

Graphical abstract
Abbreviations: MD, molecular dynamics; Sc, Saccharomyces cerevisiae; Ub, ubiquitin; UBC, ubiquitin-binding domain
Keywords: Cdc34, Docking, E2 conjugating enzyme, Ubiquitin, Virtual screening, Ubiquitination
Highlights
-
•
Cdc34 is an E2 enzyme involved in protein ubiquitination and associated with some cancers.
-
•
Cdc34 activity is modulated by phosphorylation-induced conformational changes of the acidic loop.
-
•
We used computational approaches to identify potential inhibitory compounds for Cdc34.
-
•
These inhibitors as molecular hinges stabilizing the acid loop in its inactive, closed conformation.
Abstract
Among the different classes of enzymes involved in the ubiquitin pathway, E2 ubiquitin-conjugating enzymes occupy a central role in the ubiquitination cascade. Cdc34-like E2 enzymes are characterized by a 12–14 residue insertion in the proximity of the catalytic site, known as the acidic loop. Cdc34 ubiquitin-charging activity is regulated by CK2-dependent phosphorylation and the regulatory mechanism involves the acidic loop. Indeed, the phosphorylation stabilizes the loop in an open conformation that is competent for ubiquitin charging.
Cdc34 is associated with a variety of diseases, such as hepatocellular carcinomas and prostatic adenocarcinomas. In light of its role, the discovery of potential inhibitory compounds would provide the mean to effectively modulate its activity.
Here, we carried out a computational study based on molecular dynamics, virtual screening and docking to identify potential inhibitory compounds of Cdc34, modulating the acidic loop conformation. The molecules identified in this study have been designed to act as molecular hinges that can bind the acidic loop in its closed conformation, thus inhibiting the Cdc34-mediated ubiquitination cascade at the ubiquitin-charging step. In particular, we proposed a pharmacophore model featuring two amino groups in the central part of the model and two lateral aromatic chains, which respectively establish electrostatic interactions with the acidic loop (Asp 108 and Glu 109) and a hydrogen bond with Ser 139, which is one of the key residues for Cdc34 activity.
1. Introduction
Ubiquitination is a post-translational modification that was originally known as a signal for protein degradation by the 26S proteasome [1]. It is also involved in many other different signaling pathways, including cell cycle, endocytosis, transcription, DNA repair, signal transduction, apoptosis and the immune response [1–4].
In the ubiquitination pathway, E2 enzymes charged with ubiquitin (Ub) can be recruited by an E3 ligase, along with the target substrates. The C-terminal glycine of Ub can then be attached to a lysine residue on the target substrate. This can result in the transfer of only a single Ub molecule (mono-ubiquitination) or the addition of further Ub molecules to form a poly-Ub chain. Depending on the target lysine used to cross-link the Ub molecules in the chain, different poly-Ub chains can be formed, which adopt diverse three-dimensional (3D) structures and exert different biological effects [5–8].
Ubiquitination can thus be described as a molecular zip code, which is used to sort different ubiquitination products to different destinations. Errors in delivery of ubiquitinated proteins to the proteasome or other destinations are highly detrimental for the cell [3].
E2 enzymes (E2s) have a primary role in catalyzing, alone or with the cognate E3, the covalent attachment of Ub to the target proteins and they have a major role in defining the topology of the polyUb chain and thus the fate of the substrate [9]. E2s are often multi-domain proteins that all share a conserved Ub-binding domain (UBC characterized by a α/β fold) [9–11]. The highly conserved active site Cys is located in a shallow groove of the UBC domain formed by a short loop connecting α-helix 2 and α-helix 3 and a long loop (β4-α2 loop) proximal to the active site [9–11].
E2 UBC domains have been recently classified by Michelle et al. [12] into 17 families of homologs by performing phylogenetic analyses on 207 E2 genes belonging to seven different species. Family 3 members, i.e. Cdc34-like enzymes are characterized by a conserved and disordered insertion in the β4-α2 loop in the proximity of the catalytic site. The insertion is known as the acidic loop of Cdc34-like enzymes.
In a previous work, we identified two co-evolving signature elements in Cdc34-like E2 enzymes: the acidic insertion in β4-α2 loop in the proximity of the catalytic cysteine and two conserved CK2 phospho-sites within the UBC domain [13]. We previously demonstrated by combining Ub-charging assays and MD simulations that the phosphorylation at one of this sites (S130) can modulate the opening and closing of the β4-α2 loop with respect to the catalytic cleft and, in turn, it modulates the accessibility of the catalytic Cys for Ub-charging [13]. More in details, this regulatory mechanism relies on electrostatic repulsive effects between the phosphorylated serine and the acidic residues in the β4-α2 loop. The loop can undergo a substantial shift and drift away from the catalytic cleft upon phosphorylation, promoting the accessibility of the catalytic Cys.
Cdc34 is known to be involved in a variety of diseases, such as hepatocellular carcinomas and prostatic adenocarcinomas [14–19]. In fact, Cdc34 stimulates cellular proliferation by enhancing the degradation of p53 and p27, which both act as inhibitors of cell cycle progression.
In light of the above scenario, recent studies have been focused in the design and identification of inhibitory molecules of E2 enzymes and of Cdc34 in particular. A small inhibitor (CC0651) was identified for Cdc34 [20,21]. CC0651 was able to inhibit proliferation of human cancer lines and caused accumulation of the p27 substrate. Another small inhibitor was identified for Rad6 E2s [22] with the capability to inhibit the thioester formation between the E2 catalytic cysteine and the C-terminal of Ub. These studies have opened the venue to study E2 enzymes as a suitable class of drug targets in the ubiquitination pathway.
We contribute to this scenario, proposing a group of molecules with the potential to directly counteract the Ub-charging activity of Cdc34 by acting on the acidic loop (β4-α2 loop) and keeping it in a closed conformation, shielding the catalytic cysteine needed for the attachment of the Ub molecule. The task was carried out by the integration of different computational approaches, as described by Sanders et al. [22]. In particular, we used a structural ensemble that was already available thank to our previous MD studies [13,23] to isolate the most representative conformations suitable for docking simulations. Subsequently, virtual screening and docking were performed to select putative compounds from 735,758 entries of the ZINC database [24]. We selected 20 molecules by both energy-based and structural-based screening of docking simulations of 500 compounds. We then provide a pharmacophore model with the aim of inhibiting Cdc34 Ub-charging activity acting as molecular zipper to stabilize the closed and inactive conformation of the acidic loop. The results here described can provide a valuable dataset for future experimental studies in the field.
2. Materials and methods
2.1. Rmsd-matrices and clustering
The Cdc34 MD ensemble previously published [13] along with an increased sampling achieved by performing new simulations [23] was used as a reference conformational ensemble for the present investigation. In particular, we post-processed the ensemble by rmsd-based structural clustering as described in the following.
C-alpha (Cα) root mean square deviation (rmsd) were calculated pairwise for each pair of frames of the available MD ensemble, collecting values ranging from 0.2 to 0.65 nm, indicating respectively nearly identical or highly diverging structures in the ensemble. The highest rmsd values are associated to a displacement of the acidic loop conformations, whereas secondary structures were conserved in all the MD structures and characterized by rmsd values lower than 0.25 nm [13]. The Cα rmsd matrix (Fig. 1S) was then processed to obtain structural clusters of similar conformations using the Gromos algorithm implemented in Gromacs (www.gromacs.org) with a clustering cutoff of 0.35 nm.
2.2. Virtual screening and docking simulations
The virtual screening and the docking calculations were performed with DOCK Blaster [25] and Autodock version 4.2 [26], respectively. DOCK Blaster is an online server that selects and scores thousands of compounds deposited in the ZINC database [24] for a target structure uploaded by the user. The center of mass of three residues (P110, I137 and N138) was used to set the grid for the calculation. In particular, we selected the ZINC subset 11 [24], containing 735,758 entries. Indeed, compounds belonging to this subset are described as lead-like and were selected to obey to the Lipinski rule [27], according to which an orally active drug has no more than one violation of the following criteria: not more than five hydrogen-bond (H-bonds) donors (nitrogen or oxygen atoms with one or more hydrogen atoms), no more than ten H-bond acceptors (nitrogen or oxygen atoms), a molecular weight under 500 Da and an octanol–water partition coefficient log P less than 5.
Once the virtual screening procedure was completed, we employed Autodock version 4.2 [26] for docking calculations of the first 500 compounds selected by DOCK Blaster energy-rank. For each molecule, DOCK Blaster provided one binding pose characterized by the lowest energy according to the DOCK Blaster energy function. The DOCK Blaster binding pose was used as a starting structure for docking simulations with Autodock for each molecule (500 Autodock simulations overall). The parameters used for Autodock simulations are reported in the Supplementary Table 1S.
Autodock provided different binding poses for each of the 500 molecules simulated. For each molecule, the binding poses generated by Autodock are at first clusterized by the Autodock internal routines. For each cluster referred to a specific molecule, the software generally returns the binding pose with the lowest energy (cluster binding pose). We then applied a further selection procedure by the Pymol Python APIs (application programming interfaces) on all the cluster binding poses provided by Autodock for each molecule. In particular, we used both energetic and structural criteria in this final selection step. The final goal is to obtain, for each of the 500 screened molecules, a unique binding pose that is not only characterized by the lowest energy but also that can structurally act as a molecular zipper contacting both the acidic loop and the surrounding region in the catalytic cleft, as also discussed in the Results. With this aim in mind, we ranked by the Python routine with the Pymol APIs the cluster binding poses referred to the same molecule according to their energy. Among the two cluster binding poses of a molecule that are characterized by the lowest energy, the API routine selects the one that has a center of mass at the minimal distance from the binding site (defined using as reference residues P110, I137 and N138).
3. Results
3.1. Selection of the starting structure for virtual screening
To select a suitable Cdc34 structure for docking simulations, we post-processed the previously published MD ensemble of Cdc34 [13,23].
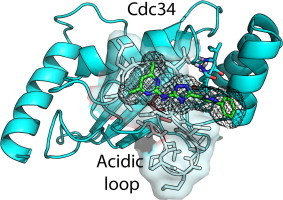
A Cα rmsd matrix (Fig. 1S) was built from the MD ensemble to evaluate the conformational variability among the collected structures. A clustering algorithm was then used to extract from the rmsd matrix different clusters of similar conformations (Fig. 1S, see Section 2 for details). In particular, we identified a highly populated cluster that also includes the conformations from the most populated basin of the previously published free energy landscape [13]. The Cdc34 structure selected for virtual screening and docking simulations is the average conformation associated with this cluster. Indeed, it can be considered representative of the closed and inactive conformation of Cdc34, i.e. with the acidic loop in a closed conformation and the catalytic cysteine in a buried position (Fig. 1).
Fig. 1.
Cdc34 three-dimensional structure. The acidic loop is represented in yellow, while the catalytic cysteine in red. The three residues (P110, I137 and N138) selected to define the binding site for virtual screening are depicted in cyan (right panel). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Selection of a binding site for the inhibitors
The binding site was selected according to the position and orientation of the acidic loop. We aimed at identifying molecules that can act as molecular zippers, i.e. able to stabilize the loop in the closed and inactive conformation so that the Ub-charging activity can be impaired. Thus, a potential inhibitor should establish strong intermolecular interactions with the acidic residues in the β4-α2 loop and, at the same time, it should be able to bind the surrounding regions of the catalytic cleft to maintain the loop in the closed conformation. In particular, we selected three residues of Cdc34 (P110, I137 and N138) whose center of mass corresponds to a cavity located in the close proximity of the catalytic cysteine and the acidic loop (Fig. 1). The selection of this binding site was essential to establish a spatial grid as an input for the docking algorithm that we employed in the first screening step (DOCK Blaster). Among the 735,758 molecules in the ZINC database (ZINC subset 11), the software returned 500 compounds ranked according to their binding free energies.
DOCK Blaster parameters are optimized for virtual screening calculations and thus they only permit a fast but coarse selection among the large number of molecules deposited in the ZINC subset 11. Thus to refine our data, we performed docking simulations with Autodock for each of the 500 molecules returned by DOCK Blaster (see Section 2). In particular, these 500 compounds were docked against Cdc34 structure, and the Autodock grid was built around the selected binding site in Fig. 2S, which also corresponds to the same search space used for the DOCK Blaster calculations.
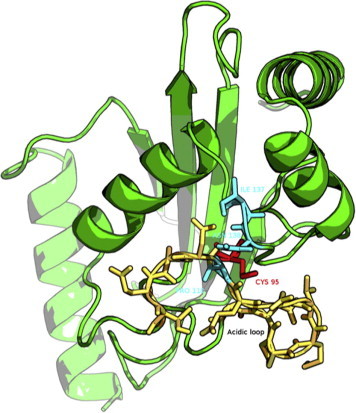
From a first visual inspection of the binding poses selected by Autodock internal routines (cluster poses, see Section 2 for details), it was not possible to detect any common feature that could be used to build a pharmacophore model (Fig. 2 and Table 1). Therefore a subsequent selection step was applied to filter out the molecules that did not properly interact with Cdc34, In particular, we analyzed the clusters calculated by Autodock for each different compound to select molecules that may either bind Cdc34 with high affinity (poses that have the lower energies in the ranking) and act as molecular hinges/zippers by both interacting with the acidic loop and the surrounding structural components (poses at the lower distances from the binding site) (see Section 2). The poses of each of the 500 molecules were classified into three bins according to their distance from the binding site (Fig. 3) so that molecules belonging to the first bin had a center of mass at a distance shorter than 0.8 nm from the center of mass of the previously defined binding site. This step is of crucial importance, since the inhibitors are expected to act as molecular hinges and therefore need to establish interactions with residues at the opposite sides of the acidic loop. According to these criteria, we selected a binding pose for each of the 500 molecules (see Section 2 for further details). After this post-clustering ranking procedure, the first 20 lower energy molecules (Fig. 4, left panel) are all located in the same area of the binding site and share recurring structural features that are discussed in details below.
Fig. 2.
Autodock results upon clustering with Autodock internal routines. The first 20 molecules from the Autodock energy-ranking list are depicted with different colors. The residues of the acidic loop are shown as yellow sticks. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
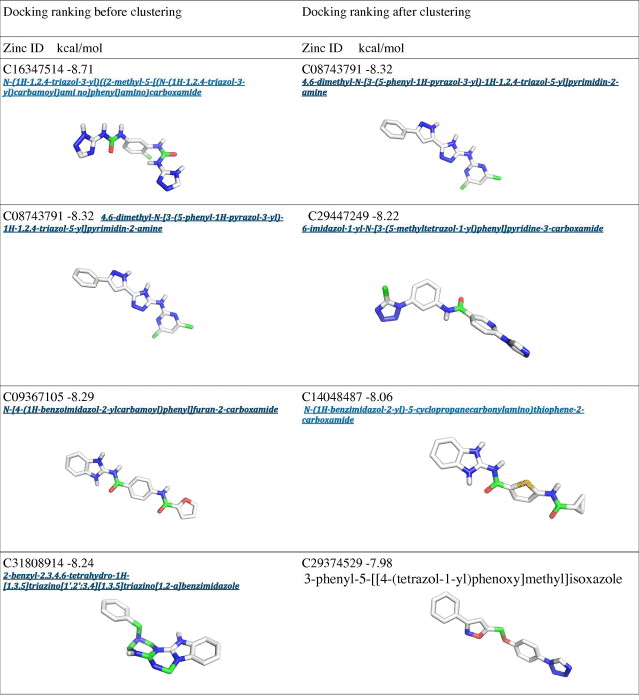
Table 1.
Docking results with Autodock before and after the clustering procedure.
 |
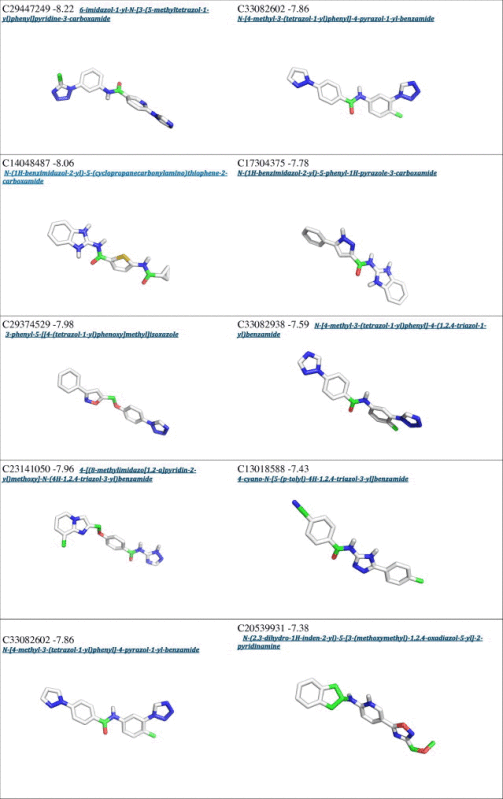 |
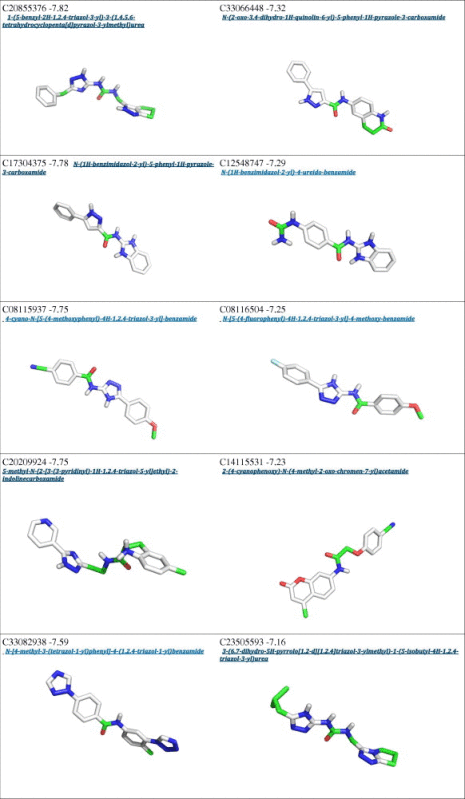 |
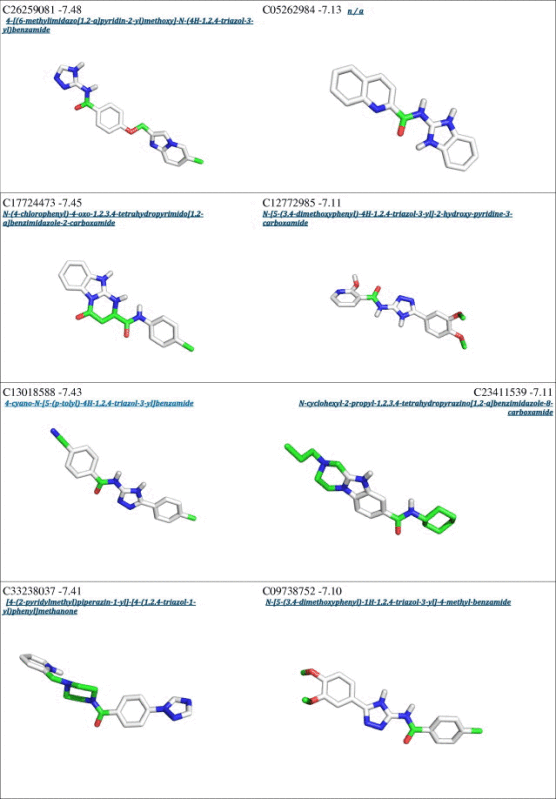 |
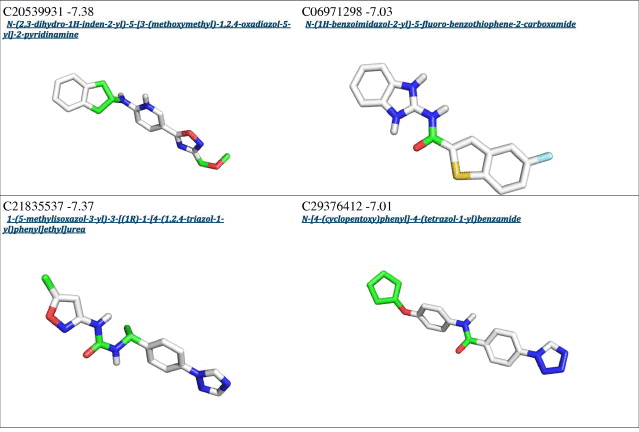 |
Fig. 3.
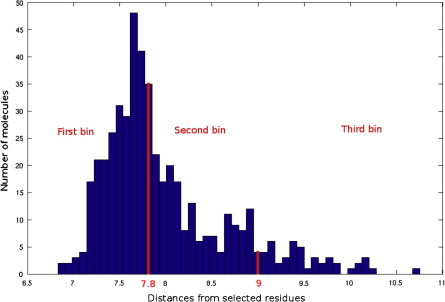
Histogram distribution of the distances calculated between the docked molecules and the residues selected for the Autodock grid construction (P110, I137 and N138).
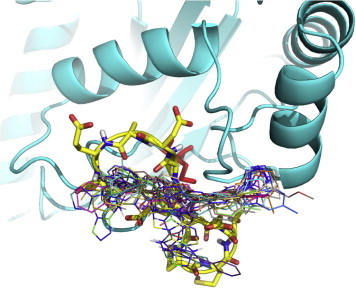
Fig. 4.
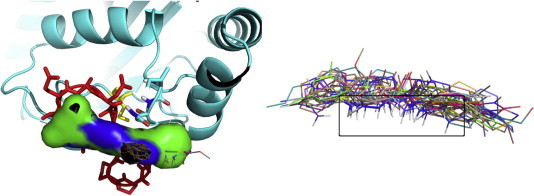
Molecules identified by the post-clustering ranking according to both structural and energetic criteria. In the left panel, the first 20 high-ranking molecules after clustering and a portion of Cdc34 are depicted with surface representation of molecule C08743791. In the right panel, the high-ranking clustered molecules are shown superimposed to underline the presence of two recurrent amino groups. The selection step has been carried out post-processing Autodock output with Phyton Pymol API tools.
Interestingly, an overlap can be observed between the Autodock clustering and the post-clustering selection procedure that we applied. Indeed, the first nine molecules belonging to the first bin of the post-clustering procedure were also found in the Autodock ranking, as shown in Table 1. Nevertheless, the ranking order after post-clustering selection with API tools is significantly modified. In particular, the molecules belonging from 9th to 20th position in the post-clustering ranking do not appear in the first 20 positions of the classical Autodock energy-based ranking (Table 1).
We emphasize the need for a spatial clustering procedure by showing in Fig. 5 the different positions for two molecules used as examples of the two possible scenarios, i.e. a case that is selected by the post-clustering ranking (left panel) and a case that is discarded upon this procedure (right panel). The left panel illustrates the orientation of the molecule C08743791 (4,6-dimethyl-N-[3-(5-phenyl-1H-pyrazol-3-yl)-1H-1,2,4-triazol-5-yl]pyrimidin-2-amine), whereas the right panel shows the molecule C20209924 (5-methyl-N-{2-[3-(3-pyridinyl)-1H-1,2,4-triazol-5-yl]ethyl}-2-indolinecarboxamide). The first molecule has an orientation suited for interaction with both the acidic loop and the surrounding structural components (V143 and V147), the latter is only interacting with the acidic loop. Though the energetic estimation for the second molecule is on average comparable to the first (∼0.5 kcal/mol), the spatial analysis suggests that it cannot act as a proper molecular hinge, since by only interacting with the acidic loop it cannot properly keep the loop in a closed conformation. For these reasons, the molecules presenting a binding mode similar to that of 5-methyl-N-{2-[3-(3-pyridinyl)-1H-1,2,4-triazol-5-yl]ethyl}-2-indolinecarboxamide were filtered out.
Fig. 5.
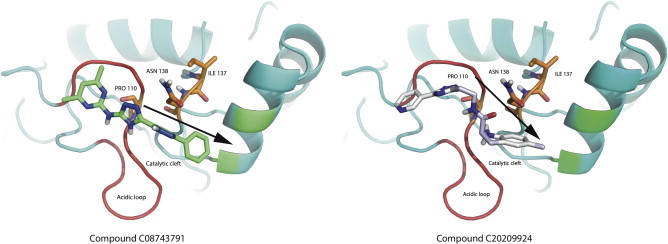
Examples of binding poses for the molecules. The left panel and right panels show the orientation of molecule C08743791 (4,6-dimethyl-N-[3-(5-phenyl-1H-pyrazol-3-yl)-1H-1,2,4-triazol-5-yl]pyrimidin-2-amine), and molecule C20209924 (5-methyl-N-{2-[3-(3-pyridinyl)-1H-1,2,4-triazol-5-yl]ethyl}-2-indolinecarboxamide), respectively. The first molecule is an example of molecule with an orientation suited for the interaction with both the acidic loop and the surrounding structural components (V143 and V147), while the latter is only interacting with the acidic loop and belongs to the subset of compounds that has been discarded upon the final selection step with API tools.
In Fig. 4 (right panel), two amino groups are shown to be a recurrent feature of the molecules identified after post-clustering selection, which (as depicted in Fig. 6 for molecule 4,6-dimethyl-N-[3-(5-phenyl-1H-pyrazol-3-yl)-1H-1,2,4-triazol-5-yl]pyrimidin-2-amine) establish two H-bonds with the acidic residues in the loop.
Fig. 6.
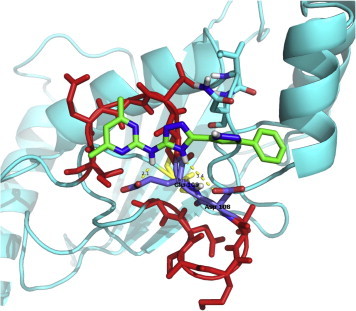
Interactions between the selected molecules and D108 and E109 residues of the acidic loop. Molecule C08743791 (4,6-dimethyl-N-[3-(5-phenyl-1H-pyrazol-3-yl)-1H-1,2,4-triazol-5-yl]pyrimidin-2-amine) is reported as an example and the hydrogen bonds with D108 and E109 of the acidic loop are highlighted.
Another recurrent feature of the selected molecules is the presence of an aromatic ring that is shown to interact with S139 (Fig. 7). Indeed, Cdc34 S139 was identified as a critical determinant for substrate mono-ubiquitination and polyubiquitination via K48-linked ubiquitination by experimental mutagenesis [28]. Moreover, it is the homologous residue of Ubc7 D127 that is known to play a crucial role in positioning the acceptor lysine of the substrate for the nucleophilic attack of the thioester bond during catalysis [29].
Fig. 7.
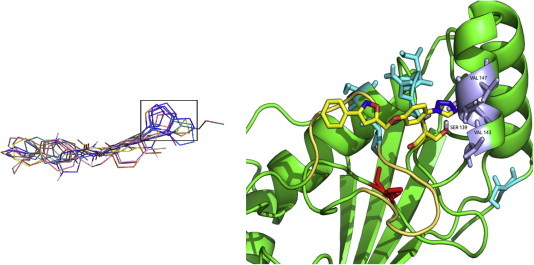
Interactions between the selected molecules and S139. (Left panel) Aromatic rings are highlighted as a recurrent feature of the high-ranking molecules. (Right panel) The aromatic ring of compound C29375629 (2-(2-bromo-4,6-difluoro-anilino)-2-oxo-ethyl]) is shown to interact with S139.
The aromatic ring of the scaffold (compound C08743791 [4,6-dimethyl-N-[3-(5-phenyl-1H-pyrazol-3-yl)-1H-1,2,4-triazol-5-yl]pyrimidin-2-amine] is reported as proof of concept in Fig. 8) is shown to interact with two hydrophobic residues in helix α3 of Cdc34: V143 and V147. This interaction might account for the specificity of the compounds. The valine steric hindrance can also limit the size of the inhibitor into the binding site.
Fig. 8.
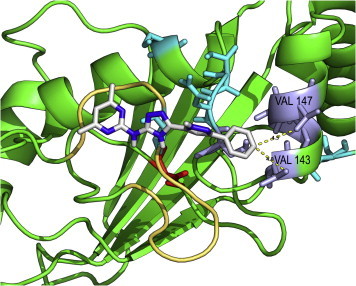
Interactions between the selected molecules and valine residues in Cdc34 catalytic cleft. The aromatic ring of molecule C08743791 (4,6-dimethyl-N-[3-(5-phenyl-1H-pyrazol-3-yl)-1H-1,2,4-triazol-5-yl]pyrimidin-2-amine) is reported as an example of interaction with V143 and V147 located on helix α3 of Cdc34.
In summary, on the basis of these common chemical features, we propose a pharmacophore model (Fig. 9) which features three main regions: a central region constituted by two amino groups which interact with the acidic residues of the loop, and two lateral regions that are characterized by the recurring presence of aromatic groups.
Fig. 9.
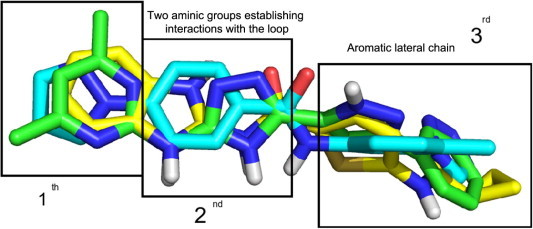
Pharmacophore model for Cdc34 inhibitors. The three main areas required for the inhibition of the acidic loop of Cdc34 are highlighted.
4. Discussion
The ubiquitin pathway is responsible for protein regulation by promoting target substrates degradation via the 26S proteasome system, and its deregulation has been implicated in a variety of diseases, including the onset of cancer and neurodegenerative disorders. In light of this crucial role, ubiquitination mechanisms have been thoroughly investigated to discover new leads to modulate cell cycle progression, as well as to find suitable molecular targets for cancer treatment.
In particular, E2 enzymes have been shown to play a central role in ubiquitination, since they act as mediators in determining the ultimate fate of the substrates. In this context, they have been recently proposed as suitable targets for inhibitory compounds [20–22].
In this context, we here focused on the human E2 enzyme Cdc34, which has been directly linked to tumorigenesis [14–19]. In particular, we used here as a model system, the homolog of Cdc34 in yeast, Saccharomyces cerevisiae Cdc34 (ScCdc34 or Ubc3). Indeed, this enzyme has been extensively characterized by our and other works [13,23,30–33] and an MD ensemble of conformations for ScCdc34 was already available [13,23]. ScCdc34 represents a valuable model since it shares a high sequence similarity with its human homolog (Fig. 3S). It has also been used as a model for studying the relationship occurring between phosphorylation and regulation of the E2 enzymatic activity [13]. We here focused on the Cdc34 acidic loop as a possible target of action for inhibitory molecules, which can constrain its position in the proximity of the catalytic cysteine, impairing Cdc34 activity. Indeed, the current data available on Cdc34 [13,23,30–33] support that Cdc34 can populate both states in which the loop is in a 'closed' conformation and the catalytic cysteine not accessible for Ub-transfer by the E1 enzyme, and active ‘open’ state in which the loop is open or partially open and the catalytic cysteine in a solvent-exposed position that can thus be compatible with Ub-charging. Those active open states, which are likely to be present even in absence of phosphorylation, are suggested to be further promoted by phosphorylation of Cdc34 catalytic domain by a population-shift mechanism [13,23]. The aim of our work is to identify molecules with the potential of entrapping the inactive states of Cdc34, i.e. a conformation of the protein in which the loop is stabilized in a closed state and the catalytic cysteine is thus buried and not accessible for Ub-transfer.
In this work, MD simulations of ScCdc34 were thus analyzed to select a conformation suitable for docking simulations. A large subset of the ZINC database was used for high-throughput virtual screening against ScCdc34 structure, followed by docking simulations with Autodock to refine the results. More in details, the pipeline that we have followed is constituted by a first step of coarse molecular selection based on shape complementarity performed by DOCK Blaster, screening the ZINC database. In a second step 500 compounds selected at the first step were re-docked in the Cdc34 binding site by Autodock and a filtering procedure was applied based on both spatial criteria and binding free energy to identify 20 molecules. It has to be noted that all the molecules selected from our in silico screening come from the ZINC database and comply with the Lipinski rule of five [27], so that for each compound the octanol–water partition coefficient is not greater than 5 and the molecular mass is less than 500 Da. These two features, along with a number of hydrogen-bond donor/acceptor groups lower than five make the selected molecules likely to diffuse inside cell membranes, so that they can exert their biological activity.
In the first part of the work, we performed a preliminary screening on thousands of compounds from a subset of the ZINC database using DOCK Blaster. One could argue that 500 molecules are not a sufficient dataset to further investigate. In this context, it has to be noted despite being less accurate than Autodock in its predictions, DOCK Blaster recapitulates the crystal ligand pose within 2 Å rmsd 50–60% of the time in common benchmarks [25]. We here analyzed in details the structural features which the majority of the 500 hundred molecules returned by DOCK Blaster have in common. These features are more clearly distinguishable after the following screening performed with Autodock and the spatial filtering (Fig. 9). Hence, the presence of distinct common chemical and structural determinants in the subset of molecules selected by DOCK Blaster supports reliability of its results, so that it is unlikely that from a random selection of ZINC molecules we could have obtained similar results.
In the 20 molecules identified in our screening, a set of common structural features can indeed be identified used to design a pharmacophore model. From an energetic point of view, the most relevant intermolecular interactions are established by two amino groups, which form two H-bonds with carboxylic groups from the acidic loop (D108 and E109). These interactions account for much of the calculated binding free energy, and therefore have been used by the energy-based ranking as the main discriminating feature for molecular selection. To quantitatively assess the importance of these hydrogen bonds on the binding free energy for each molecule, we also separately evaluated each energy component. As shown in Fig. 10, for the first four high-ranking molecules, the energy contribution of the hydrogen bonds accounts for more than 80% of the free energy of binding, as expected. In addition to the interactions established with the acidic loop, the selected compounds share a recurrent hydrogen bond with the Ser 139, that has been reported to be a crucial residue for Cdc34-mediated ubiquitination [28]. An aromatic ring able to interact with a valine cluster of Cdc34 also contributes to the molecular scaffold.
Fig. 10.
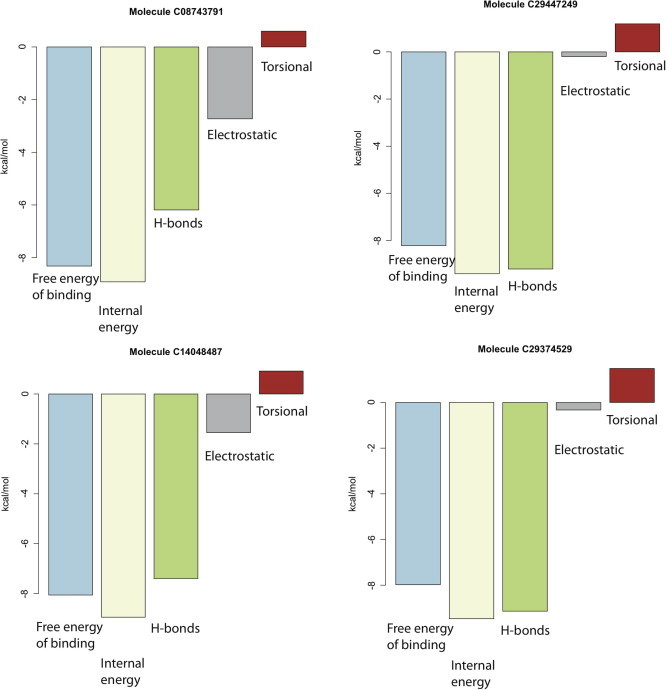
Binding free energy of the top four compounds of the post-clustering ranking. The first bar (light blue) refers to the total binding free energy (full list reported in Table 1), the second bar (light yellow) is the internal energy obtained as the sum of the H-bonds (and VdW contributions) (olive green) and the electrostatic component (gray). The last column (red) refers to the torsional energy. Binding free energy = internal energy + torsional energy. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
5. Conclusion
E2 enzymes act as mediators in determining the ultimate fate of the substrates selected for ubiquitination and for this reason they are considered privileged sites of action for inhibitory compounds.
In the present work, we performed a virtual screening procedure to select compounds, which may act as inhibitor for the catalytic activity of Cdc34, an E2 enzyme who has been directly linked to tumorigenesis [14–19]. In particular, the molecules were selected to target Cdc34 acidic loop, a structural component that has been shown to modulate Cdc34 enzymatic activity [13].
One of the problems that typically affect virtual screening procedures performed on thousands of compounds is the high number of false positives which lowers confidence of calculations results. To overcome this bias, we used at first DOCK Blaster, whose purpose is to provide a fast but coarse analysis of the entire Zinc subset, followed by Autodock calculations performed on the 500 high-ranking compounds selected by DOCK Blaster. After further refinement of the docking results, we have built a pharmacophore model from the ranked list of selected molecules, which can be a useful scaffold to develop and test inhibitory molecules for Cdc34in future experimental studies.
Acknowledgments
This work was supported by the Standard HPC Grant 2012 from CASPUR and ISCRA-Cineca Grants 2011 HP10CQEI1L and HP10COBO3E to E.P. The authors would like also to thank Paola Coccetti, Gaetano Invernizzi and Roberto Pagliarin for fruitful discussion and comments.
Contributor Information
Alberto Arrigoni, Email: arrigonialberto86@gmail.com.
Elena Papaleo, Email: elena.papaleo@unimib.it, elena.papaleo.78@gmail.com, elena.papaleo@bio.ku.dk.
Appendix A. Supplementary data
This pdf file contains Supplementary figures 1S–3S.
Parameters used for Autodock simulations.
References
- 1.Spasser L., Brik A. Chemistry and biology of the ubiquitin signal. Angew. Chem. Int. Ed. Engl. 2012;51:6840–6862. doi: 10.1002/anie.201200020. [DOI] [PubMed] [Google Scholar]
- 2.Nagy V., Dikic I. Ubiquitin ligase complexes: from substrate selectivity to conjugational specificity. Biol. Chem. 2010;391:163–169. doi: 10.1515/bc.2010.021. [DOI] [PubMed] [Google Scholar]
- 3.Harper J.W., Schulman B.A. Structural complexity in ubiquitin recognition. Cell. 2006;124(2006):1133–1136. doi: 10.1016/j.cell.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Komander D., Rape M. The ubiquitin code. Annu. Rev. Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 5.Komander D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009;37:937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 6.Sadowski M., Suryadinata R., Tan A.R., Roesley S.N.A., Sarcevic B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life. 2012;64:136–142. doi: 10.1002/iub.589. [DOI] [PubMed] [Google Scholar]
- 7.Trempe J.F. Reading the ubiquitin postal code. Curr. Opin. Struct. Biol. 2011;21:792–801. doi: 10.1016/j.sbi.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Liu J., Nussinov R. The role of allostery in the ubiquitin–proteasome system. Crit. Rev. Biochem. Mol. Biol. 2013;48:89–97. doi: 10.3109/10409238.2012.742856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye Y., Rape M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2013;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wenzel D.M., Stoll K.E., Klevit R.E. E2s: structurally economical and functionally replete. Biochem. J. 2011;433:31–42. doi: 10.1042/BJ20100985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van WijK S.J., Timmers H.T. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J. 2010;24:981–993. doi: 10.1096/fj.09-136259. [DOI] [PubMed] [Google Scholar]
- 12.Michelle C., Vourc’h P., Mignon L., Andres C.R. What was the set of ubiquitin and ubiquitin-like conjugating enzymes in the eukaryote common ancestor? J. Mol. Evol. 2009;68:616–628. doi: 10.1007/s00239-009-9225-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papaleo E., Ranzani V., Tripodi F., Vitriolo A., Cirulli C., Fantucci P., Alberghina L., Vanoni M., De Gioia L., Coccetti P. An acidic loop and cognate phosphorylation sites define a molecular switch that modulates ubiquitin charging activity in Cdc34-like enzymes. PLoS Comput. Biol. 2011;7:e1002056. doi: 10.1371/journal.pcbi.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka K., Kondoh N., Shuda M., Matsubara O., Imazeki N., Ryo A., Wakatsuki T., Hada A., Goseki N., Igari T., Hatsuse K., Aihara T., Horiuchi S., Yamamoto N., Yamamoto M. Enhanced expression of mRNAs of antisecretory factor-1, gp96, DAD1 and CDC34 in human hepatocellular carcinomas. Biochim. Biophys. Acta. 2011;1536:1–12. doi: 10.1016/s0925-4439(01)00026-6. [DOI] [PubMed] [Google Scholar]
- 15.Eliseeva E., Pati D., Diccinanni M.B., Yu A.L., Mohsin S.K., Margolin J.F., Plon S.E. Expression and localization of the CDC34 ubiquitin-conjugating enzymes in pediatric acute lymphoblastic leukemia. Cell Growth Differ. 2001;12:427–433. [PubMed] [Google Scholar]
- 16.Zhang Z., Wang Y., Yao R., Li J., Yan Y., La Regina M., Lemon W.L., Grubbs C.J., Lubet R.A., You M. Cancer chemopreventive activity of a mixture of Chinese herbs (antitumor B) in mouse lung tumor models. Oncogene. 2004;23:3841–3850. doi: 10.1038/sj.onc.1207496. [DOI] [PubMed] [Google Scholar]
- 17.Chauhan D., Li G., Hideshima T., Podar K., Shringarpure R., Mitsiades C., Munshi N., Yew P.R., Anderson K.C. Blockade of ubiquitin-conjugating enzyme CDC34 enhances anti-myeloma activity of Bortezomib/Proteasome inhibitor PS-341. Oncogene. 2004;23:3597–35602. doi: 10.1038/sj.onc.1207458. [DOI] [PubMed] [Google Scholar]
- 18.Price G.R., Armes J.E., Ramus S.J., Provenzano E., Kumar B., Cowie T.F., Ciciulla J., Hutchins A.M., Thomas M., Venter D.J. Phenotype-directed analysis of genotype in early-onset, familiar breast cancers. Pathology. 2006;38:520–527. doi: 10.1080/00313020601024052. [DOI] [PubMed] [Google Scholar]
- 19.Zeng Y., Abdallah A., Lu J.P., Wang T., Chen Y.H., Terrian D.M., Kim K., Lu Q. Delta-Catenin promotes prostate cancer cell growth and progression by altering cell cycle and survival gene profiles. Mol. Cancer. 2009;8:19. doi: 10.1186/1476-4598-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ceccarelli D.F., Tang X., Pelletier B., Orlicky S., Xie W., Plantevin V., Neculai D., Chou Y.C., Ogunjimi A., Al-Hakim A., Varelas X., Koszela J., Wasney G.A., Vedadi M., Dhe-Paganon S., Cox S., Xu S., Lopez-Girona A., Mercurio F., Wrana J., Durocher D., Meloche S., Webb D.R., Tyers M., Sicheri F. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145:1075–1087. doi: 10.1016/j.cell.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 21.Huang H., Ceccarelli D.F., Orlicky S., St-Cyr D.J., Ziemba A., Garg P., Plamondon S., Auer M., Sidhu S., Marinier A., Kleiger G., Tyers M., Sicheri F. E2 enzyme inhibition by stabilization of a low-affinity interface with ubiquitin. Nat. Chem. Biol. 2014;10:156–163. doi: 10.1038/nchembio.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders M.A., Brahemi G., Nangia-Makker P., Balan V., Morelli M., Kothayer H., Westwell A.D., Shekhar M.P. Novel inhibitors of Rad6 ubiquitin conjugating enzyme: design, synthesis, identification and functional characterization. Mol. Cancer Ther. 2013;12:373–383. doi: 10.1158/1535-7163.MCT-12-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papaleo E., Casiraghi N., Arrigoni A., Vanoni M., Coccetti P., De Gioia L. Loop 7 of E2 enzymes: an ancestral conserved functional motif involved in the E2-mediated steps of the ubiquitination cascade. PLoS ONE. 2012;7:e40786. doi: 10.1371/journal.pone.0040786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irwin J.J., Sterling T., Mysinger M.M., Bolstad E.S., Coleman R.G. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irwin J.J., Shoichet B.K., Mysinger M.M., Huang N., Colizzi F., Wassam P., Cao Y. Automated docking screens: a feasibility study. J. Med. Chem. 2009;52:5712–5720. doi: 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipinski C.A. Lead- and drug-like compounds: the rule of five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Sadowski M., Suryadinata R., Lai X., Heierhorst J., Sarcevic B. Molecular basis for lysine specificity in the yeast ubiquitin-conjugating enzyme Cdc34. Mol. Cell. Biol. 2010;30:2316–2329. doi: 10.1128/MCB.01094-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yunus A.A., Lima C.D. Lysine activation and functional analysis of E2-mediated conjugation in the SUMO pathway. Nat. Struct. Mol. Biol. 2006;13:491–499. doi: 10.1038/nsmb1104. [DOI] [PubMed] [Google Scholar]
- 30.Coccetti P., Tripodi F., Tedeschi G., Nonnis S., Marin O., Fantinato S., Cirulli C., Vanoni M., Alberghina L. The CK2 phosphorylation of catalytic domain of Cdc34 modulates its activity at the G1 to S transition in Saccharomyces cerevisiae. Cell Cycle. 2009;7:1391–1401. doi: 10.4161/cc.7.10.5825. [DOI] [PubMed] [Google Scholar]
- 31.Petroski M.D., Deshaies R.J. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123:1107–1120. doi: 10.1016/j.cell.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 32.Lass A., Cocklin R., Scaglione K.M., Skowyra M., Korolev S., Goebl M., Skowyra D. The loop-less tmCdc34 E2 mutant defective in polyubiquitination in vitro and in vivo supports yeast growth in a manner dependent on Ubp14 and Cka2. Cell Div. 2011;6:7. doi: 10.1186/1747-1028-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ziemba A., Hill S., Sandoval D., Webb K., Bennett E.J., Kleiger G. Multimodal mechanism of action for the Cdc34 acidic loop: a case study for why ubiquitin-conjugating enzymes have loops and tails. J. Biol. Chem. 2013;288:34882–34896. doi: 10.1074/jbc.M113.509190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This pdf file contains Supplementary figures 1S–3S.
Parameters used for Autodock simulations.