

Abstract
Type 1 diabetes (T1D) is a multigenic disease caused by T-cell mediated destruction of the insulin producing pancreatic islet ß-cells. The earliest sign of islet autoimmunity in NOD mice, islet leukocytic infiltration or insulitis, is obvious at around 5 weeks of age. The molecular alterations that occur in T cells prior to insulitis and that may contribute to T1D development are poorly understood. Since CD4 T-cells are essential to T1D development, we tested the hypothesis that multiple genes/molecular pathways are altered in these cells prior to insulitis. We performed a genome-wide transcriptome and pathway analysis of whole, untreated CD4 T-cells from 2, 3, and 4 week-old NOD mice in comparison to two control strains (NOR and C57BL/6). We identified many differentially expressed genes in the NOD mice at each time point. Many of these genes (herein referred to as NOD altered genes) lie within known diabetes susceptibility (insulin-dependent diabetes, Idd) regions, e.g. two diabetes resistant loci, Idd27 (tripartite motif-containing family genes) and Idd13 (several genes), and the CD4 T-cell diabetogenic activity locus, Idd9/11 (2 genes, KH domain containing, RNA binding, signal transduction associated 1 and protein tyrosine phosphatase 4a2). The biological processes associated with these altered genes included, apoptosis/cell proliferation and metabolic pathways (predominant at 2 weeks); inflammation and cell signaling/activation (predominant at 3 weeks); and innate and adaptive immune responses (predominant at 4 weeks). Pathway analysis identified several factors that may regulate these abnormalities: eight, common to all 3 ages (interferon regulatory factor 1, hepatic nuclear factor 4, alpha, transformation related protein 53, BCL2-like 1 (lies within Idd13), interferon gamma, interleukin 4, interleukin 15, and prostaglandin E2); and two each, common to 2 and 4 weeks (androgen receptor and interleukin 6); and to 3 and 4 weeks (interferon alpha and interferon regulatory factor 7). Others were unique to the various ages, e.g. myelocytomatosis oncogene, jun oncogene, and amyloid beta (A4) to 2 weeks; tumor necrosis factor, transforming growth factor, beta 1, NF?B, ERK, and p38MAPK to 3 weeks; and interleukin 12 and signal transducer and activator of transcription 4 to 4 weeks. Thus, our study demonstrated that expression of many genes that lie within several Idds (e.g. Idd27, Idd13 and Idd9/11) was altered in CD4 T-cells in the early induction phase of autoimmune diabetes and identified their associated molecular pathways. These data offer the opportunity to test hypotheses on the roles played by the altered genes/molecular pathways, to understand better the mechanisms of CD4 T-cell diabetogenesis, and to develop new therapeutic strategies for T1D.
Keywords: NOD mice, Preinsulitis, CD4 T-cells, Genome-wide gene expression profiling, Molecular pathway analysis, Type 1 diabetes susceptibility regions
Highlights
-
•
We analyzed molecular pathways in NOD CD4 T-cells in the preinsulitis stage of T1D.
-
•
Many of the altered genes lie within known Type 1 diabetes susceptibility gene loci.
-
•
Abnormal processes included apoptosis, metabolism, inflammation and immune response.
-
•
We identified several altered molecular pathways, including potential regulators.
1. Introduction
Type 1 (autoimmune) diabetes (T1D) is a multigenic disease that, in humans and NOD mice, results from T-cell mediated destruction of insulin producing beta cells in the pancreatic islets [1–4]. Insulitis, the earliest sign of autoimmune pathology in the pancreas of NOD mice, in our colony becomes obvious at around 5 weeks of age, at which stage accumulation of leukocytes is observed around the pancreatic islets. This process progressively intensifies leading to T lymphocytic infiltration of the pancreatic islets and eventual massive destruction of the insulin producing beta cells with clinical disease occurring at 12 weeks of age or later. The molecular alterations that occur in T cells prior to insulitis and that may contribute to T1D pathogenesis are poorly understood.
It is well established that genetic predisposition is a major factor in the etiology of T1D. The major histocompatibility (MHC) class II molecule H2-Ag7 in NOD mice (or HLA-DQ2 and -DQ8 in humans) is the strongest genetic determinant for T1D development [1–4]. The MHC class II-restricted CD4 T-cells are essential for the development of autoimmune diabetes [2,5]. To this end, (1) CD4 T-cells from spleens of NOD mice are reactive to pancreatic beta cell antigens [6]; (2) Spleen CD4 T-cells from NOD mice can transfer diabetes to young NOD and NOD.scid mice [7,8]; (3) NOD mice lacking CD4 T-cells do not develop diabetes [9]. Reconstitution of these mice with NOD spleen CD4 T-cells leads to development of diabetes [5,10]; and (4) transgenic NOD mice harboring CD4 T-cells with T cell receptor reactive to islet antigens develop insulitis and diabetes [8]. The chromosomal regions that modulate T1D susceptibility in NOD mice are designated insulin-dependent diabetes regions (Idd). In conjunction with the MHC locus, which is required but alone insufficient for T1D development, over 20 non-MHC Idd loci also contribute to the disease process in NOD mice [3,4,11–14]. Although, the identities of some of the non-MHC Idd genes that interactively contribute to the diabetogenic process in CD4 T-cells of NOD mice have been revealed [5,10,15–19], most of these genes and/or their interactions remain unknown.
The conventional approach in the field to understanding the pathogenesis of T1D has been mainly via targeted analysis at the individual gene or loci level. Identification of all the genes that together cause diabetes (a multigenic disease) via these approaches can be tortuous as each gene may only contribute weakly to the pathology. While these approaches have yielded useful information on how identified genes may interact with each other to confer disease susceptibility and/or protection, a whole cellular and/or molecular systems analysis (non-targeted approach) provides the opportunity to simultaneously interrogate the genes/pathways that are involved in the disease process [20]. A comprehensive understanding of these molecular interactions is important because it is now clear that the best targets for development of novel prevention and/or treatment interventions for complex trait diseases may not be the disease associated genes per se but rather their interaction partners, upstream regulators or downstream targets, or the molecular network [21–24]. Thus, to gain insights into the molecular networks that might play a role in the diabetogenic activity of CD4 T-cells in the early induction phase of T1D, we evaluated the transcriptomes of untreated, whole CD4 T-cells collected from the spleens of NOD mice in the period prior to overt insulitis and inferred the associated altered molecular networks using a suite of complementary bioinformatics tools.
2. Materials and methods
2.1. Mice, sample collection and microarray procedures
Animal procedures were approved by the University of Tennessee (UT) Health Science Center and Veteran Affairs (VA) Medical Center Animal Care and Use Committee (Protocol Numbers: UT 1159/VA 00157). Breeder mice were purchased from the Jackson Laboratory and housed at the VA animal facility. Spleen leukocytes were collected, as described previously [25,26] from female NOD mice at 2, 3 and 4 weeks of age (representing the period prior to overt insulitis) and from two matched control strains, NOR and C57BL/6 (C57); n = 5 for each strain and age group, except NOD 2 week, where n = 4. CD4 T-cells were then negatively separated by magnetic beads according to the manufacturer’s protocol (Miltenyi Biotec). Purity was assessed by flow cytometry using FITC-conjugated anti-mouse CD4 monoclonal antibodies (Becton Dickinson); only samples of > 90% purity were used in the study. Total RNA was extracted from untreated, whole CD4 T-cells, as described previously [26,27]. A total of 1–1.5 µg of total RNA was processed with Two-Cycle Target labeling protocol and hybridized on Affymetrix, Mouse430_2 expression arrays, according to the manufacturer’s instructions. Normalization, scaling, and basic evaluation of the quality of the expression data from each chip were conducted using the GCOS software (Affymetrix), as described previously [25,26]. The microarray data sets are available in the gene expression omnibus repository [GSE46600]. Microarray results were validated by quantitative Real-time PCR, as described previously [25] (Fig. S1). Expression values were normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). The target genes, primers and probes are listed in Table S1.
2.2. Statistical analysis
Statistical analysis of the microarray data was conducted as previously described [25,26]. Filtration of the probe sets present on the chip array (~60,000) identified ~31,000 probe sets that had a present/marginal expression flag in at least one of the samples. We then performed one-way ANOVA (at various statistical stringencies) on the filtered probe sets for each age separately in order to define lists of age-specific genes that were differentially expressed between strains. Lists generated at either p < 0.005 (“smaller” list) or p < 0.05 (“larger” list) adjusted with Benjamini–Hochberg multiple test correction (corresponding to a false discovery rate (FDR) of 0.5% or 5%, respectively) were used in further analysis. Finally, we conducted hierarchical clustering on these lists as previously described [27] to identify genes that were uniquely differentially expressed in NOD mice relative to both control strains. These genes are herein referred to as NOD altered genes.
2.3. Data mining analyses
We subjected the lists of NOD altered genes to data mining using a suite of modern bioinformatics tools. Enriched gene ontology (GO) categories and KEGGs pathways were determined using WebGestalt Gene Set Analysis Toolkit (http://bioinfo.vanderbilt.edu/webgestalt [28]), as described previously [25]. To identify transcription regulators whose binding sites were significantly over-represented in the promoters of the NOD altered gene sets, we used the PRIMA (PRomoter Integration in Microarray Analysis) program of the EXPANDER suite (EXpression Analyzer and DisplayER; http://acgt.cs.tau.ac.il/expander/overview.html; Tel Aviv University, Israel). PRIMA achieves this by utilizing known models for transcription factor binding sites. Corresponding transcription regulators are considered to be candidate regulators of the corresponding set of genes. We used Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com) as described in detail previously [25] to identify de novo molecular networks that are associated with the NOD altered genes.
3. Results
3.1. Genes differentially expressed in NOD CD4 T-cells at the preinsulitis stage of autoimmune diabetes
We compared the whole genome mRNA expression in CD4 T-cells from 2-, 3-, and 4-week old NOD mice to that of two matched control strains, NOR and C57BL/6 (C57). NOR shares ~88% of its genome with NOD mice, including the diabetogenic H2g7 MHC haplotype and several important non-MHC T1D susceptibility loci [1–4,29]. C57, on the other hand, is a more genetically distantly related strain to NOD. Yet, like C57, NOR is both insulitis- and diabetes-free.
We identified 362, 982, and 581 probe sets (genes) with highly significant expression differences between strains (p < 0.005, Benjamini–Hochberg; FDR of 0.5%) at 2, 3, and 4 weeks, respectively. As expected, the majority of these genes had a similar pattern of expression in NOD and NOR compared to C57 (Fig. 1). The focus of our study was to identify genes in NOR mice whose expression was similar to that in C57 but different from that in NOD, i.e. genes differentially expressed in NOD relative to both NOR and C57, and herein referred to as NOD altered genes. These constitute prospective candidate genes for protection of NOR mice against diabetes. Thus, we identified a total of 58, 115, and 65 probe sets whose expression was altered in NOD at 2, 3 and 4 weeks, respectively, compared to both controls (clusters of lower or higher expression in NOD are indicated by arrows in Fig. 1). These represented 56, 107, and 60 different genes, respectively, shown in Tables 1–4. The great majority of these genes were of lower expression in NOD mice compared to controls: ~70% at 2 weeks; ~72% at 3 weeks; and ~87% at 4 weeks. Twenty-five genes (72% of which was of lower expression in NOD mice) were common to all 3 ages. Results for qRT-PCR validation of eight genes are shown in Fig. S1 (3 of higher expression in NOD mice: protein tyrosine phosphatase 4a2 (Ptp4a2), biogenesis of organelles complex-1, subunit 6 (Bloc1s6, pallidin) and transmembrane protein 87A (Tmem87a); and 5 of lower expression in NOD mice: tripartite motif-containing 5 (Trim5), tripartite motif-containing 12A (Trim12a), Gatm (glycine amidinotransferase (l-arginine:glycine amidinotransferase)), lymphocyte antigen 6 complex, locus C1 (Ly6c1), and receptor transporter protein 4 (Rtp4) compared to control mice). The major selected functional categories (as determined by the authors based on information obtained from literature searches) included immune response, apoptosis/cell proliferation, transcription, zinc-ion binding, protein/nucleic acid binding, “Other enzymes” for enzymes not already assigned to a specific category, and “Other” for genes that did not fall in one major category. Probe sets of unknown identification/function are listed under “Unknown”. Based on these categories, a higher percentage of NOD altered genes (~22%) were involved in apoptosis/cellular proliferation at each of 2 or 3 weeks than at 4 weeks (12%). Conversely, a higher percentage of genes (22%) were involved in immune response at 4 weeks than at 2 or 3 weeks (4.1% and 12%, respectively). Overall, a high percentage of genes at all 3 ages were involved in enzymatic activity, including several kinases/phosphatases at 3 weeks. Moreover, a large number of the CD4 T-cell NOD altered genes lie within known Idds, 52%, 47% and 38% at 2, 3, and 4 weeks, respectively (Tables 1–4). A total of 14 (25%), 20 (~19%) and 10 (~17%) genes at 2, 3, and 4 weeks, respectively, lie within Idd13, a region located on chromosome (Chr) 2 and previously identified to confer resistance to diabetes in NOR mice [4,15–17,30]. These genes included 6 that were common to all 3 ages: Bloc1s6, Trp53bp1 (transformation related protein 53 binding protein 1), Tmem87a, Ctdspl2 (CTD (carboxy-terminal domain, RNA polymerase II, polypeptide A) small phosphatase like 2), Gatm and Raly (hnRNP-associated with lethal yellow). Two genes (Khdrbs1 - KH domain containing, RNA binding, signal transduction associated 1 and Ptp4a2, both altered at 2 and 3 weeks, but only Khdrbs1 at 4 weeks) lie within Idd9/11, a region on Chr4 that also confers resistance to diabetes in NOR. This region has also been demonstrated previously by several groups to regulate the diabetogenic activity of CD4 T-cells [5,10,18,19].
Fig. 1.
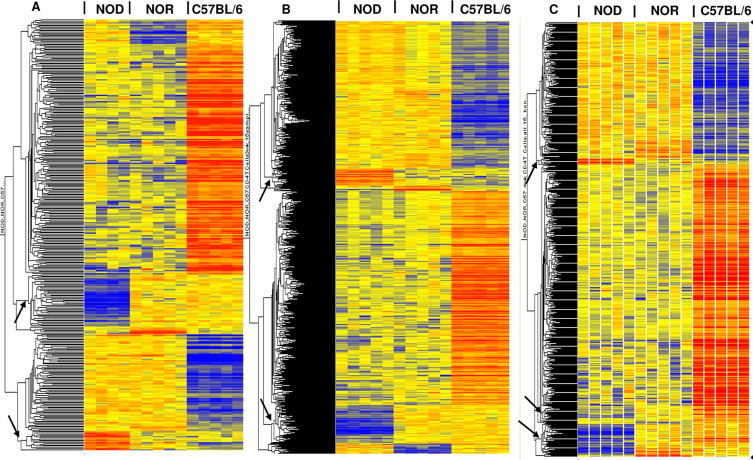
Hierarchical clusters of genes whose expression was altered in CD4 T-cells. 362 (A), 982 (B) and 581 (C) genes were differentially expressed between strains at 2, 3, and 4 weeks of age, respectively. The lists were identified by a one-way ANOVA of ~31,000 filtered probe sets at p < 0.005, with Benjamini–Hochberg multiple test correction. A total of 58, 115, and 65 probe sets were differentially expressed in NOD relative to both controls (NOR and C57BL/6) at 2-, 3- and 4-weeks, respectively; clusters of these NOD altered genes are indicated by arrows. The color intensity of the rectangles representing each gene for each sample (n = 5 for each strain/age, except NOD 2 week, where n = 4) indicates the degree of increase (red) or decrease (blue) of the gene expression signal relative to the mean signal intensity (yellow).
Table 1.
Genes differentially expressed in CD4 T-cells from 2 week-old NOD mice.
| Probe set ID | Gene symbol | Fold change | Adjusted p-value | Chromosome (Chr) | T1D susceptibility region |
|---|---|---|---|---|---|
| Immune response | |||||
| 1418642_at (L) | Lcp2 | –9.65 | 0.00048 | Chr11 | - |
| 1457088_at (H) | Pldn | 5.48 | 0.00128 | Chr2 | Idd13 |
| Apoptosis/cell proliferation | |||||
| 1440493_at (L) | Galnt10 | –37.09 | 0.000217 | Chr11 | - |
| 1457812_at (L) | Trp53bp1 | –30.33 | 0.000291 | Chr2 | Idd13 |
| 1438462_x_at (L) | Khdrbs1 | –7.42 | 0.000662 | Chr4 | Idd9/11 |
| 1417714_x_at (H) | Hba-a1 | 5.36 | 0.0031 | Chr11 | - |
| 1449716_s_at (H) | Nrd1 | 2.77 | 0.00321 | Chr4 | - |
| 1439650_at (L) | Rtn4 | –5.74 | 0.00296 | Chr11 | - |
| 1440358_at (H) | Arhgef15 | 4.82 | 0.00184 | Chr11 | - |
| 1428361_x_at (L) | Hba-a1 | –4.11 | 0.00104 | Chr11 | - |
| 1444890_at (L) | Mprip | –4.08 | 0.00368 | Chr11 | - |
| 1449052_a_at (L) | Dnmt3b | –3.24 | 0.00048 | Chr2 | Idd13 |
| 1433681_x_at (L) | Capn3 | –2.30 | 0.00214 | Chr2 | Idd13 |
| Transcription | |||||
| 1458094_at (L) | Zfp407 | –6.66 | 0.00224 | Chr18 | Idd21.1 |
| 1458274_at (H) | Zfp69 | 3.06 | 0.00288 | Chr4 | - |
| 1446147_at (H) | Rbm39 | 4.15 | 0.00129 | Chr2 | Idd13 |
| Transport | |||||
| 1455735_at (H) | Ap1s3 | 4.28 | 0.000512 | Chr1 | Idd5.4a/5.4 |
| 1417963_at (H) | Pltp | 3.00 | 0.00419 | Chr2 | - |
| 1420897_at (L) | Snap23 | –1.81 | 0.000668 | Chr2 | Idd13 |
| Zinc-ion binding | |||||
| 1437432_a_at (L) | Trim12a | –769.41 | 2.45E-08 | Chr7 | Idd27 |
| 1443858_at (L) | Trim/12c (Trim5) | –238.91 | 3.05E-07 | Chr7 | Idd27 |
| Protein/nucleic acid binding | |||||
| 1424454_at (H) | Tmem87a | 13.40 | 0.00114 | Chr2 | Idd13 |
| 1455863_at (L) | Spata5l1 | –5.40 | 0.00217 | Chr2 | - |
| 1419276_at (H) | Enpp1 | 2.07 | 0.00105 | Chr10 | - |
| 1453065_at (H) | Aldh5a1 | 1.78 | 0.00337 | Chr13 | - |
| Other enzymes | |||||
| 1442424_at (L) | Ctdspl2 | –46.26 | 0.000831 | Chr2 | Idd13 |
| 1418035_a_at (L) | Prim2 | –11.25 | 5.55E-05 | Chr1 | Idd26 |
| 1423569_at (L) | Gatm | –7.41 | 0.000112 | Chr2 | Idd13 |
| 1453009_at (L) | Cpm | –6.18 | 0.00158 | Chr10 | - |
| 1416494_at (L) | Ndufs5 | –5.21 | 0.000538 | Chr4 | - |
| 1427943_at (H) | Acyp2 | 2.75 | 4.07E-05 | Chr11 | - |
| 1427302_at (H) | Enpp3 | 2.66 | 0.0027 | Chr10 | - |
| 1417826_at (L) | Akr1e1 | –6.52 | 0.000273 | Chr13 | - |
| 1455219_at (L) | Slx1b | –5.00 | 0.00184 | Chr7 | Not Assigned |
| 1442466_a_at (L) | Ppip5k1 | –4.14 | 0.00211 | Chr2 | Idd13 |
| 1444377_at (L) | Psmb2 | –3.41 | 0.0028 | Chr4 | Idd11 |
| 1435129_at (H) | Ptp4a2 | 2.37 | 0.00487 | Chr4 | Idd9/11 |
| 1454772_at (L) | Snrnp200 | –2.32 | 0.00189 | Chr2 | Idd13 |
| 1415878_at (L) | Rrm1 | –1.90 | 0.00407 | Chr7 | - |
| 1451998_at (L) | Tasp1 | –1.78 | 0.000499 | Chr2 | Idd13 |
| Other (unknown transcripts are indicated below in the legend) | |||||
| 1444741_at (L) | Dock2 | –144.49 | 3.18E-06 | Chr11 | - |
| 1436061_at (L) | Chaf1a | –21.85 | 0.000538 | Chr17 | - |
| 1442824_at (L) | Raly | –13.69 | 0.000313 | Chr2 | Idd13 |
| 1452426_x_at (L) | LOC433762 | –12.00 | 0.000549 | Chr4 | - |
| 1452359_at (L) | Rell1 | –2.53 | 0.000662 | Chr5 | - |
| 1457822_at (L) | Tmem131 | –13.55 | 0.00438 | Chr1 | Idd26 |
| 1445214_at (L) | Rex2 | –9.26 | 0.000783 | Chr4 | Idd9.2 |
| 1456635_at (L) | Sp110 | –8.01 | 0.00156 | Chr1 | Idd5.4a/5.4 |
| 1428587_at (L) | Tmem41b | –4.36 | 0.000289 | Chr7 | Idd27 |
| 1424509_at (H) | Cd177 | 4.03 | 0.00284 | Chr7 | - |
| 1435792_at (H) | Csprs | 3.86 | 0.00199 | Chr1 | Idd5.4a/5.4 |
| 1458684_at (H) | Ss18 | 2.77 | 0.00407 | Chr18 | Idd21.3 |
| 1424721_at (L) | Mfap3 | –2.54 | 0.000895 | Chr11 | - |
| 1425331_at (L) | Zfp106 | –2.10 | 0.00183 | Chr2 | Idd13 |
Statistical analysis (1-way-ANOVA; p < 0.005, Benjamini–Hochberg) followed by hierarchical clustering identified 58 differentially expressed probe sets (representing 56 different genes) at 2 weeks; L and H, respectively, indicate lower or higher expression in NOD mice compared to control mice, NOR and C57BL/6. Two unknown transcripts (both on Chr4) included: 9930104L06Rik (L) and 2510039O18Rik (L; Idd9.2). Fold change (FC) was calculated by ratio of means of expression in NOD mice versus controls. Dashes indicate those genes are not located within a known T1D susceptibility region (Idd); all indicated Idds (except one, Idd5.4a/5.4) were identified as conferring resistance to diabetes (http://www.t1dbase.org). Genes highlighted in bold font were differentially expressed at all 3 ages, 2, 3 and 4 weeks.
Table 2.
Genes differentially expressed in CD4 T-cells from 3 week-old NOD mice (continued in Table 3).
| Probe set ID | Gene symbol | Fold change | Adjusted p-value | Chromosome (Chr) | T1D susceptibility region |
|---|---|---|---|---|---|
| Immune response | |||||
| 1418642_at (L) | Lcp2 | –12.20 | 0.00339 | Chr11 | - |
| 1457088_at (H) | Pldn | 5.67 | 0.000481 | Chr2 | Idd13 |
| 1448550_at (L) | Lbp | –3.98 | 0.000176 | Chr2 | Idd13 |
| 1435529_at (L) | Ifit1lb | –2.91 | 0.00242 | Chr19 | - |
| 1460394_a_at (H) | Inppl1 | 2.76 | 0.00318 | Chr7 | - |
| 1450783_at (L) | Ifit1 | –2.73 | 0.00395 | Chr19 | - |
| 1424775_at (L) | Oas1a | –2.20 | 0.00115 | Chr5 | - |
| 1418580_at (L) | Rtp4 | –2.10 | 0.00317 | Chr16 | - |
| 1448940_at (L) | Trim21 | –2.03 | 0.00399 | Chr7 | Idd27 |
| 1435560_at (L) | Itgal | –1.84 | 0.00242 | Chr7 | Not Assigned |
| 1452956_a_at (L) | Ifi27 | –1.74 | 0.00484 | Chr6 | |
| Apoptosis/cell proliferation | |||||
| 1457812_at (L) | Trp53bp1 | –16.30 | 0.000964 | Chr2 | Idd13 |
| 1440493_at (L) | Galnt10 | –11.41 | 0.000748 | Chr11 | - |
| 1417714_x_at (H) | Hba-a1 | 10.69 | 0.000156 | Chr11 | - |
| 1438462_x_at (L) | Khdrbs1 | –5.59 | 0.000211 | Chr4 | Idd9/11 |
| 1449716_s_at (H) | Nrd1 | 2.36 | 0.00133 | Chr4 | - |
| 1452677_at (H) | Pnpt1 | 13.80 | 0.0012 | Chr11 | - |
| 1440319_at (L) | Mef2a | –13.53 | 0.00227 | Chr7 | - |
| 1444890_at (L) | Mprip | –9.37 | 0.00439 | Chr11 | - |
| 1439650_at (L) | Rtn4 | –5.24 | 0.00318 | Chr11 | - |
| 1433681_x_at (L) | Capn3 | –2.89 | 0.000442 | Chr2 | Idd13 |
| 1428819_at (L) | Mapre1 | –2.78 | 0.00242 | Chr2 | Idd13 |
| 1449052_a_at (L) | Dnmt3b | –2.77 | 1.06E-05 | Chr2 | Idd13 |
| 1436647_at (H) | Ttbk2 | 2.69 | 0.00254 | Chr2 | Idd13 |
| 1419269_at (H) | Dut | 2.54 | 0.0016 | Chr2 | Idd13 |
| 1444500_at (L) | Ahsa1 | –2.48 | 0.0042 | Chr12 | - |
| 1436025_at (H) | Ccdc88a | 2.40 | 0.000697 | Chr11 | - |
| 1441937_s_at (L) | Pink1 | –2.11 | 0.00159 | Chr4 | - |
| 1448184_at (L) | Fkbp1a | –1.94 | 0.00449 | Chr2 | Idd13 |
| 1422808_s_at (L) | Dock2 | –1.84 | 0.000999 | Chr11 | - |
| 1457813_at (H) | Trp53bp1 | 1.62 | 0.0044 | Chr2 | Idd13 |
| 1448027_at (L) | Ncoa3 | –1.47 | 0.00277 | Chr2 | - |
| 1419562_at (L) | Birc6 | –1.31 | 0.00393 | Chr17 | - |
| Transcription | |||||
| 1458094_at (L) | Zfp407 | –8.70 | 0.000878 | Chr18 | Idd21.1 |
| 1458274_at (H) | Zfp69 | 3.25 | 0.00264 | Chr4 | - |
| 1449592_at (H) | Tcf15 | 10.64 | 0.000988 | Chr2 | Idd13 |
| 1459026_at (L) | Snw1 | –10.00 | 0.00152 | Chr12 | |
| 1417961_a_at (L) | Trim30 | –7.39 | 2.84E-05 | Chr7 | Idd27 |
| 1445214_at (L) | Zfp715 | –4.30 | 0.000612 | Chr7 | - |
| 1447703_x_at (L) | Zfp593 | –4.09 | 5.47E-05 | Chr4 | - |
| 1442356_at (L) | Max | –2.24 | 0.00491 | Chr12 | - |
| 1426765_at (L) | Commd7 | –2.09 | 0.000466 | Chr2 | Idd13 |
| 1436416_x_at (L) | Fxc1 | –1.81 | 6.23E-05 | Chr7 | Idd27 |
| 1450350_a_at (L) | Jdp2 | –1.39 | 0.00475 | Chr12 | - |
| Transporters | |||||
| 1460617_s_at (L) | Rab6b | –15.69 | 0.000612 | Chr9 | - |
| 1458426_at (L) | Kif1b | –3.72 | 0.000704 | Chr4 | Idd9.2 |
| 1455735_at (H) | Ap1s3 | 3.09 | 0.00386 | Chr1 | Idd5.4a/5.4 |
| 1424615_at (L) | Pgap2 | –2.18 | 0.0019 | Chr7 | Idd27 |
| 1424211_at (H) | Slc25a33 | 1.83 | 0.0012 | Chr4 | - |
| Zinc-ion binding | |||||
| 1437432_a_at (L) | Trim12a | –252.63 | 1.20E-08 | Chr7 | Idd27 |
| 1443858_at (L) | Trim12c (Trim5) | –140.08 | 5.39E-07 | Chr7 | Idd27 |
| 1435665_at (L) | Trim30d | –3.16 | 0.00227 | Chr7 | Idd27 |
Statistical analysis (1-way-ANOVA; p < 0.005, Benjamini–Hochberg) followed by hierarchical clustering identified 115 differentially expressed probe sets (representing 107 different genes) at 3 weeks; L and H, respectively, indicate lower or higher expression in NOD mice compared to control mice, NOR and C57BL/6. This table shows a partial list of the genes; the remaining genes are shown in Table 3. Fold change (FC) was calculated by ratio of means of expression in NOD mice versus controls. Dashes indicate those genes are not located within a known T1D susceptibility region (Idd); all indicated Idds (except one, Idd5.4a/5.4) were identified as conferring resistance to diabetes (http://www.t1dbase.org). Genes highlighted in bold font were differentially expressed at all 3 ages, 2, 3 and 4 weeks.
Table 3.
Genes differentially expressed in CD4 T-cells from 3 week-old NOD mice (continued from Table 2).
| Probe set ID | Gene symbol | Fold change | Adjusted p-value | Chromosome (Chr) | T1D susceptibility region |
|---|---|---|---|---|---|
| Protein/nucleic acid binding | |||||
| 1424454_at (H) | Tmem87a | 13.41 | 0.00264 | Chr2 | Idd13 |
| 1455863_at (L) | Spata5l1 | –10.62 | 7.71E-06 | Chr2 | - |
| 1459957_at (H) | Tnrc6a | 5.53 | 0.000595 | Chr7 | Not Assigned |
| 1429247_at (L) | Anxa6 | –4.00 | 0.000999 | Chr11 | Idd4.3 |
| 1458684_at (H) | Ss18 | 2.56 | 0.00101 | Chr18 | Idd21.3 |
| 1419276_at (H) | Enpp1 | 2.48 | 0.000575 | Chr10 | - |
| 1416559_at (L) | Rrp8 | –2.32 | 0.00226 | Chr7 | Idd27 |
| 1440416_at (H) | Usp46 | 2.07 | 0.00153 | Chr5 | - |
| 1429337_at (L) | Tmem87b | –1.52 | 0.00159 | Chr2 | Idd13 |
| 1428308_at (L) | Pdrg1 | –1.36 | 0.00361 | Chr2 | Idd13 |
| Other enzymes | |||||
| 1442424_at (L) | Ctdspl2 | –53.79 | 0.000129 | Chr2 | Idd13 |
| 1453009_at (L) | Cpm | –16.36 | 0.000671 | Chr10 | - |
| 1427302_at (H) | Enpp3 | 3.45 | 0.0016 | Chr10 | - |
| 1418035_a_at (L) | Prim2 | –10.26 | 1.11E-05 | Chr1 | Idd26 |
| 1423569_at (L) | Gatm | –8.19 | 7.47E-05 | Chr2 | Idd13 |
| 1416494_at (L) | Ndufs5 | –6.65 | 1.44E-05 | Chr4 | - |
| 1427943_at (H) | Acyp2 | 3.91 | 0.000117 | Chr11 | - |
| 1444377_at (L) | Psmb2 | –2.86 | 0.00325 | Chr4 | Idd11 |
| 1449862_a_at (H) | Pi4k2b | 2.85 | 0.00324 | Chr5 | - |
| 1451277_at (L) | Zadh2 | –2.50 | 0.000108 | Chr18 | Idd21.1 |
| 1435129_at (H) | Ptp4a2 | 2.47 | 0.0033 | Chr4 | Idd9/11 |
| 1420613_at (L) | Ptp4a2 | –2.22 | 0.000306 | Chr4 | Idd9/11 |
| 1455075_at (L) | Pigv | –2.15 | 0.00133 | Chr4 | - |
| 1419125_at (L) | Ptpn18 | –1.99 | 0.00395 | Chr1 | Idd26 |
| 1453343_s_at (H) | Vrk2 | 1.97 | 0.00454 | Chr11 | - |
| Other | |||||
| 1444741_at (L) | Dock2 | –46.41 | 2.48E-05 | Chr11 | - |
| 1436061_at (L) | Chaf1a | –24.94 | 0.000216 | Chr17 | - |
| 1442824_at (L) | Raly | –23.88 | 0.000306 | Chr2 | Idd13 |
| 1452426_x_at (L) | LOC433762 | –22.39 | 0.00114 | Chr4 | - |
| 1452359_at (L) | Rell1 | –2.84 | 0.00152 | Chr5 | - |
| 1444878_at (L) | Nf1 | –10.91 | 0.000999 | Chr11 | - |
| 1428318_at (H) | Smgc | 7.02 | 0.0019 | Chr15 | - |
| 1424133_at (H) | Tmem98 | 7.32 | 4.09E-05 | Chr11 | - |
| 1432391_at (L) | Ccdc21 | –5.10 | 0.000648 | Chr4 | - |
| 1434112_at (L) | Lphn2 | –3.65 | 0.00201 | Chr3 | - |
| 1428587_at (L) | Tmem41b | –3.57 | 1.06E-05 | Chr7 | Idd27 |
| 1456685_at (L) | HMP19/Nsg2 | –3.58 | 0.000655 | Chr11 | - |
| 1451477_at (H) | Znf41-ps | 2.90 | 2.66E-05 | Chr4 | Idd9.2 |
| 1444714_at (L) | Dcdc2b | –2.77 | 0.00166 | Chr4 | - |
| 1453465_x_at (L) | Ppp1r14c | –2.30 | 0.00159 | Chr10 | - |
| 1431259_at (H) | Adal | 2.28 | 0.0044 | Chr2 | Idd13 |
| 1424721_at (L) | Mfap3 | –1.93 | 0.00143 | Chr11 | - |
| 1445186_at (H) | Stc2 | 1.78 | 0.00367 | Chr11 | - |
| 1416935_at (L) | Trpv2 | –1.61 | 0.00289 | Chr11 | - |
| Unknown | |||||
| 1428604_at (H) | 2610305D13Rik | 28.19 | 0.00125 | Chr4 | Idd9.2 |
| 1434550_at (L) | 3830406C13Rik | –16.63 | 0.00157 | Chr14 | - |
| 1436388_a_at (L) | 3830406C13Rik | –10.85 | 0.0025 | Chr14 | - |
| 1440214_at (L) | A630001G21Rik | –8.76 | 0.0044 | Chr1 | Idd5.4a/5.4 |
| 1429203_at (H) | 2410076I21Rik | 3.10 | 5.59E-05 | Chr9 | Idd2 |
| 1438646_x_at (L) | 2510039O18Rik | –2.69 | 0.00214 | Chr4 | Idd9.2 |
| 1458419_at (L) | E130215H24Rik | –2.11 | 0.00254 | Chr2 | Idd13 |
Statistical analysis (1-way-ANOVA; p < 0.005, Benjamini–Hochberg) followed by hierarchical clustering identified 115 differentially expressed probe sets (representing 107 different genes) at 3 weeks; L and H, respectively, indicate lower or higher expression in NOD mice compared to control mice, NOR and C57BL/6. This table shows a continuation of the gene list from Table 2. Fold change (FC) was calculated by ratio of means of expression in NOD mice versus controls. Dashes indicate those genes are not located within a known T1D susceptibility region (Idd); all indicated Idds (except one, Idd5.4a/5.4) were identified as conferring resistance to diabetes (http://www.t1dbase.org). Genes highlighted in bold font were differentially expressed at all 3 ages, 2, 3 and 4 weeks.
Table 4.
Genes differentially expressed in CD4 T-cells from 4 week-old NOD mice.
| Probe set ID | Gene symbol | Fold change | Adjusted p-value | Chromosome (Chr) | T1D susceptibility region |
|---|---|---|---|---|---|
| Immune response | |||||
| 1418642_at (L) | Lcp2 | –20.53 | 7.58E-06 | Chr11 | - |
| 1457088_at (H) | Pldn | 4.60 | 0.0027 | Chr2 | Idd13 |
| 1421571_a_at (L) | Ly6c1 | –9.30 | 8.63E-07 | Chr15 | - |
| 1435529_at (L) | Ifit1 | –3.69 | 9.11E-06 | Chr19 | - |
| 1419599_s_at (L) | Ms4a6d | –3.46 | 1.85E-05 | Chr19 | - |
| 1436058_at (L) | Rsad2 | –2.913 | 0.00244 | Chr12 | - |
| 1450783_at (L) | Ifit1l | –2.58 | 0.00101 | Chr19 | - |
| 1449025_at (L) | Ifit3 | –2.39 | 0.00306 | Chr19 | - |
| 1424775_at (L) | Oas1a | –2.33 | 0.0015 | Chr5 | - |
| 1422903_at (L) | Ly86 | –2.26 | 0.00158 | Chr13 | Idd14 |
| 1418580_at (L) | Rtp4 | –2.23 | 0.000429 | Chr16 | - |
| Apoptosis/cell proliferation | |||||
| 1457812_at (L) | Trp53bp1 | –20.92 | 0.00426 | Chr2 | Idd13 |
| 1440493_at (L) | Galnt10 | –10.93 | 0.00256 | Chr11 | - |
| 1438462_x_at (L) | Khdrbs1 | –9.83 | 2.51E-06 | Chr4 | Idd9/11 |
| 1417714_x_at (H) | Hba-a1 | 5.67 | 0.00182 | Chr11 | - |
| 1449716_s_at (H) | Nrd1 | 2.62 | 1.50E-05 | Chr4 | - |
| 1428371_at (H) | Ttbk2 | 1.99 | 0.0025 | Chr2 | Idd13 |
| Transcription | |||||
| 1458094_at (L) | Zfp407 | –5.59 | 0.0015 | Chr18 | Idd21.1 |
| 1458274_at (H) | Zfp69 | 2.97 | 0.00361 | Chr4 | - |
| 1459026_at (L) | Snw1 | –5.40 | 6.75E-05 | Chr12 | - |
| 1447703_x_at (L) | Zfp593 | –4.20 | 0.00209 | Chr4 | - |
| 1417961_a_at (L) | Trim30 | –3.58 | 0.000703 | Chr7 | Idd27 |
| 1421519_a_at (L) | Zfp120 | –2.30 | 0.000974 | Chr2 | Idd13 |
| Transporters | |||||
| 1434914_at (L) | Rab6b | –36.11 | 0.000308 | Chr9 | - |
| 1458426_at (L) | Kif1b | –3.49 | 0.00345 | Chr4 | Idd9.2 |
| 1421098_at (L) | Stap1 | –1.39 | 0.00361 | Chr5 | - |
| Zinc-ion binding | |||||
| 1437432_a_at (L) | Trim12a | –186.81 | 2.35E-07 | Chr7 | Idd27 |
| 1443858_at (L) | Trim12c (Trim5) | –664.23 | 1.15E-08 | Chr7 | Idd27 |
| 1435665_at (L) | Trim30d | –3.73 | 0.000726 | Chr7 | Idd27 |
| 1421550_a_at (L) | Trim34 | –2.46 | 0.00119 | Chr7 | Idd27 |
| 1451277_at (L) | Zadh2 | –2.29 | 0.00237 | Chr18 | Idd21.1 |
| 1434193_at (L) | Zmym6 | –1.60 | 0.000597 | Chr4 | Idd11 |
| Protein/nucleic acid binding | |||||
| 1424454_at (H) | Tmem87a | 9.56 | 0.00354 | Chr2 | Idd13 |
| 1455863_at (L) | Spata5l1 | –7.28 | 0.000253 | Chr2 | - |
| 1444714_at (L) | Dcdc2b | –2.90 | 0.00328 | Chr4 | - |
| 1431231_at (L) | Hist1h3f | –2.33 | 0.0028 | Chr13 | - |
| 1435312_at (L) | Paqr7 | –2.08 | 0.00243 | Chr4 | - |
| Other enzymes | |||||
| 1442424_at (L) | Ctdspl2 | –36.64 | 0.000789 | Chr2 | Idd13 |
| 1453009_at (L) | Cpm | –12.24 | 6.21E-06 | Chr10 | - |
| 1418035_a_at (L) | Prim2 | –7.69 | 4.78E-05 | Chr1 | Idd26 |
| 1423569_at (L) | Gatm | –5.45 | 0.00467 | Chr2 | Idd13 |
| 1416494_at (L) | Ndufs5 | –5.42 | 9.99E-06 | Chr4 | - |
| 1427302_at (H) | Enpp3 | 3.32 | 0.00172 | Chr10 | - |
| 1427943_at (H) | Acyp2 | 2.53 | 0.000285 | Chr11 | - |
| 1442466_a_at (L) | Ppip5k1 | –2.84 | 0.000837 | Chr2 | Idd13 |
| 1455075_at (L) | Pigv | –2.56 | 0.00188 | Chr4 | |
| 1451998_at (L) | Tasp1 | –2.53 | 0.0044 | Chr2 | Idd13 |
| 1433977_at (L) | Hs3st3b1 | –2.48 | 0.00247 | Chr11 | - |
| Other (unknown transcripts are indicated below in the legend) | |||||
| 1444741_at (L) | Dock2 | –31.19 | 0.000307 | Chr11 | - |
| 1436061_at (L) | Chaf1a | –13.09 | 0.000734 | Chr17 | - |
| 1452426_x_at (L) | LOC433762 | –13.07 | 1.08E-05 | Chr4 | - |
| 1452359_at (L) | Rell1 | –2.50 | 0.00113 | Chr5 | - |
| 1442824_at (L) | Raly | –12.31 | 0.000974 | Chr2 | Idd13 |
| 1442494_at (L) | Ubr2 | –11.47 | 6.75E-05 | Chr17 | - |
| 1456635_at (L) | Sp110 | –8.90 | 0.00437 | Chr1 | Idd5.4a/5.4 |
| 1456685_at (L) | Nsg2 | –2.96 | 0.0021 | Chr11 | - |
Statistical analysis (1-way-ANOVA; p < 0.005, Benjamini–Hochberg) followed by hierarchical clustering identified 65 differentially expressed probe sets (representing 60 different genes) at 4 weeks; L and H, respectively, indicate lower or higher expression in NOD mice compared to control mice, NOR and C57BL/6. Fold change (FC) was calculated by ratio of means of expression in NOD mice versus controls. Dashes indicate those genes are not located within a known T1D susceptibility region (Idd); all indicated Idds (except one, Idd5.4a/5.4) were identified as conferring resistance to diabetes (http://www.t1dbase.org). Two unknown transcripts included: BE373131 (L; Chr19) and A230098N10Rik (H; Chr5). Genes highlighted in bold font were differentially expressed at all 3 ages, 2, 3 and 4 weeks.
3.2. Gene ontology categories and KEGG pathways of the CD4 T-cell NOD altered genes
To gain further insights into the biological processes associated with the NOD altered genes, we performed Gene ontology (GO) and KEGG pathway analyses. Because GO analysis is better suited for larger gene lists, we also analyzed gene lists generated at a slightly lower statistical stringency. Analysis at p < 0.05, Benjamini–Hochberg (FDR of 5%) identified 134, 252, and 185 NOD altered genes at 2, 3, and 4 weeks, respectively (Tables S2, S3 and S4, respectively). The topmost ranked major biological process of these gene lists was metabolism (Table 5). Although common to all 3 ages, this functional category was most significantly enriched at 2 weeks. Several biological processes unique to 3 weeks were also identified, including biopolymer modification, localization and transport, and T cell activation. Under “molecular function”, hydrolase activity and binding were the common top ranked categories. “Intracellular” was the most significantly enriched cellular component for all the 3 lists. Nucleus and cytoplasm/endoplasmic reticulum were uniquely enriched at 2 and 3 weeks, respectively. The immune system process ranked at the top of the biological process at 4 weeks in the analysis of the gene list generated at the higher stringency level (p < 0.005, Benjamini–Hochberg; FDR of 0.5). Findings of the GO analyses were consistent with those from our “manual” literature searches. We then performed KEGG pathway analysis to identify well characterized molecular pathways that were significantly over-represented in the gene lists of NOD altered genes. Results of the smaller and larger gene lists of NOD altered genes were similar; only those of the smaller lists are presented (Table 6). Consistent with the GO analysis, the predominantly enriched category was metabolic pathways, which still was most highly significantly enriched at 2 weeks as compared to the other two ages. Five NOD altered genes common to all 3 ages were identified in these metabolic pathways: Enpp3 (ectonucleotide pyrophosphatase/phosphodiesterase 3), Ndufs5 (NADH dehydrogenase (ubiquinone) Fe-S protein 5), Galnt10 (UDP-N-acetyl-alpha-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase 10), Prim2 (DNA primase, p58 subunit) and Gatm. These findings suggest that these genes may contribute to the metabolic abnormalities that affect the immune system and predispose NOD mice to autoimmune diabetes. Overall, these data suggest that CD4 T-cells from NOD mice already have a defect in metabolism (most prominent at 2 weeks), cellular activation and endoplasmic reticulum function (both evident at 3 weeks) and T-cell/immune response (evident at 3–4 weeks) at the preinsulitis stage.
Table 5.
Enriched Gene ontology categories for the list of CD4 T-cell NOD altered genes.
| GO category | 2 wk (134 genes) |
3 wk (252 genes) |
4 wk (185 genes) |
|||
|---|---|---|---|---|---|---|
| No. of genes in category | p-value | No. of genes in category | p-value | No. of genes in category | p-value | |
| Biological process | ||||||
| Cellular metabolism | 53 | 4.24e-5 | 88 | 1.13e-3 | 67 | 1.88e-3 |
| Primary metabolism | 52 | 2.82e-5 | 86 | 7.06e-4 | - | - |
| Nucleobase, nucleoside, nucleotide and nucleic acid metabolism | 28 | 2.02e-3 | - | - | 35 | 8.88e-3 |
| Biopolymer modification | - | - | 25 | 6.53e-3 | - | - |
| Localization | - | - | 43 | 1.13e-3 | - | - |
| Cytoskeletal organization and biogenesis | - | - | 11 | 9.07e-3 | - | - |
| Transport | - | - | 36 | 7.49e-3 | - | - |
| Intracellular transport | - | - | 16 | 1.64e-3 | - | - |
| Secretion | - | - | 8 | 5.70e-3 | - | - |
| Leukocyte activation | - | - | 6 | 8.80e-3 | - | - |
| T cell activation | - | - | 5 | 3.17e-3 | - | - |
| Regulation of hydrolase activity | - | - | 4 | 8.07e-3 | - | - |
| Molecular function | ||||||
| Catalytic activity | - | - | - | - | 60 | 2.89e-3 |
| Hydrolase activity | 22 | 2.84e-3 | 40 | 7.88e-4 | 30 | 2.30e-3 |
| Hydrolase activity, acting on ester bonds | 10 | 3.01e-3 | 15 | 6.23e-3 | 14 | 8.65e-4 |
| Phosphoric ester hydrolase activity | 7 | 1.32e-3 | 10 | 1.99e-3 | 10 | 1.75e-4 |
| Hydrolase activity, acting on acid anhydrides | - | - | 16 | 4.48e-4 | - | - |
| Pyrophosphatase activity | - | - | 15 | 1.12e-3 | - | - |
| GTPase activity | - | - | 6 | 3.49e-3 | - | - |
| Nucleotidyltransferase activity | - | - | 5 | 7.76e-3 | 6 | 2.82e-4 |
| Inositol or phosphatidylinositol phosphatase activity | - | - | - | - | 4 | 1.63e-4 |
| Binding | - | - | 140 | 3.87e-5 | 99 | 4.64e-3 |
| Metal ion binding | - | - | 55 | 2.85e-3 | 43 | 2.02e-3 |
| Protein binding | - | - | 69 | 1.89e-3 | ||
| Cation binding | 28 | 8.09e-3 | 39 | 6.74e-3 | ||
| Transition metal ion binding | 26 | 2.49e-3 | 34 | 4.44e-4 | ||
| Zinc ion binding | 24 | 6.04e-5 | 29 | 5.59e-4 | ||
| Nucleic acid binding | 28 | 4.70e-3 | ||||
| Cellular component | ||||||
| Intracellular | 65 | 2.81e-5 | 113 | 2.35e-7 | 79 | 5.64e-4 |
| Intracellular organelle | 53 | 1.22e-3 | 88 | 9.33e-4 | - | - |
| Intracellular membrane-bound organelle | 48 | 1.39e-3 | 80 | 6.71e-4 | - | - |
| Nucleus | 33 | 8.06e-3 | - | - | - | - |
| Cytoplasm | - | - | 58 | 1.38e-4 | - | - |
| Endoplasmic reticulum | - | - | 15 | 1.72e-3 | - | - |
One-way ANOVA (p < 0.05, Benjamini–Hochberg) of mRNA expression data followed by hierarchical clustering identified 134, 252, and 185 different genes at 2, 3, and 4 weeks, respectively, whose expression was altered in CD4 T-cells from NOD mice compared to two control strains (NOR and C57BL/6). These lists of NOD altered genes were analyzed in WebGestalt (http://bioinfo.vanderbilt.edu/webgestalt) using the hypergeometric test (p < 0.01). Categories represented by = 4 genes are shown. Dashes indicate that the corresponding categories were not significantly enriched at the respective ages. Categories indicated in bold font were common to all 3 age groups.
Table 6.
Enriched KEGG pathways for the list of CD4 T-cell NOD altered genes.
| KEGG pathway | Adjusted p-value | No. of genes | Genes in the pathway |
|---|---|---|---|
| 2 wk | |||
| Metabolic pathways | 1.01e-5 | 9 | Enpp3, Ndufs5, Galnt10, Prim2, Gatm, Dnmt3b, Akr1e1, Enpp1, Rrm1 |
| Purine metabolism | 5.22e-5 | 4 | Enpp3, Prim2, Enpp1, Rrm1 |
| Pantothenate and CoA biosynthesis | 0.0001 | 2 | Enpp3, Enpp1 |
| Riboflavin metabolism | 0.0001 | 2 | Enpp3, Enpp1 |
| Nicotinate and nicotinamide metabolism | 0.0003 | 2 | Enpp3, Enpp1 |
| Starch and sucrose metabolism | 0.0008 | 2 | Enpp3, Enpp1 |
| Pyrimidine metabolism | 0.0039 | 2 | Prim2, Rrm1 |
| 3 wk | |||
| Metabolic pathways | 0.0005 | 10 | Enpp3, Ndufs5, Galnt10, Prim2, Gatm, Inppl1, Dnmt3b, Akr1e1, Dut, Enpp1 |
| Riboflavin metabolism | 0.0008 | 2 | Enpp3, Enpp1 |
| Purine metabolism | 0.0008 | 4 | Enpp3, Prim2, Enpp1 |
| Pantothenate and CoA biosynthesis | 0.0008 | 2 | Enpp3, Enpp1 |
| Pyrimidine metabolism | 0.0013 | 3 | Prim2, Pnpt1, Dut |
| Nicotinate and nicotinamide metabolism | 0.0008 | 2 | Enpp3, Enpp1 |
| Spliceosome | 0.0029 | 3 | Crnkl1, Srnp200, Snw1 |
| Starch and sucrose metabolism | 0.0031 | 2 | Enpp3, Enpp1 |
| 4 wk | |||
| Metabolic pathways | 0.0002 | 7 | Enpp3, Ndufs5, Galnt10, Prim2, Gatm, Pigv, Mdh1 |
| Pyruvate metabolism | 0.0008 | 2 | Acyp2, Mdh1 |
One-way ANOVA (p < 0.005, Benjamini–Hochberg) of mRNA expression data followed by hierarchical clustering identified 58, 115, and 65 probe sets at 2, 3 and 4 weeks, respectively, whose expression was altered in CD4 T-cells from NOD mice compared to two control strains (NOR and C57BL/6). The lists of differentially expressed genes were analyzed in WebGestalt (http://bioinfo.vanderbilt.edu/webgestalt) for enriched KEGG pathways using the hypergeometric test (p < 0.01, Benjamini–Hochberg). Pathways represented by =2 genes are shown. Pathways or genes indicated in bold font were common to the 3 age groups.
3.3. Transcriptional regulatory pathways of the CD4 T-cell NOD altered genes
The 4 topmost significantly enriched transcription factor bindings sites (Table 7) were for androgen receptor (Ar), significantly enriched at 2 and 4 weeks; Interferon regulatory factor 1 (Irf1), significantly enriched at all 3 ages; and Interferon regulatory factor 7 (Irf7) and Interferon sensitive response elements (ISRE) both significantly enriched at 3 and 4 weeks. ISRE are present in the promoters of interferon stimulated genes (ISGs), also known as antiviral or innate immune response genes. All 4 binding sites were most highly significantly enriched at 4 weeks. Interestingly, each putative transcription regulator correlated with a different set of NOD altered genes at the various stages it was significantly enriched, including both age-common and age-specific NOD altered genes. This suggests a dynamic (possibly coordinated) regulation of gene expression. The promoter analysis data suggest that expression of the NOD altered genes was significantly more likely to be regulated by Ar, Irf1, Irf7, and type I interferon than by other transcription regulators.
Table 7.
Transcription factor binding sites enriched in the promoters of CD4 T-cell NOD altered genes.
| Transcription factor binding sites | p-value | No. of genes | Gene names |
|---|---|---|---|
| 2 wk | |||
| Ar | 0.008 | 9 | Enpp3, Trim34, Nrd1, Rell1, Hisppd2a, Rrm1, Sp140, Trib3, A530032D15Rik |
| Irf1 | 0.015 | 11 | Galnt10, Trim 34, Khdrbs1, Pldn, Tmem87a, Trp53bp1, Enpp1, Mfap3, Psmb2, Rtn4, Snrnp200 |
| 3 wk | |||
| Irf1 | 3.74E-04 | 21 | Galnt10, Trim34, Khdrbs1, Pldn, Tmem87a, Trp53bp1, Trim21, Ahsa1, Capn3, Enpp1, Ifi27l1, Ifit1, Mfap3, Pi4k2b, Psmb2, Ptpn18, Rtn4, Snrp200, Tnrc6a, 1700029I01Rik, 2510039O18Rik |
| Irf7 | 0.002 | 17 | Khdrbs1, Tmem87a, Zfp69, Trim21, Trim30, Anxa6, Hisppd2a, Ifi27l1, Ifit1, Kif1b, Nsg2, Pdrg1, Pi4k2b, Pnpt1, Zadh2, AI451617, 4930402H24Rik |
| ISRE | 4.48E-04 | 17 | Enpp3, Ndufs5, Trim12, Trim34, Khdrbs1, Zfp69, Trim21, Trim30, Dyx1c1, Ifi27l1, Ifit1, Pi4k2b, Pldn, Pnpt1, Snrp200, Tnrc6a, AI451617 |
| 4 wk | |||
| Ar | 6.73E-04 | 11 | Enpp3, Trim34, Nrd1, Rell1, Hisppd2a, Ly6c1, Ly86, Oas1a, Sp140, Tasp1, A530032D15Rik |
| Irf1 | 8.13E-05 | 15 | Galnt10, Trim34, Khdrbs1, Pldn, Tmem87a, Trp53bp1, Ifit1, Ifit3, Ly86, Oas1a, Pigv, Rsad2, Rtp4, Stap1, Zfp120 |
| Irf7 | 2.50E-06 | 15 | Khdrbs1, Tmem87a, Zfp69, Trim30, Hisppd2a, Ifit1, Ifit3, Kif1b, Nsg2, Oas1a, Rsad2, Rtp4, Stap1, Zadh2, AI451617 |
| ISRE | 5.58E-08 | 16 | Enpp3, Ndufs5, Trim12, Trim34, Khdrbs1, Zfp69, Trim30, Ifit1, Ifit3, Ly86, Oas1a, Pldn, Rsad2, Rtp4, Zmym6, AI451617 |
One-way ANOVA (p < 0.005, Benjamini–Hochberg) of mRNA expression data followed by hierarchical clustering identified 58, 115, and 65 probe sets at 2, 3 and 4 weeks, respectively, whose expression was altered in CD4 T-cells from NOD mice compared to two control strains (NOR and C57BL/6). The lists of differentially expressed genes were analyzed for enriched transcription factor binding sites in their promoters by using the PRIMA software of the EXPANDER suite (p < 0.05). Binding sites or genes indicated in bold font were common to the 3 age groups. ISRE – interferon sensitive response elements.
3.4. Ingenuity Pathway Networks of the CD4 T-cell NOD altered genes
In addition to the transcriptional regulatory pathway analysis, we performed Ingenuity pathway analysis (IPA; 17,18) to gain further insights into the genes/molecules that may play a role in regulating the expression of the NOD CD4 T-cell altered genes and/or molecular pathways. IPA analyses of the 3 “smaller” lists of NOD altered genes (Tables 1–4) generated several networks each. The major biological functions significantly represented by the networks included cell cycle, cellular growth/proliferation, and cell death at 2 weeks; molecular transport, cell-to-cell signaling and interaction, and immune system development and function at 3 weeks; and antiviral (innate immune) function and immune response at 4 weeks. The merged networks for each dataset clustered around several central genes (Figs. 2–4). To facilitate an objective comparison between networks and to gain further insights, we ranked the central genes according to the total number of connections linked to each one of them – the more the number of connections, the higher the rank. The top 15 central genes and their directly linked focus genes are shown in Table 8. This analysis revealed that seven central genes were common to all 3 ages, including transcription factors, hepatic nuclear factor 4, alpha (Hnf4a) and transformation related protein 53 (Trp53); anti-apoptotic factor, BCL2-like 1 (Bcl2l1); cytokines, interferon gamma (Ifng), interleukin 4 (Il4) and interleukin 15 (IL15); and the pro-inflammatory factor, prostaglandin E2. The pro-inflammatory cytokine, interleukin 6 (Il6) was common to ages 2 and 4 weeks. Other genes were unique to the respective age groups, e.g. to 2 weeks: transcription factors, myelocytomatosis oncogene (Myc), Jun oncogene (Jun), and amyloid beta (A4) precursor protein (App); to 3 weeks: the pro-inflammatory cytokine, tumor necrosis factor (Tnf), the anti-inflammatory cytokine, Tgfb1 (transforming growth factor, beta 1), and cell signaling molecules NF?B, ERK, p38MAPK, and insulin-like growth factor 1 (Igf1); and to 4 weeks: cytokines, interferon alpha and IL12 (interleukin 12), the transcription factor signal transducer and activator of transcription 4 (Stat4), and Rous sarcoma oncogene (Src). Interestingly, the central genes Bcl2l1 and Src are also located within Idd13. The biological processes associated with the central genes that were common to all 3 ages included apoptosis/cellular proliferation, Th1–Th2 balance, cytokine signaling, and inflammation. Those associated with the age-specific central genes included apoptosis/cell proliferation (at 2 weeks), cell signaling/cellular activation and inflammation (at 3 weeks), and innate immunity/link to adaptive immune response (at 4 weeks). Similar to the promoter analysis, each of the common central genes was connected to a different set of focus genes at the various ages, consisting of both age-common and age-specific genes, again suggesting a dynamic coordinated regulation of the molecular processes over time. These data suggest that abnormalities in apoptosis/cellular proliferation predominate at 2 weeks of age, while those in cell signaling/cellular activation and inflammation predominate at 3 weeks of age. Abnormalities in the innate immune response and its link to adaptive immunity predominate at 4 weeks of age.
Fig. 2.
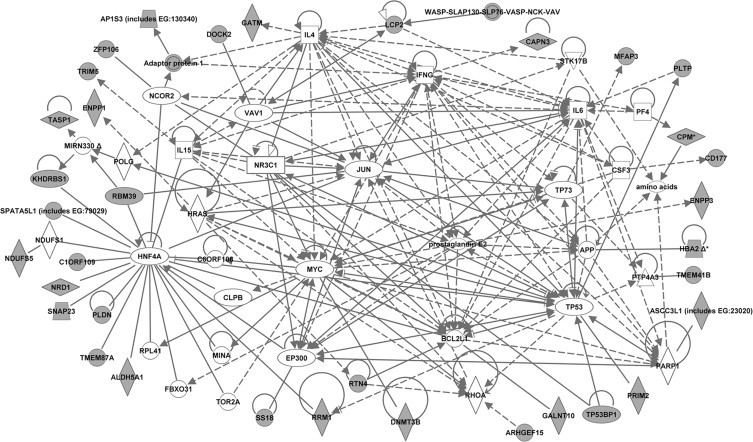
Molecular network generated by ingenuity pathway analysis (IPA) from the dataset of 2 week-old mice. The merged network was generated from the list of genes differentially expressed in CD4 T-cells from 2-week old NOD mice compared to both control strains (NOR and C57BL/6). The list was selected from the hierarchical cluster of 362 genes that had highly significant (p < 0.005, Benjamini–Hochberg) expression differences between strains at 2 weeks of age. The genes derived from our uploaded gene list (known as focus genes) are represented by gray icons while genes (or endogenous chemicals) derived from the IPA knowledge base that could be algorithmically connected to the focus genes are represented by white icons. Different shapes of the symbols represent the different classes of genes/molecules, e.g. squares = cytokines/growth factors; ovals = transcription factors, etc.
Fig. 3.
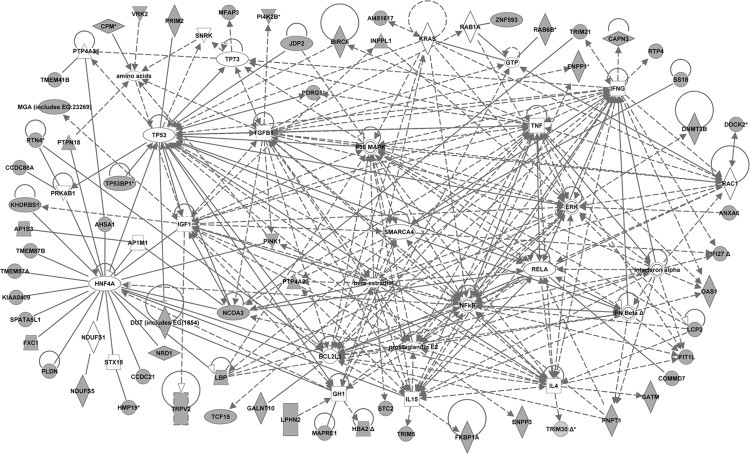
Molecular network generated by ingenuity pathway analysis (IPA) from the dataset of 3 week-old mice. The merged network was generated from the list of genes differentially expressed in CD4 T-cells from 3-week old NOD mice compared to both control strains (NOR and C57BL/6). The list was selected from the hierarchical cluster of 982 genes that had highly significant (p < 0.005, Benjamini–Hochberg) expression differences between strains at 3 weeks of age. The genes derived from our uploaded gene list (known as focus genes) are represented by gray icons while genes (or endogenous chemicals) derived from the IPA knowledge base that could be algorithmically connected to the focus genes are represented by white icons. Different shapes of the symbols represent the different classes of genes/molecules, e.g. squares = cytokines/growth factors; ovals = transcription factors, etc.
Fig. 4.
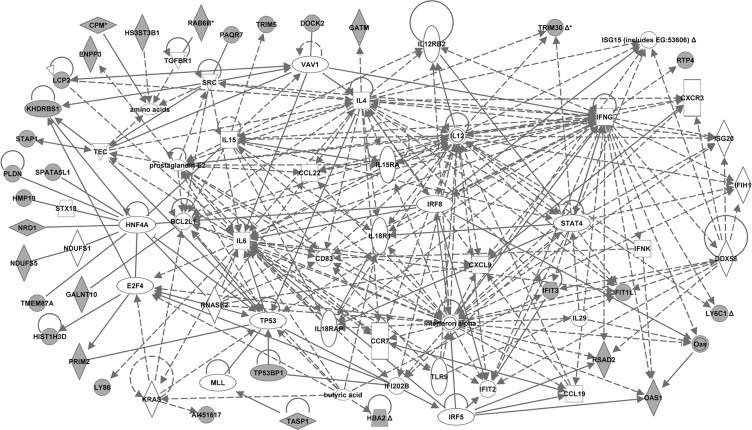
Molecular network generated by ingenuity pathway analysis (IPA) from the dataset of 4 week-old mice. The merged network was generated from the list of genes differentially expressed in CD4 T-cells from 4-week old NOD mice compared to both control strains (NOR and C57BL/6). The list was selected from the hierarchical cluster of 581 genes that had highly significant (p < 0.005, Benjamini–Hochberg) expression differences between strains at 4 weeks of age. The genes derived from our uploaded gene list (known as focus genes) are represented by gray icons while genes (or endogenous chemicals) derived from the IPA knowledge base that could be algorithmically connected to the focus genes are represented by white icons. Different shapes of the symbols represent the different classes of genes/molecules, e.g. squares = cytokines/growth factors; ovals = transcription factors, etc..
Table 8.
Central genes in the Ingenuity Pathway Networks of the CD4 T-cell NOD altered genes.
| Central Gene (IPA symbol) | Gene symbol | Total connections (no. of Focus genesa) | Connected Focus genesa (shown as IPA symbols) |
|---|---|---|---|
| 2 wk | |||
| HNF4A | Hnf4a | 27 (10) | PLDN, NRD1, KHDRBS1, TMEM87A, SPATA5L1, RRM1, ALDH5A1, RBM39, SNAP23, C1ORF109 |
| IFNG | Ifng | 26 (2) | CAPN3, Adaptor protein 1 |
| IL4 | IL4 | 26 (3) | LCP2, GATM, Adaptor protein 1 |
| TP53 | Trp53 | 24 (4) | TP53BP1, PRIM2, RRM1, PLTP |
| IL6 | IL6 | 24 (2) | LCP2, PLTP |
| MYC | Myc | 24 (1) | DNMT3B |
| BCL2L1b | Bcl2l1b | 16 (2) | GALNT10, RTN4 |
| JUN | Jun | 16 (1) | RBM39 |
| IL15 | IL15 | 13 (1) | TRIM5 |
| Prostaglandin E2 | N/A | 12 (1) | ENPP3 |
| NR3C1 | Nr3c1 | 12 (1) | TP53BP1 |
| PARP1 | Parp1 | 11 (1) | ASCC3L1 |
| APP | App | 11 (1) | HBA2 |
| EP300 | Ep300 | 11 (1) | SS18 |
| HRAS | Hras | 9 (2) | ENPP1, RTN4 |
| 3 wk | |||
| TNF | Tnf | 30 (6) | OAS1, IFIT1L, LBP, NCOA3, PDRG1, BIRC6 |
| TP53 | Trp53 | 30 (7) | TP53BP1, PRIM2, JDP2, PDRG1, BIRC6, DUT, NCOA3 |
| IFNG | Ifng | 28 (5) | CAPN3, RTP4, TRIM21, IFI27, OAS1 |
| NF?B | N/A | 28 (4) | ENPP1, LCP2, PNPT1, LBP |
| HNF4A | Hnf4a | 27 (14) | PLDN, NRD1, KHDRBS1, TMEM87A, SPATA5L1, AHSA1, NCOA3, LBP, TMEM87B, FXC1, KIAAO409, CCDC21, PTPN18, PINK1 |
| TGFB1 | Tgfb1 | 26 (4) | PI4K2B, ENPP1, NCOA3, PINK1 |
| ERK | Ephb2 | 25 (5) | LCP2, DNMT3B, ANXA6, INPPL1, LBP |
| Beta-estradiol | N/A | 25 (6) | IFIT1L, STC2, TCF15, NCOA3, PTP4A2, PINK1 |
| p38MAPK | Mapk14 | 23 (2) | LBP, JDP2 |
| IL4 | IL4 | 22 (5) | LCP2, GATM, IFIT1L, TRIM30, NCOA3 |
| IGF1 | Igf1 | 22 (5) | KHDRBS1, INPPL1, NCOA3, TRPV2, MGA |
| RELA | Rela | 21 (3) | COMMD7, PNPT1, NCOA3 |
| IL15 | IL15 | 19 (3) | TRIM5, FKBP1A, PTP4A2 |
| BCL2L1b | Bcl2l1b | 19 (3) | GALNT10, RTN4, PNPT1 |
| Prostaglandin E2 | N/A | 18 (1) | ENPP3 |
| 4 wk | |||
| Interferon alpha | Interferon alpha | 34 (4) | Oas, IFIT1L, RSAD2, OAS1 |
| IFNG | Ifng | 33 (6) | RTP4, LY6C1, Oas, OAS1, RSAD2, IFIT3 |
| IL12 | IL12 | 31 (3) | TRIM30, LY6C1, IFIT1L |
| IL4 | IL4 | 30 (5) | LCP2, GATM, TRIM30, IFIT3, IFIT1L |
| IL6 | IL6 | 27 (3) | LCP2, IFIT1L, LY86 |
| STAT4 | Stat4 | 18 (2) | IFIT1L, TRIM30 |
| IL15 | IL15 | 17 (1) | TRIM5 |
| Prostaglandin E2 | N/A | 16 (1) | ENPP3 |
| TP53 | Trp53 | 14 (2) | TP53BP1, PRIM2 |
| BCL2L1b | Bcl2l1b | 14 (1) | GALNT10 |
| HNF4A | Hnf4a | 13 (5) | PLDN, NRD1, KHDRBS1, TMEM87A, SPATA5L1 |
| DDX58 | Ddx58 | 11 (2) | IFIT3, IFIT1L |
| SRCb | Srcb | 10 (2) | KHDRBS1, PAQR7 |
| E2F4 | E2F4 | 10 (2) | KHDRBS1, PRIM2 |
| IFIT2 | Ifit2 | 9 (1) | IFIT3 |
Genes that came from the list of NOD altered genes. The connections to each gene (i.e. edges representing gene-gene relationships) in the molecular networks were manually counted. The central genes were then ranked from highest to lowest based on the number of total connections; the total # of connections represents the initial # of edges prior to editing of the networks for better image visibility. The table shows the 15 topmost central genes and their immediately connected focus genes.
BCL2L1 and SRC are located within the diabetes resistance region Idd13 (Chr2); BCL2L1 linked NOD altered genes (GALNT10, RTN4 and PNPT1) are all located on Chr11 (not in a known Idd) while SRC linked genes (PAQR7 and KHDRBS1) are both located on Chr4 (KHDRBS1 within the CD4 T-cell diabetogenic activity region Idd9/11 while PAQR7 is not in a known Idd). Central or focus genes/molecules indicated in bold font were common to all 3 age groups. N/A – not applicable.
4. Discussion
Type 1 diabetes is a multigenic disease whose study would benefit from a non-targeted approach to simultaneously identify the genes/pathways that are involved in the disease process [20]. Here, we employed a novel 3-way analysis where NOD was compared to two diabetes-resistant strains, C57BL/6 and NOR. Comparison with NOR mice (a strain that is ~88% identical to NOD mice, including sharing the diabetogenic H2g7 MHC haplotype) afforded us the ability to identify gene expression changes (and their associated molecular networks) that might contribute to T1D susceptibility in NOD or, conversely, resistance in NOR mice in the context of a permissive MHC haplotype. This study confirmed as well as advanced our recently published studies on unfractionated spleen leukocytes [25] in that, in addition to using the NOR control strain, we focused on a specific leukocyte subset (CD4 T-cells) and included an additional time point (3 weeks of age), all of which allowed us to identify novel altered pathways.
NOR mice remain diabetes-free due to resistance alleles within the ~12% portion of their genome derived from BKs [4,17,29,30]. These BKs-derived genomic regions are present on chromosomes 1, 2, 4, 5, 7, 10, 11, 12, 14 and 18 [17,29,30]. Unsurprisingly, virtually all the NOD altered genes identified in the current study are located on these chromosomes (Tables 1–4). Strikingly, a large number of these genes lie within known diabetes susceptibility regions (Tables 1–4). Linkage/congenic studies have provided evidence for the presence of NOR resistance genes on Chr1 (Idd5.2), Chr2 (Idd13) and Chr4 (Idd9/11), and potentially on Chr11 [15–17,30]. However, several studies also suggest that genes in other genetic regions distinguishing NOR from NOD may also act interactively with genes in these loci to protect NOR mice from autoimmune diabetes [5,10,30], thus providing further support for the significance of investigations at the whole cellular or molecular systems level. In further support for whole molecular systems studies, F2 segregation studies and subsequent subcongenic analyses have found that Idd13 (Chr2) and Idd9/11 (Chr4) each consist of multiple genes that contribute to NOR resistance [10,16,17,30]. Our study revealed that a total of 26 different NOD CD4 T-cell altered genes lie within Idd13, including 6 (Bloc1s6, Trp53bp1, Tmem87a, Ctdspl2, Gatm and Raly) that were common to the 3 ages studied (Tables 1–4). Interestingly, two central genes in the IPA networks, Bcl2l1 and Src (Table 8), also lie within this locus. Bcl2l1 is linked to 3 genes, Galnt10, Rtn4 (reticulon 4) and Pnpt1 (polyribonucleotide nucleotidyltransferase 1) that are located on Chr11, while Src is linked to 2 genes, Paqr7 (progestin and adipoQ receptor family member VII) and Khdrbs1 that are located on Chr4. Of note, Khdrbs1 is located within a known Idd9/11 (discussed below) while the 3 Bcl2l1 linked genes and Paqr7 are not located within a known Idd, and thus may define novel T1D susceptibility loci. These results attest to the utility of molecular network analyses in identifying novel Idds or genes not in known Idds that could act interactively upstream or downstream of Idd regions to contribute to diabetes development. The role of these genes needs to be confirmed in future studies. Several groups have determined that resistance genes in Idd9/11 (Chr4) regulate the diabetogenic activity of CD4 T-cells [5,10,18,19]. Interestingly, two of the NOD altered genes (Khdrbs1 and Ptp4a2) lie within Idd9/11, with Khdrbs1 (an adaptor protein involved in signal transduction cascades of several receptor systems, including T-cell signaling) being common to all 3 ages. In support of possible involvement of interaction of genes on several genetic regions in suppressing the diabetogenic activity of NOR CD4 T-cells, Chen et al. [5] reported that CD4 T-cells from NOR mice were somewhat more protective against diabetes than CD4 T-cells from NOD mice congenic for just the NOR-derived Idd9/11. Our study provides support suggesting that resistance genes within Idd13 (and their downstream genes) may act interactively with those in Idd9/11 (and possibly in unidentified Idd on Chr11) to regulate the diabetogenic activity of CD4 T-cells.
In addition to the NOD CD4 T-cell altered genes discussed above, several other altered genes also lie within Idds (Tables 1–4). All (except Idd5.4a/5.4) have been identified as conferring resistance to diabetes (http://www.t1dbase.org). An interesting family of genes highlighted is the tripartite-motif (Trim) family. Compared to the leukocyte study, the current study expanded the list of Trim family members. These genes, whose expression was repressed in NOD mice, with that of Trim5/12c and 12a virtually undetectable, all lie within Idd27 on Chr7. Trim proteins, which bear several domains, including three zinc-binding domains, constitute a family of ~60 molecules with diverse biological functions, including regulation of inflammation and innate immunity [31,32]. The order of the domains is conserved throughout evolution supporting a common molecular property for these proteins. We propose that one (or several) of the Trim family members identified in our studies may play an important role in immune cells in initiation of autoimmune diabetes. To this end, Trim21, a gene that for a long time has been implicated in human autoimmune diseases Sjögren’s syndrome and systemic lupus erythematosus, in which patients exhibit Trim21 autoreactivity [33], has been reported recently to regulate the innate immune response to intracellular dsDNA [34]. Another Trim (Trim28) has also been reported recently to be involved in the global regulation of CD4 T-cells [35]. Mice with a conditional T cell-specific deletion of Trim28 had a spontaneous autoimmune phenotype characterized by mononuclear tissue infiltration and altered cytokine balance leading to nonfunctional T regulatory cells (Tregs).
Thus, the present study provides new insights into the genes/pathways in CD4 T-cells that collectively might play a role in diabetes development – resistance in NOR mice or susceptibility in NOD mice. The BCL2L1 pathway and the genes within Idd13, Idd9/11 and Idd27 that are altered at all 3 ages (Pldn, Trp53bp1, Tmem87a, Ctdspl2, Gatm, Raly, Khdrbs1, and Trim12a and Trim5/12c) make particularly interesting candidate genes/pathways as they most likely represent basic genetic defects in the NOD mouse. While our study has highlighted prime candidate genes, future studies will confirm which of the NOD altered genes and/or their interacting partners or regulators act interactively to effect the diabetogenic activity of NOD CD4 T-cells. In our lab, we are investigating the genetic loci underlying the expression of the altered CD4 T-cell molecular network to define key regulatory loci.
The majority of the CD4 T-cell NOD altered genes were repressed, similar to the results of the spleen leukocyte study [25] and supported by many other studies in both mice and humans [36–42]. Kodama et al. [36] reported global repression of genes in various tissues of NOD mice (including spleen cells) in comparison to NOD.B10 controls, a strain in which the NOD MHC haplotype is replaced with that from the nondiabetic B10 mice. Liston et al. [37] also found a global dampening in gene expression in NOD thymocytes of the genes involved in T cell negative selection. They identified several differentially expressed type 1 diabetes candidate genes, among them four genes (Ly6c, Prim2, Trim12, and Trim30) whose expression was also dampened in our study, thus providing supporting evidence for possible involvement of these candidate genes in CD4 T-cells as well. Heinig et al. [38], in a systems-genetics study investigating rat tissues and macrophages and human monocytes, discovered that the IRF7-driven inflammatory network (which is enriched for innate immune response genes) is associated with type 1 diabetes risk. Interestingly, homologs of 9 of the genes belonging to this network (Lcp2, Oas1a, Rtp4, Ifit1l, Ifit1, Ly6c1, Ifit3, Sp110, and Trim21) were also repressed in NOD mice, suggesting a similar network may also be altered in CD4 T-cells of NOD mice and might contribute to diabetes development in the mouse. Similarly, in human T1D studies, Elo et al. [39] found early suppression of immune response gene expression in whole blood samples of children in the prediabetic phase who eventually developed clinical diabetes. Furthermore, Orban et al. [40] also found repression of all genes that were differentially expressed in whole untreated human peripheral blood CD4 T-cells from new onset T1D patients, including genes involved in key immune functions, such as adhesion molecules lymphocyte function-associated antigen 1 (LFA-1) and P-selectin. Of note, L-selectin (the mouse homolog of P-selectin) was also of significantly lower expression in NOD compared to C57 (p = 3.2e-06 and 0.0065 at 3 and 4 weeks, respectively). Functional analyses by Orban et al. further indicated that the CD4 T-cells were hyporesponsive to stimulation and exhibited inhibition of cell cycle entry. In as much as the Orban et al. microarray study investigated whole untreated CD4 T-cell populations rather than antigen specific suggests that the observed expression defect involves the polygenic CD4 T-cell population, and thus may signify a global CD4 T-cell repression. In support of this notion, other studies have also showed that CD4 T-cells from T1D patients have impaired activation (decreased proliferation) to non-specific primary but not diabetes-specific antigens [41]. It is worth noting that the human studies cited above used peripheral blood samples providing further support for the potential usefulness of CD4 T-cells from spleen (a peripheral organ) in yielding results that may be of relevance to T1D. Thus, future studies can test whether human peripheral blood cells (which are readily accessible) carry similar defects as those detected in our spleen CD4 T-cell study.
Similar to the study by Orban et al., we also investigated whole untreated CD4 T-cells rather than antigen specific T cells. The frequency of T cells with a given antigen specificity is usually very low, estimated to be in the range of 1 or fewer in every 20,000 cells [40]. Therefore, we argue that the defect detected in our study also involves polygenic CD4 T-cells and is likely a basic genetic defect, in the least for those changes that were observed at all 3 age points. We cannot rule out the possibility that the differences observed, especially the age-specific differences, may be due to influences other than genetic defects, e.g. activation of islet specific CD4 T-cells, especially at 4 weeks of age. However, to this end, evaluation of our expression data revealed no evidence of an activated phenotype in NOD mice, suggesting that the changes in the gene expression may not be attributed to auto-reactive cells. For example, expression of two activation markers CD69 and CD38 was significantly lower in NOD (and NOR) compared to C57 (p = 8.26e-05 and 0.0056, respectively, at 4 weeks). CD69 expression was particularly interesting in that it showed no significant difference between the three species at 2 weeks (p = 0.19) but became highly significantly upregulated at 3 and 4 weeks in C57 (3-fold at each age, p = 4.57e-04 and 8.26e-05, respectively) compared to both NOD and NOR. Obviously this result was not reported in the “Section 3” as these types of changes were not the focus of this paper. Notwithstanding, it gives further support to the notion of a deficiency in T cell activation in the NOD mice, the effects of which NOR overcomes likely due to presence of resistance genes.
Whereas the idea of a generalized repression of the immune system may seem counterintuitive (since later stages of T1D are dominated by inappropriate activation of autoreactive T-cells), it may help explain previous observations that immunosuppression exacerbates autoimmune diabetes in NOD mice [42] while immunostimulation circumvents it [43]. Furthermore, it may not be surprising that relative reduction of gene expression was seen at the early stages studied here, at which time tolerance would be induced. Of note, tolerance to self antigens requires the activation of self-reactive lymphocytes and their elimination by apoptosis as a result of this activation. Furthermore, elimination of self-reactive lymphocytes in the periphery also requires activation of the regulatory mechanisms (such as regulatory T cells). Thus, both central and peripheral tolerances are active processes that require normal mitochondrial and metabolic function. Our gene Ontology and KEGG Pathway analyses (Tables 5 and 6), together with the leukocyte study [25], provided evidence for defects in mitochondria, metabolism, antigen processing/presentation and T cell activation/function and immune response. Furthermore, our preliminary functional studies investigating the mitochondrial/metabolic defect found impaired mitochondrial potential in NOD spleen leukocytes. All these data, together with the literature discussed above, support the idea of a global immune repression, which may lead to the breakdown of self-tolerance in autoimmune diabetes. Indeed, a recent study, using a mouse model of spontaneous autoimmune arthritis [44], suggested that efficient suppression of autoimmune diseases requires polyclonal regulatory T cell specificities rather than single antigen-specificities. Thus, we propose the following hypothesis. A genetic defect in metabolism/mitochondria results in a global repression of the immune system leading to a deficiency in immune tolerance, thus predisposing NOD mice to autoimmunity. Analysis of changes in gene expression and molecular pathways in NOD mice between different ages will shade further light on the defects that directly accompany initiation of insulitis, and subsequently development of diabetes. Furthermore, the defects in antigen presenting cells (such as B cells, macrophages and dendritic cells) may synergize with defects in the regulatory and effector T-cells to create dyshomeostasis in the early stages of autoimmune diabetes. Thus, investigation of the APC cell subsets is also warranted to provide a more comprehensive picture of the molecular pathophysiology of autoimmune diabetes.
The promoter and molecular pathway analyses (Tables 7 and 8) identified several factors that may play a role in regulating the above discussed defect. Several of these factors (Hnf4a, Ifng, Trp53, Myc, IL15, Tnf, Tgfb1, beta-estradiol, IL6 and Ar) were also identified by the spleen leukocyte study [25] indicating a strong involvement of the CD4 T-cells in the unfractionated immune cells. In addition, the current study identified novel regulators, including Interferon alpha/beta, Irf1, Irf7, ISRE, IL12, and Stat4. Type 1 interferons regulate 3 (Irf1, Irf7, ISRE) of the 4 transcription regulators identified in this study, suggesting a critical role for these cytokines in regulating gene expression abnormalities in NOD CD4 T-cells at the preinsulitis stage. Irf1 is well known to control immune response gene expression [45] and has been demonstrated to be an essential element (in addition to Ifng and IL12) in the differentiation of naïve T cells [46,47]. Irf1 also functions as a transcription activator of the Tnf receptor and of genes induced by Ifng and type 1 interferons (including Irf7 and other ISGs) [45]. Together with the literature, our data provide support for a role for Irf1 in regulating self-tolerance in autoimmune diabetes. Overall, our study captured new information, which, combined with future confirmatory studies, will facilitate a CD4 T-cell systems-based understanding of autoimmune diabetes and could ultimately lead to the development of novel therapeutic strategies.
Conflicts of interest
The authors declare that they have no competing interests pertaining to this manuscript.
Acknowledgments
This work was supported by grants to ICG from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (DK62103; including a research supplement to this grant to DNK) and American Diabetes Association (ADA 7-11-BS-49). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Dr. David Brand, University of Tennessee Health Science Center/Veterans Affairs Medical Center, Memphis, for access to the cell sorting and isolation equipment and intellectual input on the cell isolation protocols. The authors also acknowledge research facilities and software made available by the Veterans Administration’s Research Service, Memphis and by Dr. John Stuart.
Appendix A. Supplementary material
Supplementary material associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.rinim.2014.05.001.
Appendix A. Supplementary materials
Validation of microarray data by quantitative real-time PCR. Quantitative real-time PCR was performed for eight genes (Ptp4a2, Pldn, Tmem87a, Trim5, Trim12, Gatm, Ly6c1, and Rtp4). Fold change values represent expression levels of genes in control strains (NOR and C57BL/6 mice) relative to NOD mice normalized to Gapdh. The results are consistent with the microarray data results. (.ppt)
{kind=link}
List of genes and primers/probes used for quantitative Real-time PCR. (.doc)
Transcripts significantly differentially expressed in CD4 T-cells of 2 week old NOD mice (Benjamini–Hochberg, p < 0.05). (A) 98 probesets of significantly lower expression in NOD mice; and (B) 49 probesets of significantly higher expression in NOD mice relative to two control strains, NOR and C57BL/6. (.xls)
Transcripts significantly differentially expressed in CD4 T-cells of 3 week old NOD mice (Benjamini–Hochberg, p < 0.05). (A) 212 probesets of significantly lower expression in NOD mice; and (B) 73 probesets of significantly higher expression in NOD mice relative to two control strains, NOR and C57BL/6. (.xls)
Transcripts significantly differentially expressed in CD4 T-cells of 4 week old NOD mice with their ANOVA p-values (Benjamini–Hochberg, p < 0.05). A. 150 probesets of significantly lower expression in NOD mice; and B. 62 probesets of significantly higher expression in NOD mice and fold-changes relative to two control strains, NOR and C57BL/6. (.xls)
References
- 1.Anderson M.S., Bluestone J.A. The NOD mouse:a model of immune dysregulation. Annual Review of Immunology. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. 15771578 [DOI] [PubMed] [Google Scholar]
- 2.Serreze D.V., Leiter E.H. Genes and cellular requirements for autoimmune diabetes susceptibility in nonobese diabetic mice. Current Directions in Autoimmunity. 2001;4:31–67. doi: 10.1159/000060527. 11569409 [DOI] [PubMed] [Google Scholar]
- 3.Todd J.A., Wicker L.S. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity. 2001;15:387–395. doi: 10.1016/s1074-7613(01)00202-3. 11567629 [DOI] [PubMed] [Google Scholar]
- 4.Driver J.P., Serreze D.V., Chen Y.-G. Mouse models for the study of autoimmune type 1 diabetes:a NOD to similarities and differences to human disease. Seminars in Immunopathology. 2011;33:67–87. doi: 10.1007/s00281-010-0204-1. 20424843 [DOI] [PubMed] [Google Scholar]
- 5.Chen Y.-G., Scheuplein F., Osborne M.A., Tsaih S.-W., Chapman H.D., Serreze D.V. Idd9/11 genetic locus regulates diabetogenic activity of CD4 T-cells in nonobese diabetic (NOD) mice. Diabetes. 2008;57:3273–3280. doi: 10.2337/db08-0767. 18776136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tisch R., Yang X.D., Singer S.M., Liblau R.S., Fugger L., McDevitt H.O. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 1993;366:72–75. doi: 10.1038/366072a0. 8232539 [DOI] [PubMed] [Google Scholar]
- 7.Haskins K., Portas M., Bradley B., Wegmann D., Lafferty K. T-lymphocyte clone specific for pancreatic islet antigen. Diabetes. 1988;37:1444–1448. doi: 10.2337/diab.37.10.1444. 2458291 [DOI] [PubMed] [Google Scholar]
- 8.Katz J.D., Benoist C., Mathis D. T helper cell subsets in insulin-dependent diabetes. Science. 1995;268:1185–1188. doi: 10.1126/science.7761837. 7761837 [DOI] [PubMed] [Google Scholar]
- 9.Prochazka M., Gaskins H.R., Shultz L.D., Leiter E.H. The non-obese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:3290–3294. doi: 10.1073/pnas.89.8.3290. 1373493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stolp J., Chen Y.G., Cox S.L., Henck V., Zhang W., Tsaih S.-W. Subcongenic analyses reveal complex interactions between distal chromosome 4 genes controlling diabetogenic B cells and CD4 T cells in nonobese diabetic mice. Journal of Immunology. 2012;189:1406–1417. doi: 10.4049/jimmunol.1200120. 22732593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leiter E.H. Genetics and immunogenetics of NOD mice and related strains. In: Leiter E.H., Atkinson M.A., editors. NOD Mice and Related Strains: Research Applications in Diabetes, AIDS, Cancer, and Other Diseases. R.G. Landes; Austin: 1998. p. 37. [Google Scholar]
- 12.McAleer M.A., Reifsnyder P., Palmer S.M., Prochazka M., Love J.M., Copeman J.B. Crosses of NOD mice with the related NON strain:a polygenic model for IDDM. Diabetes. 1995;44:1186–1195. doi: 10.2337/diab.44.10.1186. 7556956 [DOI] [PubMed] [Google Scholar]
- 13.Gao P., Jiao Y., Xiong Q., Wang C.-Y., Gerling I.C., Gu W. Genetic and molecular basis of QTL of diabetes in mouse: genes and polymorphisms. Current Genomics. 2008;9:324–337. doi: 10.2174/138920208785133253. 19471607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leiter E.H., Reifsnyder P.C., Wallace R., Li R., King B., Churchill G.C. NOD x 129.H2(g7) backcross delineates 129S1/SvImJ-derived genomic regions modulating type 1 diabetes development in mice. Diabetes. 2009;58:1700–1703. doi: 10.2337/db09-0120. 19336673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox C.J., Paterson A.D., Mortin-Toth S.M., Danska J.S. Two genetic loci regulate T cell-dependent islet inflammation and drive autoimmune diabetes pathogenesis. American Journal of Human Genetics. 2000;67:67–81. doi: 10.1086/302995. 10848492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serreze D.V., Bidgett M., Chapman H.D., Chen E., Richard S.D., Leiter E.H. Subcongenic analysis of the Idd13 locus in NOD/Lt mice: evidence for several susceptibility genes including a possible diabetogenic role for beta 2-microglobulin. Journal of Immunology. 1998;160:1472–1478. 9570569 [PubMed] [Google Scholar]
- 17.Serreze D.V., Prochazka M., Reifsnyder P.C., Bridgett M.M., Leiter E.H. Use of recombinant congenic and congenic strains of NOD mice to identify a new insulin-dependent diabetes resistance gene. Journal of Experimental Medicine. 1994;180:1553–1558. doi: 10.1084/jem.180.4.1553. 7931087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamanouchi J., Puertas M.-C., Verdaguer J., Lyons P.A., Rainbow D.B., Chamberlain G. Idd9.1 locus controls the suppressive activity of FoxP3+CD4+CD25+ regulatory T-cells. Diabetes. 2010;59:272–281. doi: 10.2337/db09-0648. 19833887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamilton-Williams E.E., Serreze D.V., Charlton B., Johnson E.A., Marron M.P., Mullbacher A. Transgenic rescue implicates ß2-microglobulin as a diabetes susceptibility gene in nonobese diabetic (NOD) mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11533–11538. doi: 10.1073/pnas.191383798. 11572996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eizirik D.L., Moore F., Flamez D., Ortis F. Use of a systems biology approach to understand pancreatic ß-cell death in Type 1 diabetes. Biochemical Society Transactions. 2008;36:321–327. doi: 10.1042/BST0360321. 18481950 [DOI] [PubMed] [Google Scholar]
- 21.Chen Y., Zhu J., Lum P.Y., Yang X., Pinto S., MacNeil D.J. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. 18344982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emilsson V., Thorleifsson G., Zhang B., Leonardson A.S., Zink F., Zhu J. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. 18344981 [DOI] [PubMed] [Google Scholar]
- 23.Bergholdt R., Brorsson C., Lage K., Nielsen J.H., Brunak S., Pociot F. Expression profiling of human genetic and protein interaction networks in Type 1 diabetes. PloS One. 2009;4:e6250. doi: 10.1371/journal.pone.0006250. 19609442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schadt E.E. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454. 19741703 [DOI] [PubMed] [Google Scholar]
- 25.Wu J., Kakoola D.N., Lenchik N.I., Desiderio D.M., Marshall D.R., Gerling I.C. Molecular phenotyping of immune cells from young NOD mice reveals abnormal metabolic pathways in the early induction phase of autoimmune diabetes. PloS One. 2012;7:e46941. doi: 10.1371/journal.pone.0046941. 23071669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerling I.C., Singh S., Lenchik N.I., Marshall D.R., Wu J. New data analysis and mining approaches identify unique proteome and transcriptome markers of susceptibility to autoimmune diabetes. Molecular and Cellular Proteomics. 2006;5:293–305. doi: 10.1074/mcp.M500197-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Wu J., Lenchik N.I., Gerling I.C. Approaches to reduce false positives and false negatives in the analysis of microarray data: applications in Type 1 diabetes research. BMC Genomics. 2008;9(Suppl. 2):S12. doi: 10.1186/1471-2164-9-S2-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang B., Kirov S., Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Research. 2005;33:W741–W748. doi: 10.1093/nar/gki475. 15980575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prochazka M., Serreze D.V., Frankel W.N., Leiter E.H. NOR/Lt mice: MHC-matched diabetes-resistant control strain for NOD mice. Diabetes. 1992;41:98–106. doi: 10.2337/diab.41.1.98. [DOI] [PubMed] [Google Scholar]
- 30.Reifsynder P.C., Li R., Silveira P.A., Churchill G., Serreze D.V., Leiter E.H. Conditioning the genome identifies additional diabetes resistance loci in type I diabetes resistant NOR/Lt mice. Genes and Immunity. 2005;6:528–538. doi: 10.1038/sj.gene.6364241. 16015371 [DOI] [PubMed] [Google Scholar]
- 31.Reymond A., Meroni G., Fantozzi A., Merla G., Cairo S., Luzi L. The tripartite motif family identifies cell compartments. EMBO Journal. 2001;20:2140–2151. doi: 10.1093/emboj/20.9.2140. 11331580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uchil P.D., Hinz A., Siegel S., Coenen-Stass A., Perte T., Luban J. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. Journal of Virology. 2013;87:257–272. doi: 10.1128/JVI.01804-12. 23077300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshimi R., Ishigatsubo Y., Ozato K. Autoantigen TRIM21/Ro52 as a possible target for treatment of systemic lupus erythematosus. International Journal of Rheumatology. 2012;2012 doi: 10.1155/2012/718237. 22701487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z., Bao M., Lu N., Weng L., Yuan B., Liu W.J. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nature Immunology. 2013;14:172–178. doi: 10.1038/ni.2492. 23222971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chikuma S., Suita N., Okazaki I.M., Shibayama S., Honjo T. TRIM28 prevents autoinflammatory T cell development in vivo. Nature Immunology. 2012;13:596–603. doi: 10.1038/ni.2293. 22544392 [DOI] [PubMed] [Google Scholar]
- 36.Kodama K., Butte A.J., Creusot R.J., Su L., Sheng D., Hartnett M. Tissue- and age-specific changes in gene expression during disease induction and progression in NOD mice. Clinical Immunology. 2008;129:195–201. doi: 10.1016/j.clim.2008.07.028. 18801706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liston A., Hardy K., Pittelkow Y., Wilson S.R., Makaroff L.E., Fahrer A.M. Impairment of organ-specific T cell negative selection by diabetes susceptibility genes: genomic analysis by mRNA profiling. Genome Biology. 2007;8:R12. doi: 10.1186/gb-2007-8-1-r12. 17239257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heinig M., Petretto E., Wallace C., Bottolo L., Rotival M., Lu H. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature. 2010;467:460–464. doi: 10.1038/nature09386. 20827270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elo L.L., Mykkanen J., Nikula T., Jarvenpaa H., Simell S., Aittokallio T. Early suppression of immune response pathways characterizes children with prediabetes in genome-wide gene expression profiling. Journal of Autoimmunity. 2010;35:70–76. doi: 10.1016/j.jaut.2010.03.001. 20356713 [DOI] [PubMed] [Google Scholar]
- 40.Orban T., Kis J., Szereday L., Engelmann P., Farkas K., Jalahej H. Reduced CD4+ T-cell-specific gene expression in human type 1 diabetes mellitus. Journal of Autoimmunity. 2007;28:177–187. doi: 10.1016/j.jaut.2007.01.002. 17320348 [DOI] [PubMed] [Google Scholar]
- 41.Eibl N., Spatz M., Fischer G.F., Mayr W.R., Samstag A., Wolf H.M. Impaired primary immune response in type-1 diabetes: results from a controlled vaccination study. Clinical Immunology (Orlando, Fla.) 2002;103:249–259. doi: 10.1006/clim.2002.5220. 12173299 [DOI] [PubMed] [Google Scholar]
- 42.Kaminitz A., Mizrahi K., Yaniv I., Stein J., Askenasy N. Immunosuppressive therapy exacerbates autoimmunity in NOD mice and diminishes the protective activity of regulatory T cells. Journal of Autoimmunity. 2010;35:145–152. doi: 10.1016/j.jaut.2010.06.002. 20638242 [DOI] [PubMed] [Google Scholar]
- 43.Serreze D.V., Hamaguchi K., Leiter E.H. Immunostimulation circumvents diabetes in NOD/Lt mice. Journal of Autoimmunity. 1989;2:759–776. doi: 10.1016/0896-8411(89)90003-6. 2533502 [DOI] [PubMed] [Google Scholar]
- 44.Oh S., Aitken M., Simons D.M., Basehoar A., Garcia V., Kropf E. Requirement for diverse TCR specificities determines regulatory T cell activity in a mouse model of autoimmune arthritis. Journal of Immunology (Baltimore, Md.: 1950) 2012;188:4171–4180. doi: 10.4049/jimmunol.1103598. 22450809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taniguchi T., Ogasawara K., Takaoka A., Tanaka N. IRF family of transcription factors as regulators of host defense. Annual Review of Immunology. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. 11244049 [DOI] [PubMed] [Google Scholar]
- 46.Kano S., Sato K., Morishita Y., Vollstedt S., Kim S., Bishop K. The contribution of transcription factor IRF1 to the interferon-?-interleukin 12 signaling axis and TH1 versus TH-17 differentiation of CD4+ T cells. Nature Immunology. 2008;9:34–41. doi: 10.1038/ni1538. 18059273 [DOI] [PubMed] [Google Scholar]
- 47.Unutmaz D., Vilcek J. IRF1:a deus ex machina in TH1 differentiation. Nature Immunology. 2008;9:9–10. doi: 10.1038/ni0108-9. 18087248 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of microarray data by quantitative real-time PCR. Quantitative real-time PCR was performed for eight genes (Ptp4a2, Pldn, Tmem87a, Trim5, Trim12, Gatm, Ly6c1, and Rtp4). Fold change values represent expression levels of genes in control strains (NOR and C57BL/6 mice) relative to NOD mice normalized to Gapdh. The results are consistent with the microarray data results. (.ppt)
List of genes and primers/probes used for quantitative Real-time PCR. (.doc)
Transcripts significantly differentially expressed in CD4 T-cells of 2 week old NOD mice (Benjamini–Hochberg, p < 0.05). (A) 98 probesets of significantly lower expression in NOD mice; and (B) 49 probesets of significantly higher expression in NOD mice relative to two control strains, NOR and C57BL/6. (.xls)
Transcripts significantly differentially expressed in CD4 T-cells of 3 week old NOD mice (Benjamini–Hochberg, p < 0.05). (A) 212 probesets of significantly lower expression in NOD mice; and (B) 73 probesets of significantly higher expression in NOD mice relative to two control strains, NOR and C57BL/6. (.xls)
Transcripts significantly differentially expressed in CD4 T-cells of 4 week old NOD mice with their ANOVA p-values (Benjamini–Hochberg, p < 0.05). A. 150 probesets of significantly lower expression in NOD mice; and B. 62 probesets of significantly higher expression in NOD mice and fold-changes relative to two control strains, NOR and C57BL/6. (.xls)