

Highlights
-
•
Selected long non-coding RNA are aberrantly expressed in hepatocellular cancers.
-
•
linc-ROR is a highly upregulated lncRNA that is expressed in response to TGFβ.
-
•
linc-ROR contributes to chemoresistance of HCC cells.
-
•
Intercellular transfer of linc-ROR occurs within extracellular vesicles.
Abbreviations: EV, extracellular vesicle; HCC, hepatocellular carcinoma; miRNA, microRNA; VD, vesicle-depleted; lncRNA, long non-coding RNA; linc-ROR, long intergenic non-coding RNA; TGFβ, transforming growth factor β; CT, cycle threshold; siRNA, small interfering RNA
Keywords: Liver cancer, Chemoresistance, Exosomes, Gene expression, RNA genes
Abstract
Hepatocellular cancers (HCC) are highly resistant to chemotherapy. TGFβ has been associated with chemoresistance in some human cancers but the mechanisms involved are unknown. We explored how TGFβ might contribute to altered responses to therapy by assessing the involvement and mechanistic contribution of extracellular vesicle long non-coding RNA (lncRNA) in mediating TGFβ-dependent chemoresistance. TGFβ reduced the sensitivity of HCC cells to sorafenib or doxorubicin and altered the release of both extracellular vesicles and of selected lncRNA within these vesicles. Amongst these, lincRNA-ROR (linc-ROR), a stress-responsive lncRNA was highly expressed in HCC cells and enriched within extracellular vesicles derived from tumor cells. Incubation with HCC-derived extracellular vesicles increased linc-ROR expression and reduced chemotherapy-induced cell death in recipient cells. Sorafenib increased linc-ROR expression in both tumor cells and extracellular vesicles, whereas siRNA to linc-ROR increased chemotherapy-induced apoptosis and cytotoxicity. Tumor-initiating cells that express CD133 have an increased resistance to therapy. TGFβ increased expression of CD133+ cells and colony growth in limiting dilution assays, both of which were attenuated by linc-ROR knockdown. These data provide mechanistic insights into primary chemoresistance in HCC by showing that: (a) TGFβ selectively enriches linc-RoR within extracellular vesicles, which has a potential role in intercellular signaling in response to TGFβ; (b) expression and enrichment of linc-ROR during chemotherapeutic stress plays a functional role in chemoresistance; and (c) the effects of TGFβ on chemoresistance in HCC may involve linc-RoR-dependent effects on tumor-initiating cells. These findings implicate extracellular vesicle lncRNA as mediators of the chemotherapeutic response, and support targeting linc-ROR to enhance chemosensitivity in HCC.
1. Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide and the third most common cause of cancer mortality [1]. These cancers are highly chemoresistant. Although a large number of therapeutic agents have been evaluated for the treatment of HCC, most have been ineffective. Sorafenib and doxorubicin are currently used for systemic or locoregional therapies for HCC but have limited efficacy [2,3]. The heterogeneous nature of HCC and the lack of targetable oncogenic driver mutations further limit the effectiveness of targeted therapies. The poor prognosis of unresectable HCC is related to the highly chemoresistant nature of this cancer [4]. Understanding the mechanisms contributing to innate or acquired resistance to therapy in HCC is therefore necessary and essential in order to develop more effective treatments.
Cellular toxicity and stress occurring during exposure to therapeutic agents such as sorafenib can elicit survival responses that eventually result in resistance to these agents. Within the liver, injury results in activation of mesenchymal cells with release of soluble mediators such as transforming growth factor β (TGFβ) [5,6]. TGFβ is a central contributor to hepatic fibrosis and has been implicated to contribute to hepatocarcinogenesis through diverse mechanisms. TGF-β can modulate the expression of genes relevant to tumor development and promote malignant transformation of progenitor cells, and thereby link hepatic injury, fibrotic responses and cancer [7–10]. Although TGFβ has been associated with chemoresistance in human cancers [11], the mechanisms remain unknown.
We postulated that TGFβ enhances local environmental changes and cellular reprogramming that facilitate chemoresistance. We have recently shown that HCC cells can release extracellular vesicles (EVs) such as exosomes [12]. These vesicles contain protein, lipids and RNA derived from their donor cell cytoplasm and can be taken up by other cells. The intercellular transfer of EV contents thus provides a mechanism by which cells can communicate with other cells in their local microenvironment. The presence of mRNA and non-coding RNA within EV is of particular interest because these RNA molecules can modulate gene expression and cellular activities in recipient cells [13–16]. Based on our previous findings that HCC cell derived EVs contain miRNAs that can modulate transformed cell behavior in target cells [12], we hypothesized that intercellular signaling by EV RNA in response to TGFβ could mediate chemoresistance. Our studies evaluated the role of EV signaling in tumor cell responses to TGFβ and identified EV long non-coding RNA signaling mediators involved in modulation of cellular responses to chemotherapy. These findings provide several new mechanistic insights into acquired chemoresistance in HCC, and identify mediators and mechanisms that could be targeted to enhance sensitivity and improve responses to conventional agents that are used for the treatment of HCC.
2. Materials and methods
2.1. Cell lines, culture, and reagents
HepG2 and PLC-PRF5 cells were obtained from American Type Culture Collection (Manassas, VA), and cultured in DMEM high glucose medium (HyClone Laboratories, Logan, UT), containing 10% fetal bovine serum and 1% antibiotic–antimycotic (Invitrogen, Grand Island, NY), at 37 °C with 5% CO2. Non-malignant human hepatocytes HH were obtained from Sciencell and cultured as recommended by the supplier. For all studies with extracellular vesicles, vesicle depleted medium was prepared by centrifuging cell-culture medium at 100,000g overnight to spin down any pre-existing vesicle content. Camptothecin and doxorubicin were obtained from Sigma-Aldrich (St. Louis, MO), and sorafenib was obtained from Selleck (Houston, TX). Compounds were dissolved in 100% DMSO (Sigma–Aldrich, St. Louis, MO) and diluted with culture media to the desired concentration with a final DMSO concentration of 0.1%. DMSO 0.1% (v/v) was used as a solvent control.
2.2. Isolation of EV
EV were isolated from HCC cells as previously described [17]. Cells (1 × 106) were plated in vesicle-depleted medium, and medium was collected after 3–4 days for EV isolation for EV isolation by sequential centrifugation. Each isolation was verified using nanoparticle tracking analysis using a Nanosight N-300 (NanoSight Ltd., Amesbury, UK) to determine size and quantity of EV isolated. Isolated EVs were used immediately, or were resuspended in 50–100 μl of PBS and stored at −80 °C.
2.3. RNA extraction and analysis
Total RNA was extracted from cells using Trizol (Invitrogen) or from EV using ExoQuick-TC (System Biosciences, Mountain View, CA). HepG2 cells (1 × 106) were plated in 11 ml of EV-depleted medium on collagen-coated 10-cm dishes. After 3–4 days, the medium was collected and sequentially centrifuged at 3000×g for 15 min to remove cells and cell debris. The supernatant was transferred to a sterile vessel and combined with 2 ml ExoQuick-TC. After an overnight precipitation at 4 °C, total RNA was extracted using SeraMir™ Exosome RNA Amplification Kit (System Biosciences, Mountain View, CA) according to the manufacturers’ instructions. RNA concentration was measured using NanoDrop ND-2000 (Nano-Drop Technologies, Wilmington, DE).
2.4. Real-time PCR analysis
RNA was treated with RNase-free DNase I (Qiagen, Valencia, CA). One microgram of RNA was reverse-transcribed to cDNA using iScript cDNA Synthesis Kit (BIO-RAD Laboratories, Inc., Hercules, CA), and Real-time quantitative RT-PCR (qRT-PCR) was performed using a Mx3000p System (Stratagene, La Jolla, CA) to detect RNU6B (U6) and lincRNA-ROR using SYBR green I (SYBR® Advantage® qPCR Premix, Clontech., Mountain View, CA). The following PCR primers were used: lincRNA-ROR primers, forward: 5′-AGGAAGCCTGAGAGTTGGC-3′, reverse: 5′- CTCAGTGGGGAAGACTCCAG-3′, U6, forward: 5′-CTCGCTTCGGCAGCACA-3′, reverse: 5′-AACGCTTCACGAATTTGCGT-3′.
2.5. Gene expression profiling
The expression of 90 lncRNA was performed using the LncProfiler™ qPCR Array Kit (System Biosciences, Mountain View, CA). RNA from EV or donor cells (n = 3 per each cell line) were treated with DNase I and 2 μg of DNase-treated RNA was reverse transcribed. Real-time PCR was performed (2X Maxima® SYBR Green with Rox, Fermentas, Glen Burnie, MD) and the cycle number at which the reaction crossed a threshold (CT) was determined for each gene. Raw CT values were normalized using a median CT value (ΔCT = CTlncRNA − CTmedian). The relative amount of each lncRNA in HCC cells relative to nonmalignant hepatocyte (fold change) was described using the equation 2−ΔΔCT where ΔΔCT = ΔCTHCC cell − ΔCTnonmalignant hepatocytes and each lncRNA in EVs relative to donor cells was described using the equation 2−ΔΔCT where ΔΔCT = ΔCTEV − ΔCTdonor cell. The expression of 84 mRNAs associated with liver cancers was examined using RT2 Profiler™ PCR Array System (Qiagen, Valencia, CA). RNA was isolated from cells and incubated with DNase I. One μg of DNase-treated RNA was reverse transcribed using RT2 First Strand Kit (Qiagen, Valencia, CA). Real-time PCR was performed (SABiosciences RT2 qPCR Master Mix, Qiagen, Valencia, CA) and mRNA expression levels were evaluated using a comparative CT method.
2.6. Transfection of siRNAs
Two different siRNA against linc-RoR (5′ to 3′); siRNA linc-ROR-1: GGAGAGGAAGCCTGAGAGT, and siRNA linc-ROR-2: GGTTAAAGACACA-GGGGAA as well as a non-targeting (NT) control siRNA (siGENOME Non-Targeting siRNA) were purchased from Dharmacon (Lafayette, CO). Validated siRNA to p53 siRNA were obtained from Life Technologies (Grand Island, NY). Cell transfections were performed using 50–100 nM siRNA using Lipofectamine 2000 (Life Technologies, Grand Island, NY).
2.7. Luciferase assay
Cells were co-transfected with p53-Luc Plasmid (Agilent Technologies, Santa Clara, CA) and pRL-TK Renilla Vector (Promega, Madison, WI) using Lipofectamine 2000 (Life Technologies, Grand Island, NY). After 24 h, total cell extracts were assayed for luciferase activity using Dual-Luciferase® Reporter Assay System (Promega), and a multiwell plate luminometer (Turner Biosystems, Sunnyvale, CA) according to the manufacturer’s instruction.
2.8. Chemosensitivity assays
For studies of chemotherapeutic stress, cells were incubated with varying concentrations of sorafenib, camptothecin, doxorubicin or the appropriate diluent (DMSO) control for 72 h. Cells were seeded (1 × 104/well) in collagen-coated 24-well or 96-well plates. Cell viability was assessed by microscopy after staining with trypan blue and the number of viable cells expressed relative to cell counts at baseline. Inhibition of proliferation was assessed using a CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay Kit MTS assay (Promega) and a FLUOstar Omega Microplate Reader (BMG Labtech, Cary, NC). Proliferation index was expressed as a percentage of absorbance values in experimental conditions compared to that for control cells.
2.9. Caspase 3/7 activity assay
Cells were seeded (1 × 104/well) into 96-well collagen-coated plates in appropriate media and incubated for 24 h. Then medium was then replaced with medium containing different concentrations of sorafenib. At selected time points, caspase-3/7 activity was assessed using Caspase-GloR3/7 Assay kit (Promega, Madison, WI) using a FLUOstar Omega Microplate Reader (BMG Labtech, Cary NC). Data were expressed as relative luminescence values relative to those of controls without chemical substance.
2.10. Flow cytometry
Flow cytometry was performed using an Accuri C6 flow cytometer (Accuri, Ann Arbor, MI). For cell cycle analysis, cells were permeabilized with 70% ethanol, and DNA was stained with 20 μg/ml propidium iodide, 0.2 mg/ml RNase A, and 0.01 mol/L PBS (pH 7.4). Analysis was performed using FCS express version 3 software (De Novo Software, Los Angeles, CA). For analysis of cell death, cells were stained with annexin V-FITC or propidium iodide (Clontech, Mountain View, CA), and the proportion of cells undergoing apoptotic death was quantitated. For CD133+ and EpCAM expression, cells were incubated with CD133/1 (AC133) pure mouse monoclonal antibody (Miltenyi Biotec, Cambridge, MA), mouse monoclonal (AUA1) antibody to EPCAM (Abcam, Cambridge, MA) or IgG1 (normal mouse IgG1 antibody, Santa Cruz, Dallas, Texas) as an isotype control, and then with goat-anti-mouse IgG1-FITC (Santa Cruz, Dallas, Texas).
2.11. In vitro limiting dilution assay
Self renewal capacity was assessed using spheroid formation assays. Single cell suspensions were seeded (100 to 2000 / well) in ultra-low attachment surface 96-well plates (Corning, Corning, NY). Each well was supplemented with 100 μl of serum-free DMEM medium or DMEM medium containing 1% FBS. The number of spheroids was examined under a light microscope after 7 days. The total number of wells containing spheroids was determined. Twelve replicates were used for each condition and the percentage of total cultures which yield positive spheroid (proportion) was analyzed using L-Calc software (StemCell Technologies, Vancouver, BC, Canada) based on Poisson distribution.
2.12. Statistical analysis
Data were expressed as the mean and standard error from at least four replicates unless noted otherwise. Comparisons between groups were performed using the two-tailed Student’s t test, and results were considered to be statistically significant when p < 0.05.
3. Results
3.1. Does TGFβ modulate chemotherapy resistance and increase EV release?
Innate or acquired resistance to therapy is a hallmark of hepatocellular cancers. In order to explore therapeutic strategies to enhance chemotherapeutic responses, or to reduce the potential for acquired resistance, we investigated the effects and mechanisms by which TGFβ could modulate responses of hepatocellular cancer to therapy. Exogenous exposure of HepG2 or PLC-PRF5 cells to TGFβ did not significantly alter the growth of either HepG2 or PLC-PRF5 HCC cells. These cells were sensitive to sorafenib (IC50 of 2.8 μM in HepG2 cells and 2.4 μM in PLC-PRF5 cells) as well as to doxorubicin (IC50 of 22 nM in HepG2 cells and 0.9 μM for PLC-PRF5 cells). TGFβ ameliorated growth inhibition with either agent, with similar effects observed in both of the cell lines (Fig. 1A). Furthermore, TGFβ reduced sorafenib induced caspase 3/7 activity, consistent with a reduction in apoptosis (Fig. 1B). Although TGFβ has been reported to have divergent roles in cancers, these data are consistent with those from other malignancies such as colorectal cancer, breast and ovarian cancers in which TGFβ has been implicated in chemoresistance [18].
Fig. 1.
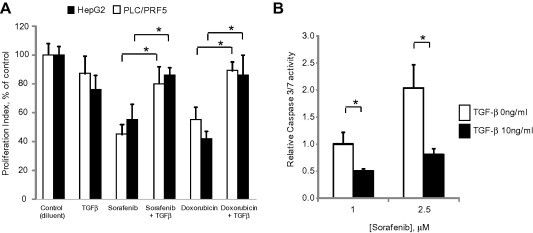
TGFβ modulates chemosensitivity of HCC cells. (A) HepG2 or PLC/PRF-5 HCC cells (1 × 104/well) were cultured in 96 well collagen-coated plates for 24 h. Cells were then exposed to diluent (controls), TGFβ (10 ng/ml), doxorubicin (25 nM for HepG2 cells or 1.0 μM for PLC/PRF-5 cells) or sorafenib (2.5 μM). Cell proliferation was assessed after 72 h using CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay. Proliferation index represents absorbance values expressed as a percentage of control cells. (B) HepG2 cells were plated (1 × 104/well) in a 96 well plate and incubated with 0 or 10 ng/ml of TGFβ for 24 h. Cells were then exposed to 1 or 2.5 μM sorafenib for 6 h. Caspase-3/7 activity was assessed using a commercial luminometric assay. Data were expressed relative to luminescence values of 1 μM sorafenib without TGFβ. Data represents the means ± standard error of the mean (SEM) of 3 separate studies, with each study conducted in quadruplicate. ∗p < 0.05.
3.2. Can intercellular transfer of extracellular vesicles (EVs) modulate chemoresistance?
To ascertain if inter-cellular signaling by EV could modulate responses to chemotherapy, we first isolated EV from HepG2 cells in culture and characterized them using electron microscopy, sedimentation characteristics and size quantitation using nanoparticle tracking analysis using Nanosight (Fig. 2A). EV isolations consisted of a homogeneously sized population of vesicles, many of which have morphological features of exosomes. The term exosome refers to a particular subset of vesicles that are defined by their biogenesis, and therefore we have used the term EV rather than exosomes in this report. The effect of exposure to tumor cell derived EV on cell responses to chemotherapy was then ascertained. HepG2 cells were cultured in EV-depleted medium in the presence or absence of HepG2-derived EVs for 24 h. Cells were then incubated with sorafenib, doxorubicin or camptothecin and cell viability was assessed after 48 h. Cell viability increased in an EV concentration dependent manner, with effects more prominent for doxorubicin and camptothecin than for sorafenib (Fig. 2B–D). These observations indicate that EV can modulate cellular stress in response to chemotherapeutic agents and enhance viability.
Fig. 2.
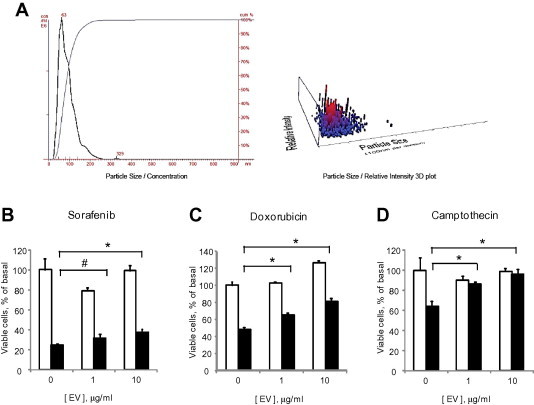
Tumor cell derived EV modulate chemosensitivity. (A) Analysis of extracellular vesicles (EVs) derived from HepG2 cells by nanoparticle tracking analysis using a Nanosight N-300. The analysis revealed EVs with a mean size of 90 ± 40 nm. (B–D) HepG2 cells (1 × 104/well) were plated in 96 well collagen-coated plates in EV depleted medium and incubated with different concentrations of EVs. After 24 h, cells were exposed to diluent (white bars) or 10 μM (black bars) of (B) sorafenib, (C) doxorubicin or (D) camptothecin and cell viability was assessed after 48 h using an MTS assay. Bars express the mean value ± SEM of 3 separate studies, each performed in quadruplicate. ∗p < 0.05, #p = 0.13.
3.3. Is the expression of long non-coding RNA altered by TGFβ?
In recent studies, we identified enrichment of non-coding RNA in EV released by HCC cells. Long non-coding RNAs (lncRNAs) are being increasingly implicated in human cancers [19], but their involvement in chemoresistance are unknown. To identify potential mediators of TGFβ mediated chemosensitivity, we examined the effect of TGFβ on exosomal lncRNA content using qRT-PCR. There were marked quantitative differences in several but not all lncRNA between expression in HepG2 cells and in EV derived from the same cells (Fig. 3 and Table 1). These data suggest that EV release of lincRNA-ROR may have specific roles in malignant cells. We identified two lncRNAs, lincRNA-ROR (linc-ROR) and linc-VLDLR that were increased in malignant hepatocytes, selectively enriched within EV with EV release further enhanced by TGFβ. Together, these observations support the presence of TGFβ mediated pathways for selective enrichment of these lncRNAs within EV, and their potential involvement in inter-cellular signaling in response to TGFβ and as potential mediators of chemoresistance. We focused our efforts on linc-ROR because the greatest quantitative differences in EV release in response to TGFβ were observed with this lncRNA, and was also highly up-regulated in malignant HepG2 cells compared with non-malignant human hepatocytes (Table 2).
Fig. 3.
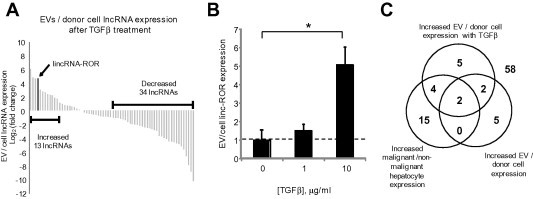
Effect of TGFβ on lncRNA enrichment within EV. (A) Expression profiling of 90 lncRNAs was performed in donor HepG2 cells and EV derived from these cells after 72 h incubation with 10 ng/ml of TGFβ from three independent samples. Sixty-eight lncRNAs were identified in EVs of which thirteen lncRNAs were increased by >2-fold change in EVs compared to their donor cells. Each column represents an independent lncRNA. (B) The expression of linc-ROR was assessed by qRT-PCR in HepG2 derived EVs following incubation of donor cells with 0, 1 or 10 ng/ml TGFβ for 72 h. Linc-ROR in EVs was expressed relative to expression in donor cells and normalized to that of RNU6B. Bars represent the mean value ± SEM of 3 separate determinants. ∗p < 0.05. (C) The Venn diagram summarizes the results of lncRNA profiling and illustrates number of lncRNA for which the ratio was greater than 2-fold in each group. The central overlap indicates two lncRNA that were selectively enriched in all three profiling studies, and includes linc-ROR and lincRNA-VLDLR.
Table 1.
Effect of TGFβ on EV long non coding-RNA released from HCC cells.
| Basal expression |
TGFβ, 10 ng/ml |
||
|---|---|---|---|
| lncRNA | EV/cell ratio Log 2 (fold change) | lncRNA | EV/cell ratio Log 2 (fold change) |
| CAR Intergenic 10 | 4.39 | HOTAIR | 6.10 |
| DISC2 (family) | 3.99 | lincRNA-VLDLR | 4.86 |
| DHFR ut (family) | 3.81 | CAR Intergenic 10 | 4.68 |
| HAR1B | 3.52 | lincRNA-RoR | 4.66 |
| lincRNA-VLDLR | 3.36 | Tsix | 3.04 |
| Tsix | 2.42 | Y RNA-1 | 2.78 |
| lincRNA-RoR | 1.99 | Nespas | 2.66 |
| lincRNA-p21 | 1.45 | LUST | 2.39 |
| NEAT1 (family) | 1.16 | Jpx | 2.31 |
| SNHG4 | 2.29 | ||
| Alpha 280 | 1.91 | ||
| anti-NOS2A | 1.79 | ||
| Zfhx2as | 1.16 | ||
Expression profiling of 90 lncRNAs was performed in HepG2 cells, and extracellular vesicles (EVs) derived from these cells under basal conditions or following incubation of cells with TGFβ 10 ng/ml for 24 h. LncRNAs increased by >2-fold are shown. Enrichment within EV of several lncRNA is noted, and amongst these, further enrichment of lincRNA-RoR occurs in response to TGFβ.
Table 2.
LncRNA expression profiling in malignant and non-malignant hepatocytes.
| lncRNA | HepG2/HH ratio Log 2 (fold change) |
|---|---|
| anti-NOS2A | 10.42 |
| lincRNA-SFMBT2 | 9.58 |
| lincRNA-RoR | 5.42 |
| Alpha 280 | 4.27 |
| lincRNA-VLDLR | 4.12 |
| E2F4 antisense | 4.10 |
| HOXA3as | 3.06 |
| snaR | 2.72 |
| LUST | 2.37 |
| UM9-5 | 2.34 |
| Zfhx2as | 2.14 |
| SNHG4 | 2.01 |
| Gomafu | 1.71 |
| Zfas1 | 1.47 |
| p53 mRNA | 1.32 |
| Air | 1.29 |
| SNHG5 | 1.10 |
| H19 antisense | 1.08 |
| Hoxa11as | 1.04 |
| LOC285194 | 1.02 |
| HAR1B | 1.01 |
Expression profiling of selected lncRNAs was performed by comparing the expression of genes in malignant human hepatocytes (HepG2 cells) and non-malignant human hepatocytes (HH cells) using PCR. LncRNAs increased by >2-fold in malignant cells compared to non-malignant cells are shown.
3.4. Does linc-ROR prevent chemotherapy-induced cell death?
To evaluate the potential end-target effects of linc-ROR in HCC cells, we next examined the effect of linc-ROR knockdown using siRNA on chemotherapeutic stress. Transfection of HepG2 cells with 100 nM linc-ROR-1 siRNA and linc-ROR-2 siRNA constructs reduced linc-ROR expression by 73 ± 17% and 70 ± 10% respectively after 48 h, compared with non-targeting siRNA controls. We then investigated the effect of linc-ROR knockdown and observed a significant concentration-dependent effect on cell viability in response to sorafenib, camptothecin, or doxorubicin with siRNA to linc-ROR-1 compared to controls (Fig. 4). Further studies showed that siRNA to linc-ROR-1 increased the percentage of cells undergoing early apoptosis from 11.8% to 25.2% compared with non-targeting control siRNA during incubation with 1 μM sorafenib. siRNA to linc-ROR-1 also increased the percentage of total apoptotic cells (Fig. 5A, B). Furthermore, caspase-3/7 activity was increased by 1.7–1.8-fold in cells transfected with siRNA to linc-ROR-1 compared to controls in HepG2 cells that were incubated with sorafenib 1 μM for 6 and 24 h (Fig. 5C). Thus, linc-ROR can functionally modulate chemotherapy-induced apoptosis and cell survival.
Fig. 4.
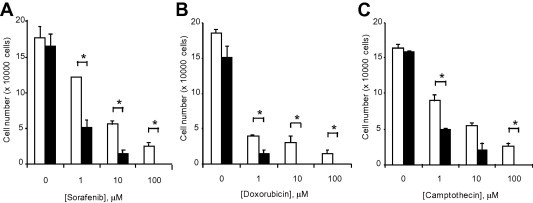
Linc-ROR knockdown modulates chemotherapeutic response. (A–C) HepG2 cells were transfected with siRNAs against linc-ROR-1 (black bars) or non-targeting control siRNA (white bars). After 48 h, cells were plated (1 × 104/well) on 96 well plates and treated with (A) sorafenib, (B) doxorubicin or (C) camptothecin at the indicated concentrations. The number of viable cells was counted after 48 h using a hemocytometer after trypan blue staining. Bars express the mean value ± SEM of 3 separate determinants. ∗p < 0.05.
Fig. 5.
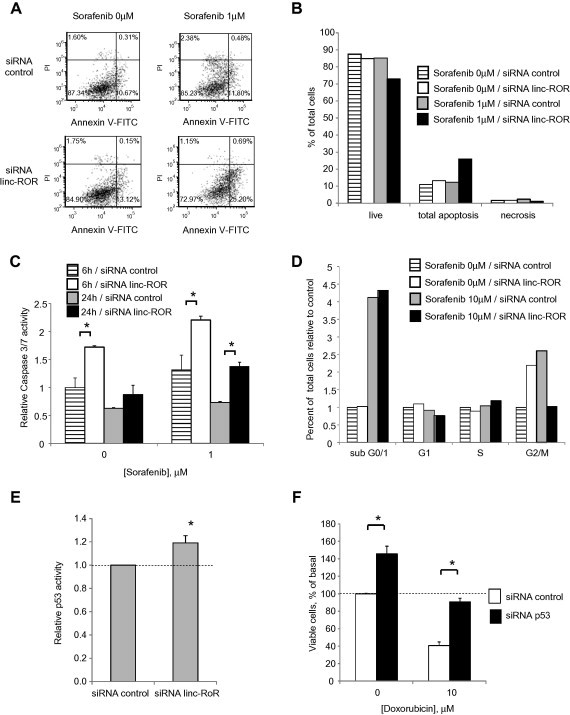
Cellular effects of linc-ROR knockdown. (A–E) HepG2 cells were transfected with either siRNA to linc-ROR-1 or non-targeting control siRNA for 48 h. (A, B) Transfected cells were incubated with 1 μM sorafenib, and analyzed 24 h later using an Accuri C6 flow cytometer after staining with annexin V/propidium iodide. Cells in live, apoptosis and necrosis group are expressed as percentages of the total cell population. (C) Transfected cells were plated (1 × 104/well) in a 96 well plate and incubated with diluent or 1 μM sorafenib for either 6 or 24 h. Caspase-3/7 activity was assessed using a commercial luminometric assay. Data were expressed relative to luminescence values of controls without sorafenib. (D) Transfected cells were incubated with 10 μM sorafenib. After 24 h, cell cycle analysis was performed using Accuri C6 flow cytometer after staining with propidium iodide. Cells in sub G0/1, G1, S, and G2/M phases of the cell cycle are expressed as percentages of the total cell population. (E) Transfected cells were then cotransfected with p53-Luc Plasmid and pRL-TK Renilla Vector. After 24 h, luciferase expression was measured. p53 luciferase activity was normalized to that of Renilla and expressed relative to control. (F) HepG2 cells were transfected with either siRNA to p53 or non-targeting control siRNA for 24 h. Cells were then incubated with diluent or 10 μM doxorubicin. After 48 h, cell viability was assessed using an MTS assay. Bars express the mean value ± SEM of 3 separate determinations. ∗p < 0.05.
To identify mechanisms by which deregulated expression of linc-ROR could contribute to tumor cell behavior, we next assessed the effect of knock-down of linc-RoR using siRNA on the mRNA expression of several genes related to liver cancer using PCR based assays. Caspase 8 and GADD45B, genes related to apoptosis and DNA damage, were increased in HepG2 cells transfected with siRNA to linc-ROR-1 compared with control siRNA (Supplementary Table 2). Moreover siRNA to linc-ROR-1 did not significantly alter cell cycle progression following incubation with 10 μM of sorafenib, with an increase in percentage of cells in subG0/1 phase from 4.12% to 4.33% with siRNA to linc-ROR-1 compared with control (Fig. 5D). As linc-ROR was reported to inhibit cell apoptosis through repression of p53 [18], we examined p53 promoter activity (Fig. 5E). siRNA to linc-RoR-1 significantly increased p53 activity. Furthermore, siRNA to p53 increased cell viability of HepG2 cells during doxorubicin exposure (Fig. 5F). Altogether, these data indicate that the effects of linc-ROR could be mediated through p53 dependent signaling.
3.5. Does sorafenib increase linc-ROR expression and release within EV?
To examine the potential that linc-ROR could contribute to acquired chemoresistance, we examined the effect of chemotherapy on linc-ROR expression. First, we identified that incubation of HepG2 cells with sorafenib significantly increased linc-ROR expression compared with control cells (Fig. 6A). Moreover, sorafenib also increased linc-ROR expression within EVs (Fig. 6B). Together, these studies show that sorafenib increases linc-ROR expression within HCC cells and also within EV released by these cells. Linc-ROR expression was assessed by qRT-PCR in recipient HepG2 cells after incubation with varying concentrations of HepG2 derived EVs for 24 h, and noted to increase in a concentration-dependent manner supporting transfer of linc-ROR to recipient cells by EV (Fig. 6C). In addition, incubation with EVs derived from linc-ROR knockdown HepG2 cells significantly reduced recipient cell viability during sorafenib exposure compared with EVs derived from cells transfected with control siRNA (Fig. 6D) These data showing a correlation between resistance to chemotherapy and linc-ROR in EV’s are consistent with inter-cellular transfer of linc-ROR similar to the inter-cellular transfer of microRNA by EV that we have previously shown [12,17].
Fig. 6.
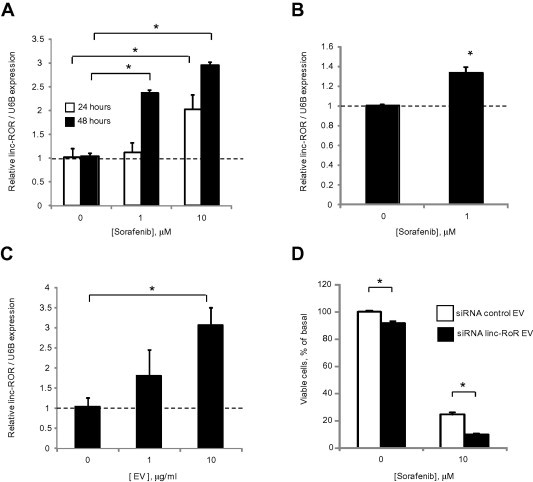
Cellular and extracellular vesicle linc-ROR in response to sorafenib. (A) HepG2 cells were incubated with varying concentrations of sorafenib, and linc-ROR expression was examined by qRT-PCR after 24 or 48 h. (B) HepG2 cells were incubated with 1 μM sorafenib or diluent controls. After 24 h, EVs were isolated and EV linc-ROR expression examined by qRT-PCR. (C) HepG2 cells were incubated with different concentrations of isolated EVs. After 24 h incubation, linc-ROR expression was assessed by qRT-PCR in recipient HepG2 cells. Expression of linc-ROR was normalized using the expression of RNU6B and expressed relative to controls. (D) HepG2 cells were transfected with either siRNA to linc-ROR-1 or non-targeting control siRNA for 24 h, then cultured in vesicle-depleted medium. After 72 h, EVs were collected and added to recipient HepG2 cells in 96 well plate. Recipient cells were then incubated with diluent or 10 μM sorafenib and cell viability assessed after 48 h using an MTS assay. Bars express the mean value ± SEM of 3 separate determinations. ∗p < 0.05.
3.6. Does linc-ROR enhance expression of tumor-initiating cells?
Tumor initiating cells with a stem-cell like phenotype may have an increased resistance to therapy. We first evaluated the effect of TGFβ using limiting dilution assays of self-renewal capacity. The proportion of single cells forming spheroids was increased by TGFβ (Fig. 7). Linc-ROR has been shown to be involved in epigenetic reprogramming in embryonic stem cells [20]. We therefore examined the effect of linc-ROR on spheroid formation, and observed a reduction in spheroid formation with knockdown of linc-ROR using siRNA (Fig. 7). In HCC, tumor initiating cells can be identified based on expression of cell surface markers such as CD133+ [21,22]. TGFβ increased expression of CD133+ cells (Fig. 8). These data indicate that TGFβ can enhance the growth of tumor initiating cells in vitro. We next examined the effect of linc-ROR on expression of CD133 expressing subpopulation using siRNA to linc-ROR-1 or NT controls. A decrease in CD133+ cells was noted with knockdown of linc-ROR compared to controls (Fig. 8). Moreover, siRNA to linc-ROR-1 attenuated the effects of TGFβ on CD133+ expression and colony formation. Thus, linc-ROR dependent expression of tumor-initiating cells may contribute to the effects of TGFβ on chemoresistance in HCC.
Fig. 7.
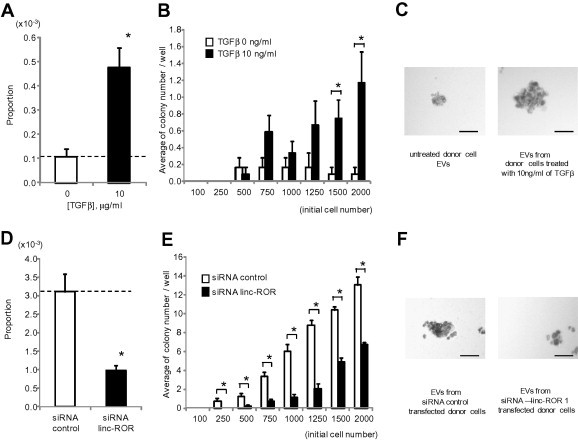
Effect of TGFβ and linc-ROR on spheroid formation. Stromal independent growth was examined in single cells in a limiting dilution assay. (A–C) EVs were isolated from donor HepG2 cells incubated with diluent or 10 ng/ml of TGFβ for 72 h. Recipient HepG2 cells were incubated with these EVs for 48 h. Cells were then collected and plated on Ultra-Low Attachment 96 well plates with serum free DMEM medium. (D–F) HepG2 cells were transfected with either siRNA to linc-ROR-1 or non-targeting control siRNA. After 48 h, cells were collected and plated on Ultra-Low Attachment 96 well plates with DMEM medium containing 1% FBS. Spheroid formation assays were performed as described in Section 2 after 7 days. (A, D) Proportion of cells forming spheroids. (B, E) Relationship between initial cell number and average number of colonies per well. Bars express the mean value ± SEM of 12 separate determinants based on Poisson distribution. ∗p < 0.05. (C, F) Representative photographs of spheroids at day 7 with 2000 cells/well incubated with EVs isolated from cells incubated with diluent or 10 ng/ml TGFβ, or transfected with control siRNA or siRNA to linc-ROR-1. Bar represents 100 μm.
Fig. 8.
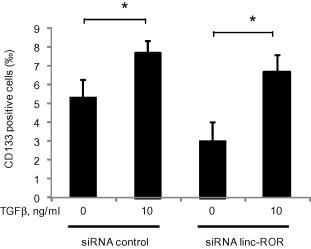
Effect of TGFβ and EV linc-ROR on CD133 tumor-initiating cells. HepG2 cells were transfected with either siRNA to linc-ROR-1 or non-targeting control siRNAs. After 48 h, cells were collected and plated in 10 cm dishes in EV-depleted medium followed by incubation for 72 h with 0 or 10 ng/ml TGFβ. EVs were then isolated from each cell. Recipient HepG2 cells were incubated with EV from each group for 48 h, and CD133 expression was assessed by flow cytometry. Bar graphs represent the mean and standard error of the permillage of CD133 positive cells from 3 separate determinants. ∗p < 0.05.
4. Discussion
The poor prognosis of advanced HCC is, in part, related to the lack of effective therapeutic agents for unresectable cancers. The only agent currently approved by the FDA is sorafenib, a multi-kinase inhibitor that exerts antiangiogenic and anti-tumor effects by blocking multiple growth factor pathways [23]. Modest survival benefits have been reported with sorafenib in two phase III randomized trials in patients with advanced HCC [2,24]. The use of this agent is associated with acquired chemoresistance which limits further benefit. Our findings are of importance in understanding potential mechanisms of acquired therapeutic resistance by identifying mediators and pathways by which exposure to chemotherapy modulates the local environment to limit toxicity.
Several lncRNA have been implicated in human liver diseases. A critical role for lncRNA in several diverse aspects of human disease is emerging but their specific involvement in targetable processes remains mostly unexplored. Linc-ROR is amongst the most significantly upregulated lncRNA in malignant hepatocytes. This lncRNA has been recognized to contribute to epigenetic regulators involved in pluripotency and lineage commitment [25]. Recent studies reported that linc-ROR plays a role in promoting survival in iPSCs and ESCs by preventing the activation of cellular stress pathways [20]. Our study shows that survival effects can be mediated in tumor cells following exposure to chemotherapy. The involvement of a long non-coding RNA in therapeutic responses adds to the current literature on functional capabilities of these non-coding RNA genes. The demonstration of a functional contribution to a clinically important effect provides a justification for future efforts to therapeutically target the expression of this lncRNA, through strategies similar to those proposed for targeting other non-coding RNAs [26].
Our studies have also identified a previously unrecognized role of linc-ROR as a mediator of cell-to-cell communication through the transfer of extracellular vesicles. This represents an important mechanism by which stressed cells can signal to other cells within the local microenvironment to orchestrate responses such as activation of survival pathways. These may then result in acquired chemoresistance within tissues and contribute to loss of therapeutic effect of agents such as sorafenib.
Accumulating evidence implicates cancer stem cells in cancer resistance to therapy as well as cancer growth and spread [22]. Cancer stem cells or tumor initiating cells are thought to represent the least sensitive cell subpopulation to chemotherapy [27]. Deregulated expression and activity of TGFβ has been characterized in liver cancer stem cells [28]. Targeting liver cancer stem cells is therefore an attractive strategy to improve therapeutic responses for liver cancers and other tumors.[21,29] In order to do this, an understanding of the mechanisms by which liver cancer stem cells can mediate chemoresistance is essential. Within the liver, stem cells can be identified on the basis of expression of cell surface markers such as CD133, CD90, CD44 and EpCAM [22,30,31]. CD133+ liver cancer stem cells have the capacity for self-renewal and ability to differentiate, and have been identified to confer resistance to chemotherapy. Tumors arising from these cells are difficult to treat and lethal [28]. An alternate mechanism contributing to a chemoresistant phenotype in poorly differentiated tumors may involve the loss of tumor cell differentiation with acquisition of epithelial mesenchymal transition. The involvement of TGFβ on this potential mechanism was not directly addressed in our study.
In conclusion, these findings provide several new mechanistic insights into acquired chemoresistance in HCC. The role of EV signaling in tumor cell responses to TGFβ was evaluated and specific EV lncRNA mediators such as linc-ROR that are involved in modulation of cellular responses to chemotherapy were identified. Targeting these inter-cellular signaling mechanisms and mediators may be useful in enhancing sensitivity and improving responses to conventional therapeutic agents that are used for the treatment of HCC.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
This project was supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Numbers R01DK069370 and under award UH2TR000884 supported by the NIH Common Fund through the Office of Strategic Coordination/Office of the NIH Director. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Financial support: Supported in part by Grants R01DK069370 and UH2TR000884 from the National Institutes of Health.
Appendix A. Supplementary data
This document contains Supplementary tables.
References
- 1.El-Serag H.B., Rudolph K.L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.F. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 3.Asghar U., Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J. Hepatol. 2012;56:686–695. doi: 10.1016/j.jhep.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Hernandez-Gea V., Toffanin S., Friedman S.L., Llovet J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512–527. doi: 10.1053/j.gastro.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y., Wang J., Asahina K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial–mesenchymal transition in liver injury. Proc. Natl. Acad. Sci. U.S.A. 2013;110:2324–2329. doi: 10.1073/pnas.1214136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshida K., Matsuzaki K., Mori S., Tahashi Y., Yamagata H., Furukawa F. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am. J. Pathol. 2005;166:1029–1039. doi: 10.1016/s0002-9440(10)62324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giannelli G., Bergamini C., Fransvea E., Sgarra C., Antonaci S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129:1375–1383. doi: 10.1053/j.gastro.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 8.Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Padua D., Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 10.Majumdar A., Curley S.A., Wu X., Brown P., Hwang J.P., Shetty K. Hepatic stem cells and transforming growth factor beta in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2012;9:530–538. doi: 10.1038/nrgastro.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biswas S., Guix M., Rinehart C., Dugger T.C., Chytil A., Moses H.L., Freeman M.L., Arteaga C.L. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Invest. 2007;117:1305–1313. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kogure T., Lin W.L., Yan I.K., Braconi C., Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011;54:1237–1248. doi: 10.1002/hep.24504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thery C., Ostrowski M., Segura E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 14.Saunderson S.C., Schuberth P.C., Dunn A.C., Miller L., Hock B.D., MacKay P.A. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J. Immunol. 2008;180:8146–8152. doi: 10.4049/jimmunol.180.12.8146. [DOI] [PubMed] [Google Scholar]
- 15.Skog J., Wurdinger T., van Rijn S., Meijer D.H., Gainche L., Sena-Esteves M. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valadi H., Ekstrom K., Bossios A., Sjostrand M., Lee J.J., Lotvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 17.Kogure T., Patel T. Isolation of extracellular nanovesicle microRNA from liver cancer cells in culture. Methods Mol. Biol. 2013;1024:11–18. doi: 10.1007/978-1-62703-453-1_2. [DOI] [PubMed] [Google Scholar]
- 18.Zhang A., Zhou N., Huang J., Liu Q., Fukuda K., Ma D. The human long non-coding RNA-RoR is a p53 repressor in response to DNA damage. Cell Res. 2013;23:340–350. doi: 10.1038/cr.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibb E.A., Brown C.J., Lam W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loewer S., Cabili M.N., Guttman M., Loh Y.H., Thomas K., Park I.H. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010;42:1113–1117. doi: 10.1038/ng.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L., Sun H., Zhao F., Lu P., Ge C., Li H. BMP4 administration induces differentiation of CD133+ hepatic cancer stem cells, blocking their contributions to hepatocellular carcinoma. Cancer Res. 2012;72:4276–4285. doi: 10.1158/0008-5472.CAN-12-1013. [DOI] [PubMed] [Google Scholar]
- 22.Ma S. Biology and clinical implications of CD133(+) liver cancer stem cells. Exp. Cell Res. 2013;319:126–132. doi: 10.1016/j.yexcr.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Wilhelm S.M., Carter C., Tang L., Wilkie D., McNabola A., Rong H. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 24.Cheng A.L., Kang Y.K., Chen Z., Tsao C.J., Qin S., Kim J.S. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 25.Boyer L.A., Plath K., Zeitlinger J., Brambrink T., Medeiros L.A., Lee T.I. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 26.Braconi C., Patel T. Non-coding RNAs as therapeutic targets in hepatocellular cancer. Curr. Cancer Drug Targets. 2012;12:1073–1080. [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X., Lingala S., Khoobyari S., Nolta J., Zern M.A., Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J. Hepatol. 2011;55:838–845. doi: 10.1016/j.jhep.2010.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.You H., Ding W., Rountree C.B. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology. 2010;51:1635–1644. doi: 10.1002/hep.23544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma H.I., Chiou S.H., Hueng D.Y., Tai L.K., Huang P.I., Kao C.L., Chen Y.W., Sytwu H.K. Celecoxib and radioresistant glioblastoma-derived CD133+ cells: improvement in radiotherapeutic effects. Laboratory investigation. J. Neurosurg. 2011;114:651–662. doi: 10.3171/2009.11.JNS091396. [DOI] [PubMed] [Google Scholar]
- 30.Yamashita T., Honda M., Nakamoto Y., Baba M., Nio K., Hara Y. Discrete nature of EpCAM+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology. 2013;57:1484–1497. doi: 10.1002/hep.26168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu Z., Hao X., Yan M., Yao M., Ge C., Gu J., Li J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int. J. Cancer. 2010;126:2067–2078. doi: 10.1002/ijc.24868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This document contains Supplementary tables.