the P2 domain of LepH3 (Nout–Cin orientation) [35,36]. To investigate the efficiency of translocon-mediated membrane insertion, two N-linked glycosylation sites were engineered in LepH2 (G1, G2) and LepH3 (G2, G3) (Fig. 2A). In cases where a TMD-segment replaces the H2 TM helix (LepH2 constructs), a fraction of the molecules is often cleaved into a smaller fragment (fc) in the presence of CRMs (Fig. 2B–D). Cleavage is prevented by addition of a signal peptidase inhibitor during translation, resulting in a concomitant increase in the fraction of doubly glycosylated molecules [36]. The signal peptidase active site is located close to the lumenal surface of the ER membrane [36] and the processed form thus originates from molecules in which the TMD-segment is inserted into the membrane and the P2 domain is in the lumen [44]. Fractions of singly (f1x) and doubly (f2x) glycosylated species were quantified in order to calculate an apparent equilibrium constant Kapp, for the membrane insertion of the given TMD-segment. For LepH3, Kapp is calculated as and for LepH2, the fractions of cleaved fc molecules were taken into account as well (if necessary) as . Kapp can be converted into apparent free energies, , between the inserted and non-inserted states [35,36,41]. We predicted the ΔGapp value for PS1 TMD1-9, including their flanking residues of PS1 by a computational model provided by the ΔG prediction server (http://www.cbr.su.se/DGpred/). When possible, 20 residues of the flanking region were included followed by two tetrapeptides (GGPG…GPGG) in order to avoid influence from the Lep sequence (Table 1). Our results show that all TMDs of PS1 integrate efficiently into the membrane by the translocon (TMD1: 86%; TMD2: 100%; TMD3: 84%; TMD4: 88%; TMD5: 90%; TMD6: 81%; TMD8: 100%; TMD9: 84%), except for the hydrophobic domain 7 (H7) in the large hydrophilic loop 6 (HL6) (H7: 6%) and TMD7 (47%) (Table 1, Fig. 2C). Both H7 and TMD7 code for low integration efficiency in their proposed natural orientation and therefore, we also determined their insertion efficiency in the other orientation (LepH2 vs LepH3), and some variations in translocon mediated insertion efficiency was found (H7 increased from 6% to 33%, TMD7 decreased from 47% to 14%). It seems likely that H7 in HL6 is involved in the endoproteolysis [45,46], probably as a reentrant loop [47,48], and integrates more efficiently in the H3 Nout–Cin orientation (increases to 33%). By contrast, TMD7, which harbours one of the two catalytic aspartate residues, integrates less efficiently in the H3 Nout–Cin orientation (decreases to 14%). The preferred orientation and the difference in insertion efficiency could be due to the charged residues flanking the TMD and the length and the hydrophobicity of the PS1 segment [28,32]. Importantly, the partial integration of TMD7 is consistent with results reporting that only half the TMD7 is integrated as an α-helix [49,50]. Moreover, as a control, glycosylation of TMD7 was confirmed by Endo H-treatment (Fig. 2D).
Fig. 2.
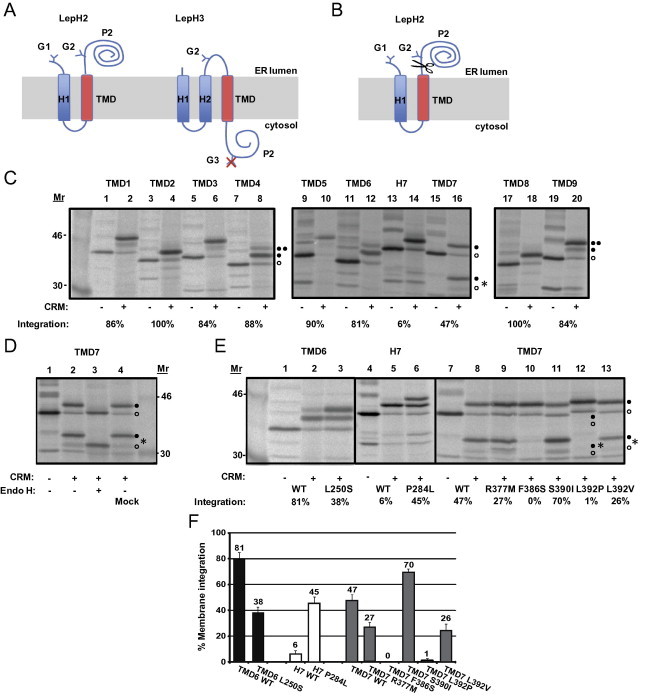
Model protein Lep and FAD-linked mutations within the TM domains. (A) The leader peptidase (Lep) model proteins, LepH2 and LepH3. Wild type Lep has two transmembrane helices (H1 and H2) and a large lumenal domain (P2). Lep integrates into dog pancreas rough microsomes (rough ER-vesicles, RMs) in an Nlum–Clum orientation. RMs are used because during translation membrane proteins are efficiently targeted to the ER membrane with the help of SRP and the SRP receptor. The proteins are integrated into the membrane by the translocon and glycosylated during translation by the oligosaccharyl transferase. PS1 TMDs were either introduced into the H2 TM helix (LepH2) or into the P2 domain of Lep (LepH3) to conserve the natural orientation of the PS1 TMDs. Odd numbered helices were inserted into the LepH2 construct and even numbered helices into the LepH3 construct. Engineered Asn-X-Thr glycosylation acceptor sites were introduced into the N-terminal tail (G1) and/or into the P2-domain (G2 and G3). Constructs in which the PS1 TMD-segment is integrated into the endoplasmic reticulum (ER) membrane become glycosylated on the G1 and G2 sites (LepH2 constructs) or on the G2 site (LepH3 constructs). (B) Membrane integration of TMD7 and H7 in the LepH2 construct result in cleaved glycosylated and unglycosylated smaller protein species, cleaved by the signal peptidase (showed as a pairs of scissors). (C) In vitro translation in the presence of column-washed dog pancreas rough microsomes (rough ER-vesicles, CRMs) [68] of constructs containing different PS1 TMDs. (D) In vitro translation in the presence of CRMs and Endo H-treatment to verify N-linked glycosylation of TMD7. (E) In vitro translation of selected FAD-linked mutations within the TMDs of TMD6, H7 and TMD7. In panels (C)–(E), unglycosylated, singly glycosylated, and doubly glycosylated forms of the protein are indicated by one open circle, one filled circle and two filled circles, respectively. A signal peptidase-cleaved fragment is indicated by an asterisk (∗). The percentage of molecules in which the TMD is integrated into the membrane is given below the lanes (average of at least three independent experiments). (F) Membrane integration of PS1 TMD-segments with and without FAD-linked mutations. The percentage of molecules retained in the membrane is shown. Bars represent mean of three experiments with error bars indicating S.E.
Table 1.
Predicted vs. measured ΔG values for each of the hydrophobic domains in PS1 wild type. Predicted TMD is indicated in red and the number in () correlate with the TM segment together with the flanking region and the two-tetra peptides, GGPG…GPGG. The integration is calculated from at least three independent experiments. N405Q mutation is indicated in blue.
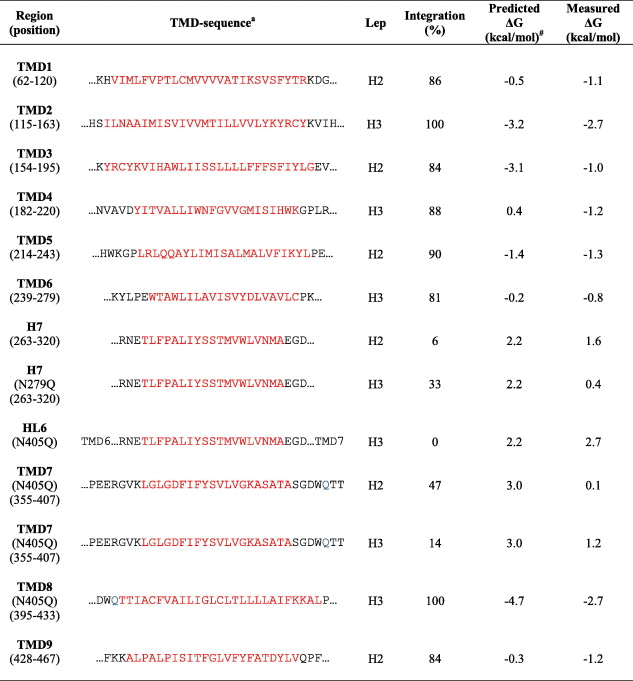 |
a TMD segments were predicted from full length PS1 with ΔG predictor.
# The predicted ΔG values were determined by the ΔG prediction server (http://www.cbr.su.se/DGpred/).
2.3. FAD mutations around the catalytic site influence membrane integration efficiency
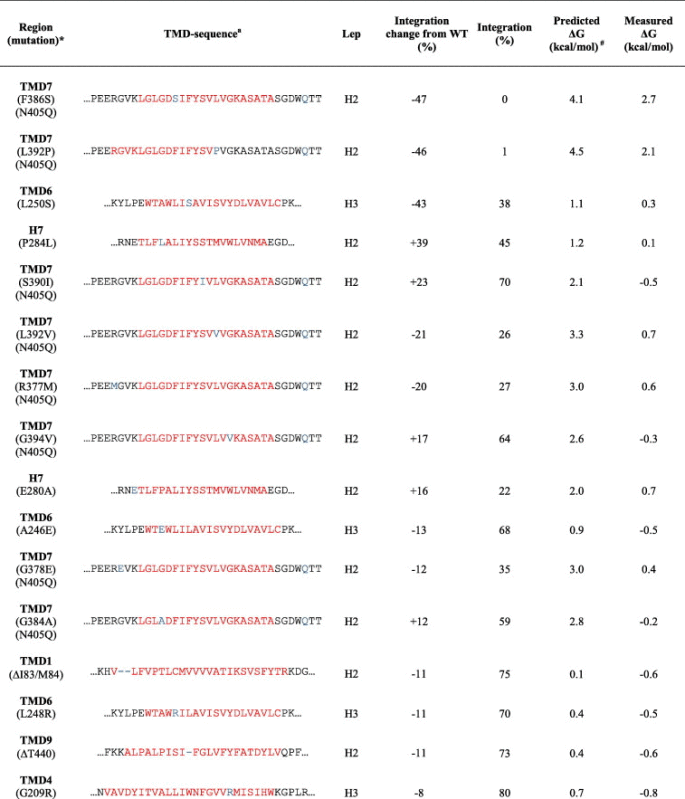
To further investigate how PS1 FAD mutations influence the insertion into the membrane by the translocon, we analyzed 35 PS1 FAD mutations located in the 10 hydrophobic domains of PS1. Several non-conservative TM mutations of PS1 belong to the eight hydrophobic TMDs (TMD1-6, 8-9) that code for efficient insertion into the membrane, but some are found in hydrophobic domains that are not inserted efficiently into the membrane (H7, TMD7) (Table 2). Our result showed, that seven PS1 FAD mutations affected the insertion efficiency with ⩾20%. Six of the seven substitutions were located in two of the marginally hydrophobic segments, H7 (P284L), and TMD7 (R377M, F386S, S390I, L392P, L392V), and one was located in the more efficiently integrated TMD6 (L250S) (Fig. 2E and F). The most prominent changes were: L250S (TMD6) which had a change in membrane integration by 43% (from 81% to 38%); P284L (H7) which had an increase from 6% to 45%; and two mutations in TMD7, F386S and L392P, which gave rise to a decrease from 47% to 0% and 1%, respectively (Table 2, Fig. 2E and F). Taken together, this experiment shows that the TMDs facing the catalytic site are affected by FAD mutations in terms of membrane integration.
Table 2.
Predicted vs. measured ΔG values for each FAD mutations of the hydrophobic domains of PS1. Predicted TMD is indicated in red and FAD mutation is indicated in blue. The integration is calculated from double samples and the most prominent ones from at least three independent experiments.
 |
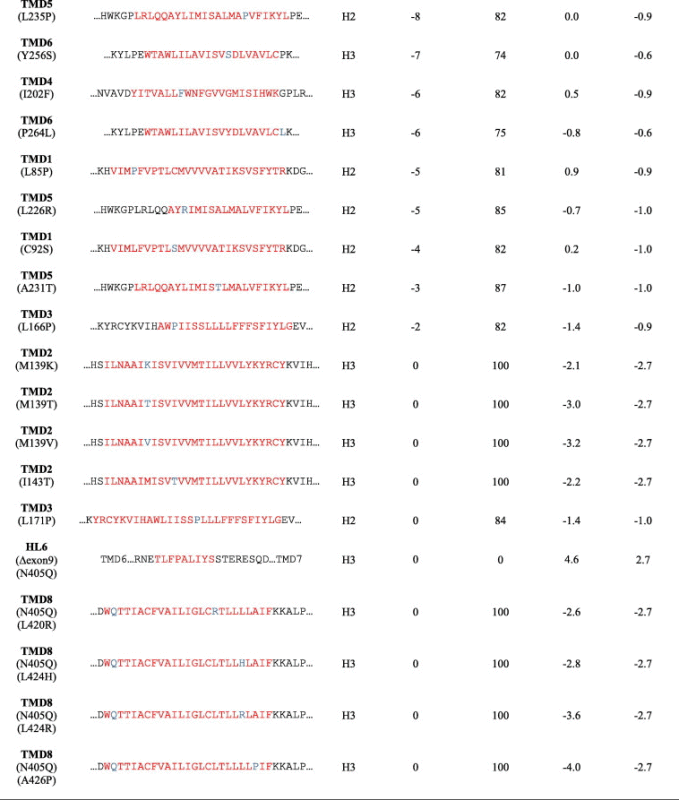 |
a TMD segments were predicted from full length PS1 with ΔG predictor.
∗ Mutations are from the Alzheimer Disease and Frontotemporal Dementia Mutation Database (http://www.molgen.ua.ac.be/admutations/).
# The predicted ΔG values were determined by the ΔG prediction server (http://www.cbr.su.se/DGpred/).
According to our results, I143T (TMD2), L166P (TMD3) and G384A (TMD7) substitutions did not show any effect in membrane integration in contrast to the results from the Aβ42/Aβ40 ratio experiments, which showed a 4–7 fold increase in the ratio (Fig. 1B and Table 3). We hypothesize that, while these mutations do not disrupt or change the TM domains’ efficiency in anchoring to the membrane, they might accommodate differently in the lipid environment. To test our assumption, we introduced acceptor sites at different positions relative to the membrane in the wild type TM and its mutant based on the well-defined distance between the oligosaccharyl transferase active site and the end of the TM segment [51]. The minimal glycosylation distance (MGD) that is required for half-maximal glycosylation is approximately 14 residues away from the N-terminal end and 10 residues away from the C-terminal end of the TM helix [47,52].
Table 3.
PS1 FAD mutations with their age of onset, Aβ42/Aβ40 ratio and% membrane integration.
| Region | Mutation | Age of onset (years)⁎ | Aβ42/Aβ40 ratio change from WT | Membrane integration change from WT (%) |
|---|---|---|---|---|
| TMD2 | I143T | 34.0 | 4.4 | 0 |
| TMD3 | L166P | 24.0 | 7.3 | −2 |
| TMD6 | A246E | 52.5 | 1.2 | −13 |
| TMD6 | L250S | 52.2 | 1.7 | −43 |
| H7 | E280A | 47.4 | 2.0 | +16 |
| H7 | P284L | 32.0 | 1.9 | +39 |
| HL6 | Δexon9 | 45.5 | 3.2 | 0 |
| TMD7 | R377M | 39 | 1.4 | −20 |
| TMD7 | G384A | 34.9 | 6.0 | +12 |
| TMD7 | F386S | 37–58 [70] | 2.6 | −47 |
| TMD7 | S390I | 39–49 [71] | 1.9 | +23 |
| TMD7 | L392P | 38.3 | 3.6 | −46 |
| TMD7 | L392V | 42.5 | 2.3 | −21 |
Mutations and data are from the Alzheimer Disease and Frontotemporal Dementia Mutation Database (http://www.molgen.ua.ac.be/admutations/), except for the indicated publications.
Furthermore, the proline (L166P in TMD3), that could cause a shift in the membrane, is located in the cytosolic side of the membrane [53]. In order to capture any effect on that side we introduced TMD3 and its mutant in the other orientation, in LepH3. The MGD values were determined for each construct and none of the mutations showed a difference in position relative to the membrane surface compared to the wild type TM segment. This indicates that the mutations do not affect accommodation of the TMDs in the lipid environment (Fig. 3A–D).
Fig. 3.
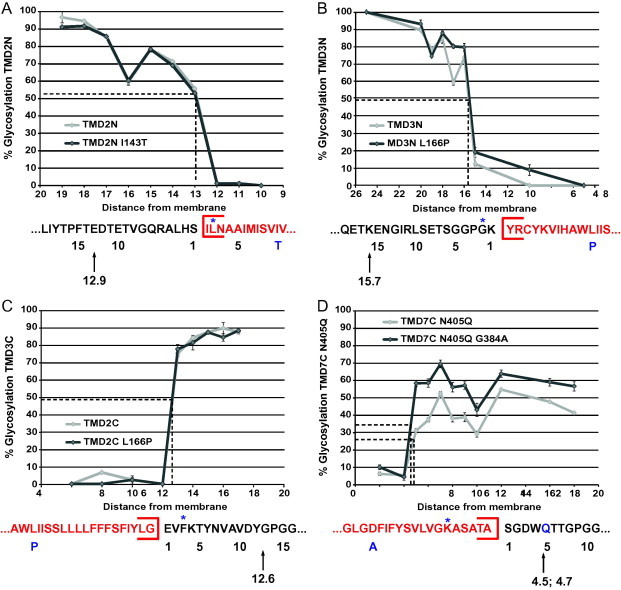
Topology mapping based on the predicted TM segments of (A) TMD2N and TMD2N (I143T), (B) TMD3N and TMD3N (L166P), (C) TMD3C and TMD3C (L166P), (D) TMD7C and TMD7C (G384A). The dotted line shows the number of residues required between the acceptor site and either end of the TM segment (12.9 residues for TMD2N/TMD2N I143T, 15.7 residues for TMD3N/TMD3N L166P, 12.6 residues for TMD3C/TMD3C L166P, 4.5 residues for TMD7C and 4.7 residues for TMD7C G384A) for half-maximal glycosylation, also indicated by an arrow in the sequence below the graph. The residues in red are the predicted membrane embedded TMDs and the star indicates the experimentally result where the TM segments starts or ends. The letter in blue is the residues that have been mutated and its position in the sequence. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.4. PS1 FAD mutations cause changes in the catalytic site
Since not all PS1 FAD mutations caused a change in membrane integration, especially the mutants with a high Aβ42/Aβ40 ratio (>4), (I143T, L166P and G384A), other mechanisms are probably also involved in the increased Aβ42/Aβ40 ratio. One mechanism could be changed conformation in the catalytic site and one way to investigate this is to use affinity capture with a γ-secretase transition state analogue inhibitor such as L-685,458. Membranes from BD8 cells expressing six selected PS1 FAD mutations were incubated in the presence of L685,458 coupled to a cleavable biotin group (GCB) followed by incubation with streptavidin beads to capture the active γ-secretase complexes. L-685,458-GCB has previously been shown to block Aβ production with a similar IC50 as L-685,458 [54]. As a negative control, L-685,458 was added prior to the biotinylated inhibitor. If the catalytic site was affected by the PS1 FAD mutants, fewer active γ-secretase complexes would be pulled down compared to PS1 wild type. We selected three mutations that showed no altered membrane integration (I143T, L166P, G384A) and three that gave rise to a substantial change in membrane integration, either an increase (P284L) or a reduction (L250S and L392P). Interestingly, after normalizing the pulled-down fragment to input we found that all six mutations affected the catalytic site of γ-secretase, by a reduction between 40 and 70%, and that the differences do not depend on membrane integration (Fig. 4A). These results are consistent with a previous study using a similar approach [40], as well as with reported results from Berezovska et al. that used a FLIM/FRET assay [39].
Fig. 4.
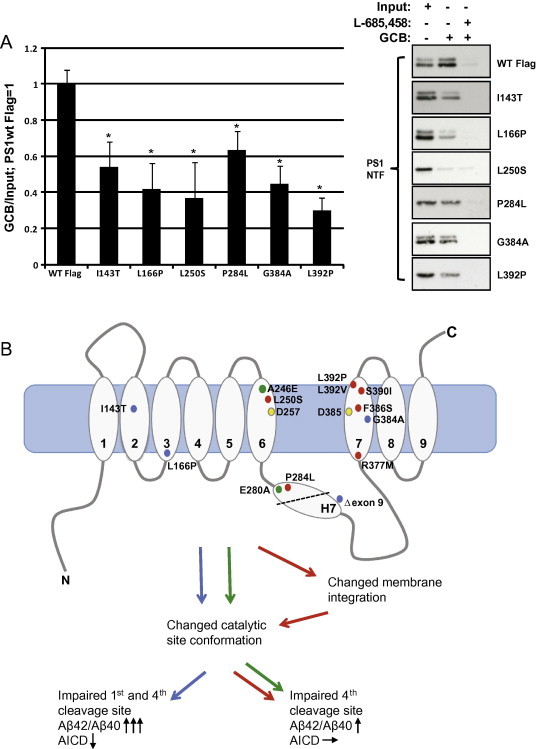
(A) PS1 FAD-linked mutations change conformation of the catalytic site of PS1. Six selected FAD mutations, three mutations that did not cause (I143T, L166P, G384A) and three that caused (L250S, P284L and L392P) altered membrane integration, were subjected to affinity capture with the γ-secretase transition state analogue inhibitor L-685,458 coupled to a cleavable biotin group (GCB). As a negative control L-685,458 was added prior to the biotinylated inhibitor. Pulled-down fragments were normalized with input and related to PS1 WT Flag. Bars represent mean of four experiments with error bars indicating S.E. Statistical significance is calculated by the non-parametric Mann–Whitney U test. ∗p < 0.05. (B) Schematic overview of the localization of the 13 PS1 FAD mutations and their effect on membrane integration, catalytic site conformation, Aβ42/Aβ40 ratio and AICD production. Arrows reflect changes compared with PS1 WT (↓, reduced peak; ↑, increased peak; –>, no change).
3. Discussion
More than 185 FAD causing mutations have been identified in the PS1 molecule, the catalytic subunit of γ-secretase. The FAD causing mutations, distributed all over the PS1 molecule, cause an altered Aβ production and in most cases an increased Aβ42/Aβ40 ratio. However, the mechanism by which they impact the PS structure and function remains largely elusive. In this report we have made a thorough effort to compare the impact of several PS FAD mutations on structural parameters of PS1 and asked whether it relates to a specific modulation of the APP/Aβ processing cascade.
We selected 13 FAD mutations that provided a substantial coverage of several regions of PS1. Eight of the mutations caused an increase in Aβ42, eleven mutations caused a decrease in Aβ40, and twelve mutations caused a decrease in Aβ38. Together, these results suggest that impaired Aβ38, Aβ40 and increased Aβ42 production are common Aβ phenotypes of FAD mutations, irrespective of their spatial location within PS1 or their impact on TMD integration. One of the mutations, the L250S, resulted in neither an increase in Aβ42 or a decrease in Aβ38, but caused a decrease in Aβ40. Thus, considering these three Aβ peptides we identified in total three FAD mutation-induced Aβ profiles: a selective decrease in Aβ40; a decrease in both Aβ40 and Aβ38; and an increase in Aβ42 in combination with a decrease in both Aβ38 and Aβ40, respectively. Furthermore, four out of the thirteen FAD mutations caused a decrease in total Aβ and AICD formation, reflecting a FAD-induced partial loss of γ-secretase activity. Interestingly, these mutations (I143T, L166P, Δexon9, G384A) gave rise to a very high Aβ42/40 ratio, which primarily was due to an extraordinary increase in Aβ42 production and/or decrease in Aβ40 (Table 3).
Recent progress in understanding APP processing has identified several γ-secretase mediated cleavage events, which result in different Aβ peptides (Aβ30-51) as well as several 3- and 4- peptides and a pentamer and a hexamer [25,55,56]. These findings have lead to the “Aβ40 and Aβ42 product line hypothesis”, where the Aβ40 product line represent AICD50–99 and Aβ49, 46, 43, 40, 37/36 and 34, and the Aβ42 product line result in AICD49–99 and Aβ48, 45, 42, 39/38 and 34, respectively. According to this model, Aβ peptides are generated either in a precursor product manner, where longer Aβ species are immediate substrate for the production of shorter isoforms or, alternatively, the different reactions take place independently in a more random manner [56]. According to the former model, the increase in Aβ42 and decrease in Aβ40 and Aβ38 would reflect an impaired processing of Aβ43 to Aβ40 and Aβ42 to Aβ38. In support for that model we found FAD mutations causing a decrease in Aβ40 to be associated with increased Aβ43/Aβ40 ratio, and the increase in Aβ42 to be linked to a decrease in Aβ38. There were however also some exceptions. For instance, the E280A mutation caused a decrease in Aβ40 but there was no increase in Aβ43. Moreover, the L166P and A246E mutants did not affect Aβ42 levels, whereas both mutants caused a suppression of Aβ38. The obtained data are therefore largely compatible with the precursor-product mechanism of Aβ generation, but still there are data that cannot be explained by this simplistic model. It is noteworthy that recent findings have identified “cross-talk” between the Aβ40 and 42 product lines and both Aβ43 and Aβ41 have been shown to be precursors for Aβ38 [55,56]. Thus, Aβ43 and Aβ38 production and turnover may not be directly linked to Aβ40 and Aβ42, respectively, and may explain why there was a lack of correlation between some Aβ peptides as outlined above. It is interesting that the Aβ42 levels remained normal albeit the total AICD level was decreased by the L166P and I143T mutations. Since most AICD production stems from the Aβ40 product line, this Aβ phenotype may be the consequence of a selective decrease of the entire Aβ40 product line, starting at the ε-cleavage, whereas Aβ42 product line processing remains intact. Such a mechanism was recently suggested by both Chavez-Gutierrez et al, who studied other PS1 mutations, and by us and others in the context of APP FAD mutations [26,56,57]. One caveat with our analysis is that we did not analyse AICD production or smaller γ-secretase-derived peptides with mass spectrometry. Moreover, we have not studied the longer membrane bound Aβ forms such as Aβ45 and Aβ46. Such data would clarify many of the questions raised above, but is beyond the scope of this study.
Given the spread of FAD causing mutations across the entire PS1 molecule, we hypothesized that FAD mutations in general may give rise to subtle structural changes. Since PS1 is a highly integrated membrane protein, exhibiting nine TMDs, we focused our studies on exploring the impact of FAD mutations on PS1 membrane integration. Using a membrane integration assay in an in vitro transcription/translation system based on N-linked glycosylation, we were able to study how efficient each TMD of PS1 integrates independently into the lipid bilayer of the ER membrane. All TMDs but TMD7 showed a nice membrane integration as would be expected for true transmembrane spanning sequences. The less hydrophobic segment TMD7 is not predicted to be a TMD according to the ΔG predictor program, which is mostly based on the hydrophobicity. These data confirm the proposed general membrane topography of PS1 and also validate the experimental system to delineate the impact of different PS1 FAD mutations on membrane integration.
We studied in total 35 different FAD mutations, covering all TMDs, and revealed several novel insights to the structural impact of FAD mutations on PS. Not surprisingly, the TMD7 is found to be very sensitive to substitution of amino acid residues, e.g. F386S and L392P substitutions strongly reduced the insertion that made the TMD7 incapable to insert independently into the membrane. On the other hand, we could observe that the more hydrophobic segment TMD6 is also susceptible for drastic changes. For instance, substituting the hydrophobic Leu at position 250 to the polar residue Ser decreased the integration efficiency by 43%. TMD7 is part of the hydrophilic catalytic cavity, which is embedded in the hydrophobic core of the membrane, probably protected and surrounded by the strongly and stable hydrophobic domains (TMD1-6, TMD8-9). This notion is supported by the recently published crystal structure of the PS homologue from the archaeon Methanoculleus marisnigri JR1 [58]. Moreover, the fact that water must be present in order to cleave membrane protein substrates suggests that the hydrophilic residues within TMD7 are needed and important for catalysis. The hydrophilic milieu causes the substrate to unfold, making the backbone amide bonds accessible to the nucleophilic attack that allows cleavage [59,60]. The predicted topology program suggests H7 as a weak or marginally hydrophobic TMD, but experimental data shows that H7 is not recognized as a TMD by the translocon, and thereby is not inserted into the membrane. This observation agrees well with the model that H7 reaches the catalytic site in order get endoproteolysed and not to be situated in the membrane. The substitution P284L (H7) caused an increase of insertion efficiency from 6% to 45%, which is in line with that the overall hydrophobicity of the TM segment increases.
Overall, the findings from the 35 studied PS1 FAD mutations suggest that some mutations (those located in TMD6, H7 and TMD7) have either a strong positive or negative impact on membrane integration, whereas other mutations do not appear to affect membrane integration at all (Tables 1 and 2). Although these studies are based on insertion efficiency of single TMDs and PS1 is a multispanning-membrane protein possibly influenced by the other TMDs in the assembled protein, our data provide an important insight into the biogenesis of PS1. To conclude, a comparison between the FAD-induced APP processing phenotypes and the effect, or lack of effect, on membrane integration of hydrophobic sequences did not reveal any striking correlation between the phenotypes. It is therefore conceivable that the impact of FAD mutants on PS1 function is complex, and that the abnormal membrane integration identified here represents one mechanism.
Three different reports have suggested that PS1 FAD mutations could alter the catalytic site of γ-secretase. Although these studies are based on only a few PS FAD mutants, they provide an attractive common molecular rationale to the Aβ modulatory effect of FAD mutations. In order to explore whether an altered active site indeed is a more general feature associated with FAD mutations, we selected six mutations that both had a differential impact on membrane integration and modulated Aβ production at different levels. Thus, we explored both mutations that had a negative effect on AICD/total Aβ production while either exhibiting normal or increased Aβ42 levels, and mutations that did not affect total Aβ/AICD production but both caused an increase in Aβ42 and a decrease in Aβ40, or only a decrease in Aβ40. Interestingly, neither of these mutants was pulled-down to the same extent as wt PS1, using a biotinylated version of the transition state analogue L685.458. These results suggest that all FAD mutations explored affect the active site of PS1. Why this common phenotype results in different Aβ modulatory profiles remains uncertain, but it is likely that the pull-down experiment is a rather gross measurement of alterations within the active site, and that different structural changes in the active site could affect APP processing in multiple directions.
In summary, we have made a thorough analysis of different PS FAD mutants with regard to pivotal structural aspects and APP processing activity of PS1. We have found that the FAD mutants impact membrane integration in several different ways and to various extents, and we have also identified several FAD-induced APP processing phenotypes that all result in an increased Aβ42/Aβ40 ratio, which is believed to play a pivotal role in AD pathogenesis. Most importantly, while we could not identify a specific correlation between abnormal membrane integration and a particular APP processing phenotype, our data suggest that an altered active site of PS1 is a common abnormal structural parameter that links FAD mutations to pathogenic Aβ production (summarized in Fig. 4B). Future studies are warranted to reveal the linkage between FAD mutations and altered PS1 active site conformation.
4. Experimental procedures
4.1. Antibodies, chemicals and enzymes
The following antibodies were used for immunoblotting: NT-1 recognizing the PS1-NTF [61], α-loop (Chemicon) raised against PS1-CTF, N1660 (Sigma) raised against the C-terminal of nicastrin, 3891 (ProSci Inc.) raised against Pen-2 and α-GAPDH (Acris GmbH) recognizing GAPDH. For the quantification of secreted Aβ38, 40 and 42 with Meso Scale Discovery (MSD) technology, C-terminal specific antibodies (Aβ 1-x) were used and detection was performed by SULFO-TAG™ 6E10 antibody. For quantification of sectreted Aβ40 and Aβ43 using Aβ40 Wako II ELISA kit (Wako Chemicals GmbH) and FL 1-43 ELISA kit (Immuno-biological Laboratories), respectively, the capture antibody was BNT77 for Aβ40 and Aβ38-43 for Aβ43. Detection antibodies were BA27 (Aβ40) and 82E1 (Aβ43), respectively. Unless otherwise stated, all chemicals were from Sigma–Aldrich. Plasmid pGEM1, TNT® Quick transcription/translation system, and deoxynucleotides were purchased from Promega and 35S-Met from PerkinElmer. All enzymes were obtained from Fermentas except Phusion DNA polymerase that was from Finnzymes. Oligonucleotides were from Eurofins MWG Operon.
4.2. cDNA constructs
For the membrane integration assay, double-stranded oligonucleotides encoding the human PS1 segments were introduced as amplified PCR fragments into the previously described modified lepB gene [35,36]. The PS1 fragments were amplified using the Phusion DNA polymerase with primers complementary to the 5′ and 3′ ends of the selected part of the gene and then cloned into the pGEM1 vector, containing the corresponding lepB gene, on SpeI/KpnI sites. In order to remove the endogenous glycosylation acceptor sites found in the PS1 upstream of TM8, Asn405-Thr-Thr was substituted to Gln405-Thr-Thr. Site-specific mutagenesis was performed using the QuikChange™ Site-Directed Mutagenesis Kit (Stratagene). The amplified DNA products were purified using the QIAquick PCR Purification Kit (QIAGEN) and all inserted fragments and mutants were confirmed by sequencing at Eurofins MWG Operon. For the functionality assays, full-length PS1 WT with or without a Flag-tag was cloned into the pcDNA5FRT/TO vector (Invitrogen) on BamHI/NotI sites. cDNAs encoding Flag-PS1 mutants I143T, L166P, G384A were generated by site directed mutagenesis using the QuikChange Site Directed mutagenesis protocol (Stratagene). The mutations, A246E, L250S, E280A, P284L, R377M, F486S, S390I, L392P and L392V, were generated with the same protocol as above using PS1 WT without a Flag-tag as a template. The PS1 Δexon9 deletion mutant has been described elsewhere [62]. The DNA sequence of all constructs was verified using the BigDye® Terminator Version 3.1 Cycle Sequencing kit (Applied Biosystems). The reporter constructs MH100, CMV-β-gal and C99-GVP used in the Luciferase-based Reporter Gene Assay have been described previously [38,63].
4.3. Cell culture and transfection
Blastocyst-derived embryonic stem cells deficient for PS1 and PS2, BD8 cells [64], were cultured in ES medium; Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 1 mM sodium pyruvate, 0.1 mM β-mercaptoethanol, and nonessential amino acids (Invitrogen). BD8 cells stably expressing APP WT have been previously generated in our laboratory [65]. The cDNA constructs were transiently transfected into the cells using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions.
4.4. Immunoblotting
Cells were lysed 24–48 h after transfection in cell lysis buffer (10 mM Tris, pH 8.1, 1 mM EDTA, 150 mM NaCl, 0.65% IGEPAL CA-630) supplemented with Protease Inhibitor Mixture (Roche Applied Science). Protein levels were determined by the BCA™ protein assay kit (Pierce), separated on NuPAGE 4–12% gradient Bis-Tris gels with MES buffer (both Invitrogen), and transferred to nitrocellulose membranes (Bio-Rad), which were probed with primary antibody. The blots were developed using horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) followed by Immobilon™ Western chemiluminescent HRP substrate (Millipore) using Amersham Biosciences HyperfilmTM ECL (GE Healthcare) or the CCD camera LAS-3000 (FUJIFILM Life Science). In the case of quantification, the respective bands were measured using ImageJ software (National Institutes of Health).
4.5. Quantification of secreted Aβ
A sandwich immunoassay using the Meso Scale Discovery SECTOR Imager 6000 was used to quantify secreted Aβ38, Aβ40 and Aβ42 peptides in conditioned medium. All reagents were from Meso Scale Discovery. The analysis was carried out as described earlier with small modifications [65,66]. In brief, BD8 cells stably expressing human APP WT were transiently transfected with PS1 WT or different FAD mutations. Cells were incubated in fresh medium containing either 1 μM L-685,458 or vehicle (DMSO 0.5%) for 24 h before analysis of the conditioned medium. The corresponding concentrations of Aβ peptides in the samples were calculated using Aβ peptide standard curves. To adjust for differences in transfection efficiency, the β-galactosidase activity of the cell lysate was determined and normalized to the levels of secreted Aβ peptides. Experiments were performed in triplicate and repeated 4–7 times. For Aβ43 analysis FL 1-43 ELISA (Immuno-biological Laboratories) was used and at the same time Aβ40 (human/rat β-amyloid (40) ELISA kit WakoII (Wako Chemicals GmbH) was analysed to obtain a ratio between Aβ43 and Aβ40 for comparison to Aβ42/Aβ40 from Meso Scale Discovery analysis. The protocol was according to the manufacturer’s instructions but with a modification in antibody binding quantification as described previously [67]. 50 μM Amplex UltraRed Reagent (Invitrogen) was incubated for 1 h at room temperature and followed by detection of fluorescent signal using 566 nm excitation filter and 583 nm emission filter in a microplate reader (TECAN, Safire II). Samples were measured in duplicates and repeated 4 times.
4.6. Luciferase-based reporter gene assay
BD8 cells were transfected with the cDNA constructs: PS1 WT/mutations/mock, MH100, CMV-β-gal and C99-GVP, lysed 36 h after transfection and then analyzed as previously described [38]. The γ-secretase activity was normalized against the transfection efficiency by determining the β-galactosidase activity of the cell lysate. Experiments were performed in triplicate and repeated 4–5 times.
4.7. In vitro membrane integration assay
The pGEM1 vectors containing the PS1 fragments were transcribed and translated in the TNT® SP6 Quick Coupled System (Promega) using 10 μl of reticulocyte lysate, 150–200 ng DNA template, 1 μl of [35S]Met (5 μCi) and 0.5 μl column-washed dog pancreas rough microsomes (rough ER membrane vesicles, CRMs) (a gift from Arthur E. Johnson [68]) and the samples were incubated for 90 min at 30 °C. For Endo H treatment, 6 μl of the TNT reaction were mixed with 3 μl dH2O and 1 μl of 10× Glycoprotein Denaturing Buffer. Following addition of; 1 μl of Endo H (500,000 units/ml; NEB, MA, US) and 7 μl of dH2O as well as 2 μl of 10× G5 Reaction Buffer, the sample was incubated for 1 h at 37 °C. The proteins were analyzed by SDS–PAGE gels and visualized in a Fuji FLA 3000 phosphorImager (Fujifilm) using the Image Reader V1.8J/Image Gauge V 3.45 software (Fujifilm). The MultiGauge (Fujifilm) software was used to generate a profile of each gel lane and the multi-Gaussian fit program (Qtiplot, www.qtiplot.ro) was used to calculate the peak areas of the glycosylated protein bands. The membrane integration efficiency of a given mutant was determined by normalizing the peak area of the singly or doubly glycosylated band (depending on which Lep construct was used) to the total area of both singly and doubly glycosylated protein bands. On average, the glycosylation levels vary by no more than ±5% between repeated experiments.
4.8. Affinity capture of γ-secretase using GCB
To investigate if the FAD mutations affect the conformation of the catalytic site of the γ-secretase, complexes containing FAD PS1 were pulled down with a γ-secretase transition state analogue inhibitor (L-685,458) coupled to biotin via a cleavable linker (GCB). The GCB pull down, its structure, and characteristics have been described earlier [54]. BD8 cells transiently transfected with PS1 WT, I143T, L166P, G384A, L205S, P284L and L392P or untransfected cells were subjected to membrane preparations and GCB pull down, as described previously [69]. Briefly, the cells were homogenized in homogenization buffer (10 mM KCl and 10 mM MOPS, pH 7.0, supplemented with PIC) by 25 strokes at 1500 rpm using a pestle homogenizer. The homogenates were centrifuged at 1000g for 10 min at 4 °C and the obtained post-nuclear supernatants were centrifuged further at 100,000g for 1 h at 4 °C. The resultant membrane pellets were suspended in homogenization buffer supplemented with 20% glycerol, flash frozen in liquid nitrogen and stored at −80 °C prior to use. After membrane preparation, samples were concentrated using the Amicon Ultra-2 50K device (Millipore) according to the manufacturer’s instructions and then subjected to Size-exclusion chromatography in order to purify the γ-secretase components bound in the complex from unbound components. The samples were injected onto a Superose™ 6 10/300 GL column (GE Healthcare) and buffer H (20 mM Hepes, pH 7.4, 150 mM NaCl and 2 mM EDTA) supplemented with Protease Inhibitor Mixture (Roche Applied Science) and 0.5% CHAPSO was used as the mobile phase at a flow rate of 0.5 ml/min. 0.5 ml fractions were collected from 7 to 30 min, concentrated using the Amicon Ultra-2 50K device (Millipore) and analyzed by SDS–PAGE as described above. After the initial experiment, where the fractions containing γ-secretase components were determined, the four fractions containing the most γ-secretase were pooled and concentrated as described before. After removal of an aliquot to use as input, samples were incubated with 200 nM GCB before the addition of magnetic streptavidin beads (Invitrogen). As a negative control, samples were incubated with 10 μM L-685,458 prior to the addition of GCB. The captured γ-secretase components were eluted with Laemmli sample buffer, analyzed by SDS–PAGE and Western blotting and related to the amount of input.
Author contributions
Planned experiments: (JW, PL, KÖ, LT, IMN, HK), performed experiments: (JW, PL, KÖ, SM, NM, LC, IMN, HK), analyzed data: (JW, PL, KÖ, SM, LT, JL, IMN, HK), wrote the paper: (JW, PL, KÖ, LT, JL, IMN, HK).
Acknowledgements
We gratefully thank Prof. Arthur E. Johnson, Texas A&M University for providing dog pancreas microsomes (CRMs). This work was supported by grants from Swedish Brain Power (HK), Wallenberg’s Foundation (HK), Åke Wibergs stiftelse, 563634056 (HK), Stiftelsen Gamla Tjänarinnor (HK, JW, IMN, LT), KI stiftelsen för åldersforskning, 2009Ålde0032 (HK), Swedish Medical Society, SLS-94931 (HK), Gun och Bertil Stohnes Stiftelse, 2013-11-04 (HK, JW, LT), Strategic Research Area: Neuroscience (Regulation Number 2009-1077) (HK), Magn. Bergvalls Stiftelse, 24940.1.08/09.7300; 24940.1.07/08.7300 (IMN), Henrik Granholms Stiftelse, SU 33-1013-09 (IMN), Swedish Cancer Foundation, 130624 (IMN), The Swedish Alzheimer Foundation, 03-220, (LT), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT), 210/083(12); KU 2003-4674 (IMN) and the Swedish Foundation for Strategic Research, A305:200 (IMN).
Contributor Information
IngMarie Nilsson, Email: ingmarie@dbb.su.se.
Helena Karlström, Email: Helena.Karlstrom@ki.se.
References
- 1.Masters C.L., Simms G., Weinman N.A., Multhaup G., McDonald B.L., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masters C.L., Selkoe D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012;2:a006262. doi: 10.1101/cshperspect.a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esler W.P., Wolfe M.S. A portrait of Alzheimer secretases – new features and familiar faces. Science. 2001;293:1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- 4.Czirr E., Cottrell B.A., Leuchtenberger S., Kukar T., Ladd T.B., Esselmann H., Paul S., Schubenel R., Torpey J.W., Pietrzik C.U., Golde T.E., Wiltfang J., Baumann K., Koo E.H., Weggen S. Independent generation of Abeta42 and Abeta38 peptide species by gamma-secretase. J. Biol. Chem. 2008;283:17049–17054. doi: 10.1074/jbc.M802912200. [DOI] [PubMed] [Google Scholar]
- 5.Kakuda N., Funamoto S., Yagishita S., Takami M., Osawa S., Dohmae N., Ihara Y. Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular domain from beta-carboxyl-terminal fragment by gamma-secretase. J. Biol. Chem. 2006;281:14776–14786. doi: 10.1074/jbc.M513453200. [DOI] [PubMed] [Google Scholar]
- 6.Qi-Takahara Y., Morishima-Kawashima M., Tanimura Y., Dolios G., Hirotani N., Horikoshi Y., Kametani F., Maeda M., Saido T.C., Wang R., Ihara Y. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J. Neurosci. 2005;25:436–445. doi: 10.1523/JNEUROSCI.1575-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lesne S., Koh M.T., Kotilinek L., Kayed R., Glabe C.G., Yang A., Gallagher M., Ashe K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 8.Walsh D.M., Klyubin I., Fadeeva J.V., Cullen W.K., Anwyl R., Wolfe M.S., Rowan M.J., Selkoe D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 9.Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A., Regan C.M., Walsh D.M., Sabatini B.L., Selkoe D.J. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown M.S., Ye J., Rawson R.B., Goldstein J.L. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 11.Francis R., McGrath G., Zhang J., Ruddy D.A., Sym M., Apfeld J., Nicoll M., Maxwell M., Hai B., Ellis M.C., Parks A.L., Xu W., Li J., Gurney M., Myers R.L., Himes C.S., Hiebsch R., Ruble C., Nye J.S., Curtis D. Aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 12.Goutte C., Tsunozaki M., Hale V.A., Priess J.R. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. U.S.A. 2002;99:775–779. doi: 10.1073/pnas.022523499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfe M.S., Xia W., Ostaszewski B.L., Diehl T.S., Kimberly W.T., Selkoe D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 14.Yu G., Nishimura M., Arawaka S., Levitan D., Zhang L., Tandon A., Song Y.Q., Rogaeva E., Chen F., Kawarai T., Supala A., Levesque L., Yu H., Yang D.S., Holmes E., Milman P., Liang Y., Zhang D.M., Xu D.H., Sato C., Rogaev E., Smith M., Janus C., Zhang Y., Aebersold R., Farrer L.S., Sorbi S., Bruni A., Fraser P., St. George-Hyslop P. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 15.Henricson A., Kall L., Sonnhammer E.L. A novel transmembrane topology of presenilin based on reconciling experimental and computational evidence. FEBS J. 2005;272:2727–2733. doi: 10.1111/j.1742-4658.2005.04691.x. [DOI] [PubMed] [Google Scholar]
- 16.Laudon H., Hansson E.M., Melen K., Bergman A., Farmery M.R., Winblad B., Lendahl U., von Heijne G., Naslund J. A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 2005;280:35352–35360. doi: 10.1074/jbc.M507217200. [DOI] [PubMed] [Google Scholar]
- 17.Oh Y.S., Turner R.J. Topology of the C-terminal fragment of human presenilin 1. Biochemistry. 2005;44:11821–11828. doi: 10.1021/bi0509494. [DOI] [PubMed] [Google Scholar]
- 18.Spasic D., Tolia A., Dillen K., Baert V., De Strooper B., Vrijens S., Annaert W. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J. Biol. Chem. 2006;281:26569–26577. doi: 10.1074/jbc.M600592200. [DOI] [PubMed] [Google Scholar]
- 19.Thinakaran G., Borchelt D.R., Lee M.K., Slunt H.H., Spitzer L., Kim G., Ratovitsky T., Davenport F., Nordstedt C., Seeger M., Hardy J., Levey A.I., Gandy S.E., Jenkins N.A., Copeland N.G., Price D.L., Sisodia S.S. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 20.Scheuner D., Eckman C., Jensen M., Song X., Citron M., Suzuki N., Bird T.D., Hardy J., Hutton M., Kukull W., Larson E., Levy-Lahad E., Viitanen M., Peskind E., Poorkaj P., Schellenberg G., Tanzi R., Wasco W., Lannfelt L., Selkoe D., Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 21.Kumar-Singh S., Theuns J., Van Broeck B., Pirici D., Vennekens K., Corsmit E., Cruts M., Dermaut B., Wang R., Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum. Mutat. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 22.Bentahir M., Nyabi O., Verhamme J., Tolia A., Horre K., Wiltfang J., Esselmann H., De Strooper B. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem. 2006;96:732–742. doi: 10.1111/j.1471-4159.2005.03578.x. [DOI] [PubMed] [Google Scholar]
- 23.Citron M., Westaway D., Xia W., Carlson G., Diehl T., Levesque G., Johnson-Wood K., Lee M., Seubert P., Davis A., Kholodenko D., Motter R., Sherrington R., Perry B., Yao H., Strome R., Lieberburg I., Rommens J., Kim S., Schenk D., Fraser P., St. George Hyslop P., Selkoe D.J. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 24.Kretner B., Fukumori A., Gutsmiedl A., Page R.M., Luebbers T., Galley G., Baumann K., Haass C., Steiner H. Attenuated Abeta42 responses to low potency gamma-secretase modulators can be overcome for many pathogenic presenilin mutants by second-generation compounds. J. Biol. Chem. 2011;286:15240–15251. doi: 10.1074/jbc.M110.213587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takami M., Nagashima Y., Sano Y., Ishihara S., Morishima-Kawashima M., Funamoto S., Ihara Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 2009;29:13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chavez-Gutierrez L., Bammens L., Benilova I., Vandersteen A., Benurwar M., Borgers M., Lismont S., Zhou L., Van Cleynenbreugel S., Esselmann H., Wiltfang J., Serneels L., Karran E., Gijsen H., Schymkowitz J., Rousseau F., Broersen K., De Strooper B. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J., Onstead L., Randle S., Price R., Smithson L., Zwizinski C., Dickson D.W., Golde T., McGowan E. Abeta40 inhibits amyloid deposition in vivo. J. Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.von Heijne G. Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 2006;7:909–918. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- 29.Park E., Rapoport T.A. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- 30.Shao S., Hegde R.S. Membrane protein insertion at the endoplasmic reticulum. Annu. Rev. Cell Dev. Biol. 2011;27:25–56. doi: 10.1146/annurev-cellbio-092910-154125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin P.J., Jongsma C.G., Liao S., Johnson A.E. Transmembrane segments of nascent polytopic membrane proteins control cytosol/ER targeting during membrane integration. J. Cell Biol. 2011;195:41–54. doi: 10.1083/jcb.201103117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hedin L.E., Ojemalm K., Bernsel A., Hennerdal A., Illergard K., Enquist K., Kauko A., Cristobal S., von Heijne G., Lerch-Bader M., Nilsson I., Elofsson A. Membrane insertion of marginally hydrophobic transmembrane helices depends on sequence context. J. Mol. Biol. 2010;396:221–229. doi: 10.1016/j.jmb.2009.11.036. [DOI] [PubMed] [Google Scholar]
- 33.Pitonzo D., Skach W.R. Molecular mechanisms of aquaporin biogenesis by the endoplasmic reticulum Sec61 translocon. Biochim. Biophys. Acta. 2006;1758:976–988. doi: 10.1016/j.bbamem.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 34.Sadlish H., Skach W.R. Biogenesis of CFTR and other polytopic membrane proteins: new roles for the ribosome-translocon complex. J. Membr. Biol. 2004;202:115–126. doi: 10.1007/s00232-004-0715-6. [DOI] [PubMed] [Google Scholar]
- 35.Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S.H., von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 36.Lundin C., Kim H., Nilsson I., White S.H., von Heijne G. Molecular code for protein insertion in the endoplasmic reticulum membrane is similar for N(in)–C(out) and N(out)–C(in) transmembrane helices. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15702–15707. doi: 10.1073/pnas.0804842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez-Tur J., Froelich S., Prihar G., Crook R., Baker M., Duff K., Wragg M., Busfield F., Lendon C., Clark R.F. A mutation in Alzheimer’s disease destroying a splice acceptor site in the presenilin-1 gene. Neuroreport. 1995;7:297–301. [PubMed] [Google Scholar]
- 38.Karlstrom H., Bergman A., Lendahl U., Naslund J., Lundkvist J. A sensitive and quantitative assay for measuring cleavage of presenilin substrates. J. Biol. Chem. 2002;277:6763–6766. doi: 10.1074/jbc.C100649200. [DOI] [PubMed] [Google Scholar]
- 39.Berezovska O., Lleo A., Herl L.D., Frosch M.P., Stern E.A., Bacskai B.J., Hyman B.T. Familial Alzheimer’s disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J. Neurosci. 2005;25:3009–3017. doi: 10.1523/JNEUROSCI.0364-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kornilova A.Y., Bihel F., Das C., Wolfe M.S. The initial substrate-binding site of gamma-secretase is located on presenilin near the active site. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3230–3235. doi: 10.1073/pnas.0407640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hessa T., Meindl-Beinker N.M., Bernsel A., Kim H., Sato Y., Lerch-Bader M., Nilsson I., White S.H., von Heijne G. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- 42.Enquist K., Fransson M., Boekel C., Bengtsson I., Geiger K., Lang L., Pettersson A., Johansson S., von Heijne G., Nilsson I. Membrane-integration characteristics of two ABC transporters, CFTR and P-glycoprotein. J. Mol. Biol. 2009;387:1153–1164. doi: 10.1016/j.jmb.2009.02.035. [DOI] [PubMed] [Google Scholar]
- 43.Johansson M., Nilsson I., von Heijne G. Positively charged amino acids placed next to a signal sequence block protein translocation more efficiently in Escherichia coli than in mammalian microsomes. Mol. Gen. Genet. 1993;239:251–256. doi: 10.1007/BF00281625. [DOI] [PubMed] [Google Scholar]
- 44.Nilsson I., Johnson A.E., von Heijne G. Cleavage of a tail-anchored protein by signal peptidase. FEBS Lett. 2002;516:106–108. doi: 10.1016/s0014-5793(02)02511-5. [DOI] [PubMed] [Google Scholar]
- 45.Fukumori A., Fluhrer R., Steiner H., Haass C. Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of gamma-secretase-mediated intramembrane proteolysis. J. Neurosci. 2010;30:7853–7862. doi: 10.1523/JNEUROSCI.1443-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knappenberger K.S., Tian G., Ye X., Sobotka-Briner C., Ghanekar S.V., Greenberg B.D., Scott C.W. Mechanism of gamma-secretase cleavage activation: is gamma-secretase regulated through autoinhibition involving the presenilin-1 exon 9 loop? Biochemistry. 2004;43:6208–6218. doi: 10.1021/bi036072v. [DOI] [PubMed] [Google Scholar]
- 47.Kauko A., Hedin L.E., Thebaud E., Cristobal S., Elofsson A., von Heijne G. Repositioning of transmembrane alpha-helices during membrane protein folding. J. Mol. Biol. 2010;397:190–201. doi: 10.1016/j.jmb.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 48.Sadlish H., Pitonzo D., Johnson A.E., Skach W.R. Sequential triage of transmembrane segments by Sec61alpha during biogenesis of a native multispanning membrane protein. Nat. Struct. Mol. Biol. 2005;12:870–878. doi: 10.1038/nsmb994. [DOI] [PubMed] [Google Scholar]
- 49.Sato C., Morohashi Y., Tomita T., Iwatsubo T. Structure of the catalytic pore of gamma-secretase probed by the accessibility of substituted cysteines. J. Neurosci. 2006;26:12081–12088. doi: 10.1523/JNEUROSCI.3614-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sobhanifar S., Schneider B., Lohr F., Gottstein D., Ikeya T., Mlynarczyk K., Pulawski W., Ghoshdastider U., Kolinski M., Filipek S., Guntert P., Bernhard F., Dotsch V. Structural investigation of the C-terminal catalytic fragment of presenilin 1. Proc. Natl. Acad. Sci. U.S.A. 2010;107:9644–9649. doi: 10.1073/pnas.1000778107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nilsson I.M., von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 1993;268:5798–5801. [PubMed] [Google Scholar]
- 52.Stefansson A., Armulik A., Nilsson I., von Heijne G., Johansson S. Determination of N- and C-terminal borders of the transmembrane domain of integrin subunits. J. Biol. Chem. 2004;279:21200–21205. doi: 10.1074/jbc.M400771200. [DOI] [PubMed] [Google Scholar]
- 53.Nilsson I., Saaf A., Whitley P., Gafvelin G., Waller C., von Heijne G. Proline-induced disruption of a transmembrane alpha-helix in its natural environment. J. Mol. Biol. 1998;284:1165–1175. doi: 10.1006/jmbi.1998.2217. [DOI] [PubMed] [Google Scholar]
- 54.Teranishi Y., Hur J.Y., Welander H., Franberg J., Aoki M., Winblad B., Frykman S., Tjernberg L.O. Affinity pulldown of gamma-secretase and associated proteins from human and rat brain. J. Cell. Mol. Med. 2009 doi: 10.1111/j.1582-4934.2009.00907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okochi M., Tagami S., Yanagida K., Takami M., Kodama T.S., Mori K., Nakayama T., Ihara Y., Takeda M. Gamma-secretase modulators and presenilin 1 mutants act differently on presenilin/gamma-secretase function to cleave Abeta42 and Abeta43. Cell Rep. 2013;3:42–51. doi: 10.1016/j.celrep.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 56.Olsson F., Schmidt S., Althoff V., Munter L.M., Jin S., Rosqvist S., Lendahl U., Multhaup G., Lundkvist J. Characterization of intermediate steps in amyloid beta (Abeta) production under near-native conditions. J. Biol. Chem. 2013;289:1540–1550. doi: 10.1074/jbc.M113.498246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dimitrov M., Alattia J.R., Lemmin T., Lehal R., Fligier A., Houacine J., Hussain I., Radtke F., Dal Peraro M., Beher D., Fraering P.C. Alzheimer’s disease mutations in APP but not gamma-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat. Commun. 2013;4:2246. doi: 10.1038/ncomms3246. [DOI] [PubMed] [Google Scholar]
- 58.Li X., Dang S., Yan C., Gong X., Wang J., Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- 59.Wolfe M.S., Kopan R. Intramembrane proteolysis: theme and variations. Science. 2004;305:1119–1123. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y., Zhang Y., Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 61.Mathews P.M., Cataldo A.M., Kao B.H., Rudnicki A.G., Qin X., Yang J.L., Jiang Y., Picciano M., Hulette C., Lippa C.F., Bird T.D., Nochlin D., Walter J., Haass C., Levesque L., Fraser P.E., Andreadis A., Nixon R.A. Brain expression of presenilins in sporadic and early-onset, familial Alzheimer’s disease. Mol. Med. 2000;6:878–891. [PMC free article] [PubMed] [Google Scholar]
- 62.Laudon H., Mathews P.M., Karlstrom H., Bergman A., Farmery M.R., Nixon R.A., Winblad B., Gandy S.E., Lendahl U., Lundkvist J., Naslund J. Co-expressed presenilin 1 NTF and CTF form functional gamma-secretase complexes in cells devoid of full-length protein. J. Neurochem. 2004;89:44–53. doi: 10.1046/j.1471-4159.2003.02298.x. [DOI] [PubMed] [Google Scholar]
- 63.Taniguchi Y., Karlstrom H., Lundkvist J., Mizutani T., Otaka A., Vestling M., Bernstein A., Donoviel D., Lendahl U., Honjo T. Notch receptor cleavage depends on but is not directly executed by presenilins. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4014–4019. doi: 10.1073/pnas.052017699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Donoviel D.B., Hadjantonakis A.K., Ikeda M., Zheng H., Hyslop P.S., Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wanngren J., Franberg J., Svensson A.I., Laudon H., Olsson F., Winblad B., Liu F., Naslund J., Lundkvist J., Karlstrom H. The large hydrophilic loop of presenilin 1 is important for regulating gamma-secretase complex assembly and dictating the amyloid beta peptide (Abeta) Profile without affecting Notch processing. J. Biol. Chem. 2010;285:8527–8536. doi: 10.1074/jbc.M109.055590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pamren A., Wanngren J., Tjernberg L.O., Winblad B., Bhat R., Naslund J., Karlstrom H. Mutations in nicastrin protein differentially affect amyloid beta-peptide production and Notch protein processing. J. Biol. Chem. 2011;286:31153–31158. doi: 10.1074/jbc.C111.235267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hashimoto M., Bogdanovic N., Volkmann I., Aoki M., Winblad B., Tjernberg L.O. Analysis of microdissected human neurons by a sensitive ELISA reveals a correlation between elevated intracellular concentrations of Abeta42 and Alzheimer’s disease neuropathology. Acta Neuropathol. 2010;119:543–554. doi: 10.1007/s00401-010-0661-6. [DOI] [PubMed] [Google Scholar]
- 68.Walter P., Blobel G. Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol. 1983;96:84–93. doi: 10.1016/s0076-6879(83)96010-x. [DOI] [PubMed] [Google Scholar]
- 69.Franberg J., Svensson A.I., Winblad B., Karlstrom H., Frykman S. Minor contribution of presenilin 2 for gamma-secretase activity in mouse embryonic fibroblasts and adult mouse brain. Biochem. Biophys. Res. Commun. 2011;404:564–568. doi: 10.1016/j.bbrc.2010.12.029. [DOI] [PubMed] [Google Scholar]
- 70.Raux G., Guyant-Marechal L., Martin C., Bou J., Penet C., Brice A., Hannequin D., Frebourg T., Campion D. Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: an update. J. Med. Genet. 2005;42:793–795. doi: 10.1136/jmg.2005.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campion D., Dumanchin C., Hannequin D., Dubois B., Belliard S., Puel M., Thomas-Anterion C., Michon A., Martin C., Charbonnier F., Raux G., Camuzat A., Penet C., Mesnage V., Martinez M., Clerget-Darpoux F., Brice A., Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999;65:664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]