

Summary
Niemann-Pick type C (NPC) disease is a fatal inherited lipid storage disorder causing severe neurodegeneration and liver dysfunction with only limited treatment options for patients. Loss of NPC1 function causes defects in cholesterol metabolism and has recently been implicated in deregulation of autophagy. Here, we report the generation of isogenic pairs of NPC patient-specific induced pluripotent stem cells (iPSCs) using transcription activator-like effector nucleases (TALENs). We observed decreased cell viability, cholesterol accumulation, and dysfunctional autophagic flux in NPC1-deficient human hepatic and neural cells. Genetic correction of a disease-causing mutation rescued these defects and directly linked NPC1 protein function to impaired cholesterol metabolism and autophagy. Screening for autophagy-inducing compounds in disease-affected human cells showed cell type specificity. Carbamazepine was found to be cytoprotective and effective in restoring the autophagy defects in both NPC1-deficient hepatic and neuronal cells and therefore may be a promising treatment option with overall benefit for NPC disease.
Graphical Abstract
Highlights
-
•
Generation of Niemann-Pick type C (NPC) disease patient-specific iPSCs
-
•
NPC1 hepatic and neuronal cells show defects in cholesterol and autophagic flux
-
•
TALEN-mediated genetic correction rescues the cholesterol and autophagy defects
-
•
Autophagy inducers can restore functional autophagy and increase cell viability
iPSCs are valuable for studying human diseases in affected cell types and screening for therapeutic compounds. Here, Jaenisch and colleagues show that genetic correction of a disease-causing mutation in Niemann-Pick type C iPSCs directly linked NPC1 protein function to impaired cholesterol metabolism and autophagy. Stimulating autophagy with carbamazepine was cytoprotective in both hepatic and neural cells and therefore may be of therapeutic relevance.
Introduction
NPC disease is an inherited, autosomal recessive lysosomal storage disorder caused by loss-of-function mutations primarily in the NPC1 gene (∼95%), leading to severe neurodegeneration and liver dysfunction (Carstea et al., 1997; Millard et al., 2005; Vance and Peake, 2011; Vanier, 2010). NPC1 is a transmembrane protein located on the late endosomal/lysosomal (LE/L) compartments where it regulates cholesterol efflux (Abi-Mosleh et al., 2009; Carstea et al., 1997; Millard et al., 2005). So far, more than 250 different NPC1 mutations effecting protein expression, function and stability have been identified. The most common mutation associated with the classical juvenile-onset phenotype, NPC1I1061T, promotes ER-mediated degradation of the mutant protein (Gelsthorpe et al., 2008). Characteristic for NPC disease is the sequestration of low-density lipoprotein (LDL)-derived cholesterol and other lipids in the cellular LE/L compartments due to defective export (Xie et al., 1999). Loss of NPC1 function causes impaired cholesterol homeostasis that has a major impact on liver and brain (Vance and Peake, 2011; Vanier, 2010; Xie et al., 1999).
Autophagy, an intracellular degradation pathway for damaged organelles and aggregation-prone proteins, is essential for cellular homeostasis (Mizushima et al., 2008; Ravikumar et al., 2010). Autophagy regulates lipid metabolism and alterations in intracellular lipid content is likely to impact the autophagy pathway (Singh and Cuervo, 2012; Singh et al., 2009). The degenerative phenotypes in the liver and cerebellum observed in NPC patients resemble those seen in the organs of autophagy-deficient mice (Hara et al., 2006; Komatsu et al., 2006, 2007; Mizushima et al., 2008; Rosenbaum and Maxfield, 2011), suggesting a role of autophagy in the etiology of NPC disease. The dynamic process of autophagy, defined as autophagic flux, encompasses the generation of autophagosomes and its fusion with late endosomes to form amphisomes, which subsequently fuse with lysosomes forming autolysosomes where the autophagic cargo is degraded (Ravikumar et al., 2010). Although impaired autophagy has been shown to contribute to neurodegenerative and liver disorders (Komatsu, 2012; Mizushima et al., 2008; Sarkar, 2013) and is implicated in NPC disease (Elrick et al., 2012; Ordonez et al., 2012; Pacheco et al., 2007; Sarkar et al., 2013), the exact nature of autophagy dysfunction in human NPC disease-affected cells has not been clarified.
The generation of NPC patient-specific induced pluripotent stem cells (iPSCs) provides access to unlimited numbers of disease-affected cell types and a unique opportunity to gain mechanistic insights and screening for new cell type-specific therapeutic compounds (Grskovic et al., 2011; Hara et al., 2006; Kondo et al., 2013; Saha and Jaenisch, 2009; Soldner et al., 2009; Soldner and Jaenisch, 2012; Yusa et al., 2011). Isogenic iPSCs that differ exclusively in a single disease-causing genetic mutation enable studying of the disease phenotypes under highly controlled conditions and allow linking the observed defects directly to disease-causing genetic alterations (Soldner et al., 2011).
Here, we report the generation of patient-specific NPC1 iPSCs and isogenic mutant and control cell lines. NPC1 iPSC-derived hepatic and neuronal cells showed reduced cell viability compared to their controls and displayed defects in cholesterol metabolism and impairment in autophagic flux. TALEN-mediated correction of the NPC1I1061T mutation rescued these disease phenotypes, including dysfunctional autophagic flux, thus implying that the defect in autophagy is directly linked to loss of NPC1 protein function. Screening of small molecule autophagy inducers identified compounds that could rescue the block in autophagy, leading to increased cell viability in NPC1-deficient hepatic and neuronal cells.
Results
Generation and Characterization of NPC Patient-Specific iPSCs
We generated transgene-free iPSCs from fibroblasts of NPC patients (Table 1) using Cre-excisable lentiviruses (Figure S1A available online) (Soldner et al., 2009; Sommer and Mostoslavsky, 2010) and derived up to 15 independent NPC1 iPSC lines from each patient sample (Table 1). We chose those with the lowest number of viral integrations for Cre-recombinase-mediated vector excision, which was confirmed by Southern blot analysis (Figures S1B and S1C). NPC1 iPSC lines expressed transcripts of endogenous pluripotency-related genes, stained positive for pluripotency markers, displayed a normal karyotype and were capable of forming teratomas with contribution to all three embryonic germ layers (Figures S1D–S1G). NPC1 protein levels were markedly reduced in NPC1 iPS-derived cells compared to control cells (Figure S1H). To generate disease-affected cell types, we induced hepatic (Si-Tayeb et al., 2010) and neuronal differentiation (Marchetto et al., 2010). Hepatic-like cells showed characteristic morphology, stained positive for lineage-specific markers such as α-fetoprotein (AFP), HNF4-α (HNF4a) and human albumin (ALB), and expressed lineage-specific genes (Figures 1A, S1I, and S1J). Neurons expressed specific markers such as class III β-tubulin (TUJ1) and microtubule-associated protein 2 (MAP2) (Figure 1B). Cell viability was significantly reduced in NPC1 iPSC-derived hepatic-like cells and aged neuronal cultures as compared to control iPSC and hESC-derived cells (Figures 1C and 1D).
Table 1.
Overview of Generated NPC Patient-Specific iPS Cell Lines and Used ESCs
| iPS/ES Clone ID | Parental Cell Line | Donor | Age of Biopsy (years) | Reprogramming Factors | Number of Factor-free iPS Clones | Number of iPS Clones Characterized | Cell Line Designation |
|---|---|---|---|---|---|---|---|
| NPC1-1 (#4, #13) | GM18453 | Niemann-Pick disease, type C NPC1 (I1061T/I1061T) | nk | loxP-TetO-OKSM, loxP-FUW-M2rtTA | 13 | 2 | WIBR-IPS-NPC1I1061T/ I1061T |
| NPC1-2 (#9, #26) | GM03123 | Niemann-Pick disease, type C NPC1 (P237S/I1061T) | 9 | loxP-TetO-OKSM, loxP-FUW-M2rtTA | 15 | 2 | WIBR-IPS-NPC1P237S/ I1061T |
| NPC1-3 (#4, #47) | GM22870 | Niemann-Pick disease, type C NPC1 (1920 delG/1009G > A) | 4 | loxP-TetO-OKSM, loxP-FUW-M2rtTA | 15 | 2 | WIBR-IPS-NPC11920 delG/1009G > A |
| NPC1-4 (#17, #20) | GM22871 | Niemann-Pick disease, type C NPC1 (1920 delG/1009G > A) | 4 | loxP-TetO-OKSM, loxP-FUW-M2rtTA | 9 | 2 | WIBR-IPS-NPC11920 delG/1009G > A |
| Control-1 | hES Cell Line | WIBR3 | |||||
| Control-2 (#11, #13) | GM23151 | Niemann-Pick disease, type C NPC1 (1920 delG/wt) | 39 | loxP-TetO-OKSM, loxP-FUW-M2rtTA | 10 | 2 | WIBR-IPS-NPC11920 delG/wt |
nk, not known.
Figure 1.
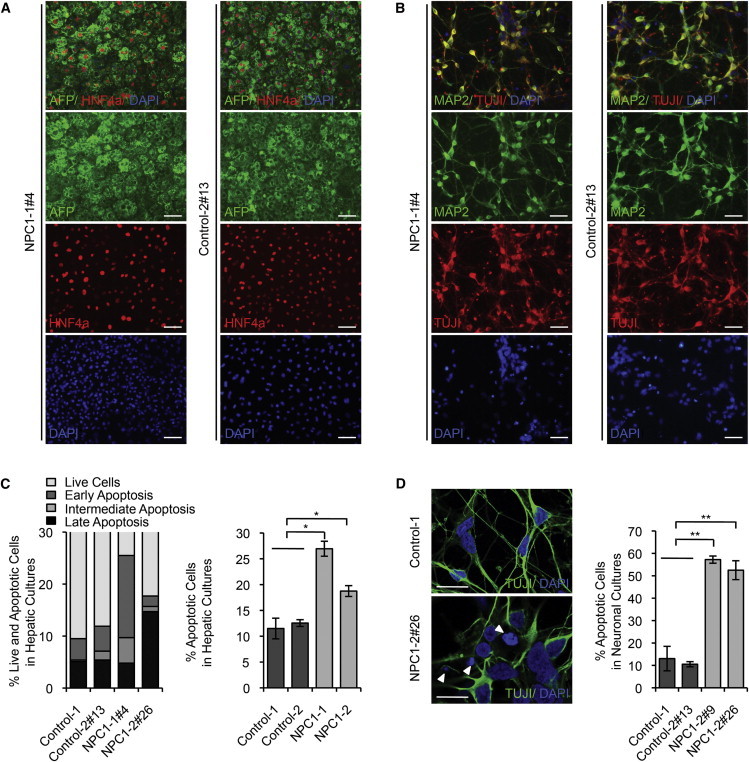
Generation and Characterization of Patient-Specific NPC1 iPSCs
(A) Immunofluorescence staining of hepatic cultures derived from representative NPC1 iPSC lines 21 days after induction of hepatocyte differentiation for Alpha-fetoprotein (AFP; green) and HNF4a (red). Nuclei were stained with DAPI (blue). Scale bar, 100 μm.
(B) Immunofluorescence staining of neuronal cultures derived from representative NPC1 iPSC lines 14 days after induction of differentiation for neuron-specific microtubule-associated protein 2 (MAP2; green) and class III β-tubulin (TUJI; red). Nuclei were stained with DAPI (blue). Scale bar, 100 μm.
(C) FACS analysis of cell viability and apoptosis in control and NPC1 iPSC-derived hepatic cultures measuring FITC-Annexin V and propidium iodide staining. Graphical data (right panel) represent mean ± SE (n = 3).
(D) Analysis of cell death in control and NPC1 iPSC-derived 5-week-old TUJI positive neurons. Nuclei stained with DAPI. Arrow shows apoptotic nuclei. Scale bar, 10 μm. Graphical data represent mean ± SE (n = 3).
Results shown are representative of at least three independent experiments using two different clones of each line unless otherwise indicated. ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, nonsignificant.
Generation of Isogenic Mutant and Control NPC1 iPSCs
Recent progress in human gene targeting using zinc finger nuclease and TALENs allows for the correction of a single disease-causing point mutation in iPSCs, and thereby the generation of isogenic disease and control cell lines (Soldner et al., 2011; Yusa et al., 2011). To repair the NPC1I1061T mutation, we designed TALEN pairs introducing a DNA double-strand break close to nt 3181C (Figures 2A, 2B, and S2A; see Supplemental Information) (Cermak et al., 2011). The donor construct contained a puromycin selection cassette (puroΔtk) flanked by piggyBac terminal repeats (Yusa et al., 2011) (Figure 2B) allowing for correction of the I1061T mutation and the complete removal of the selection cassette. We targeted a NPC patient line that is compound heterozygous and carries the NPC1I1061T mutation on one allele (NPC1-2) (Table 1). Integration of the piggyBac cassette was confirmed by Southern blot analysis and PCR (Figure S2B; data not shown). Out of 146 NPC1-2 iPSC-derived clones analyzed, five were targeted on the allele carrying the I1061T mutation (Table S1). In addition, we targeted the control line (control-2) that has one NPC1 mutant and one wild-type allele (Table 1). Two clones had the selection cassette integrated on the wild-type allele (Table S1). Integration of the piggyBac cassette on the wild-type allele in this cell line disrupted exon 21 and thereby generated a second mutant allele (Control-2-Mut). Overall, we observed a targeting efficiency of 6%. Transient expression of transposase in the targeted clones led to removal of the piggyBac selection cassette, which was confirmed by Southern blot (Figure 2C). We did not detect any reintegration of the piggyBac element (Figure S2C). Correction of the I1061T mutation in the NPC1-2-Corr line and restoration of the wild-type allele in the Control-2-Corr line were further confirmed by sequence analysis and PCR (Figures 2D and S2D). Analysis of the genomic DNA of corrected clones at the top ten predicted off-target cutting loci of TALEN pair 1 revealed no mutations (see Supplemental Information; data not shown). Genome-wide comparison of copy number variations (CNVs) of independent NPC1 iPSC lines and the isogenic pairs using Illumina sequencing showed no major changes (Figures S2E and S2F). Corrected NPC1 iPSC lines expressed pluripotency markers, were capable of forming teratomas and efficiently differentiated into hepatic-like and neuronal cells (Figures 2E, S2G, and S2H). Loss and restoration of NPC1 protein levels in the targeted NPC1 iPSCs was confirmed by western blot analysis (Figure 2F).
Figure 2.
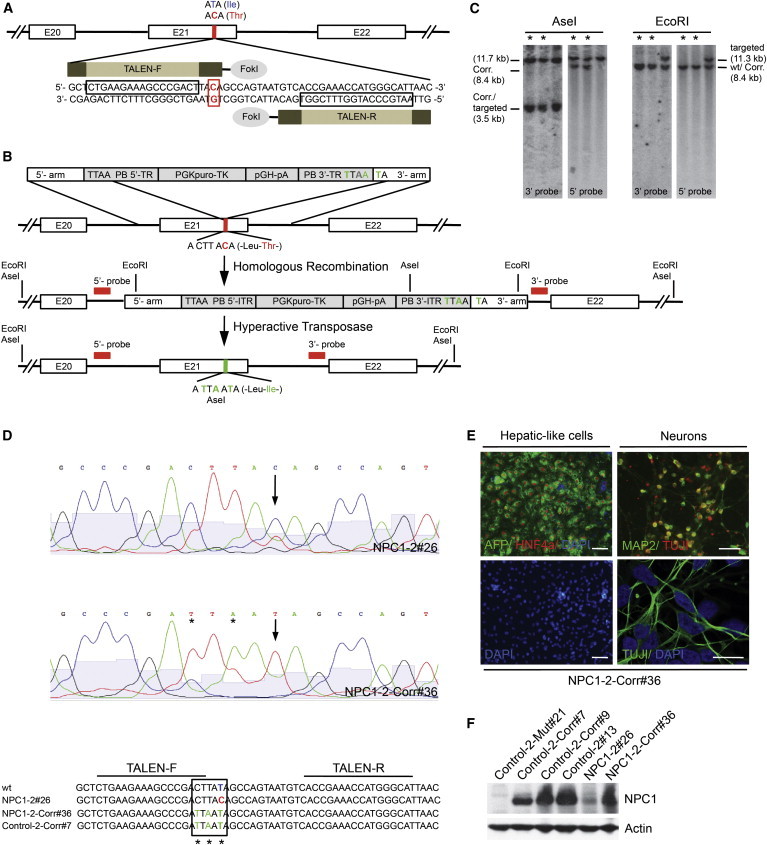
Correction of NPC1I1061T Mutation in Patient-Specific iPSCs
(A) Schematic overview of specific TALENs cutting site in the NPC1 gene. Blue letters are indicating the wild-type base and amino acid, respectively; red indicates the mutation.
(B) Schematic overview depicting the NPC1I1061T targeting strategy showing piggyBac (PB) donor plasmid design with homologous 5′- and 3′-arms, PB terminal repeats (PB-TR), and selection cassette (PGK-puroTK, pGH-pA). Exons (white boxes), restriction sites, and location of external 5′ and 3′ Southern blot probes (red bars) are indicted. Enlarged sequence indicates I1061T mutation in exon 21 (red base). Introduced changes are labeled in green.
(C) Southern blot analysis of NPC1 iPSCs after excision of PB selection cassette. Fragment sizes of wild-type (wt), corrected (Corr.), and targeted alleles are indicated. Excised clones are marked with an asterisk.
(D) Sequencing of genomic NPC1 locus in indicated NPC1 iPSC lines. The corrected I1061T mutation is marked with an arrow, induced bp changes with an asterisk.
(E) Immunofluorescence staining of hepatic [AFP (green) and HNF4a (red)] and neuronal cultures [MAP2 (green) and TUJ1 (red or green, respectively)] derived from representative NPC1-2-Corr#36 line. Nuclei were stained with DAPI (blue). Scale bar, 100 μm and 10 μm (lower right).
(F) Immunoblot analysis of indicated control and NPC1 iPSCs lines detecting NPC1 and actin protein levels.
Results shown in (C)–(F) are representative of at least three independent experiments using two different clones of each line.
NPC1 iPSC-Derived Hepatic and Neuronal Cells Show Defects in Cholesterol Metabolism
NPC disease is considered to be primarily a neurodegenerative disorder; nevertheless, hepatomegaly is one of the first detectable disease manifestations in more than 50% of NPC patients, and a significant number of infants die from liver failure (Kulinski and Vance, 2007; Rosenbaum and Maxfield, 2011; Vanier, 2010), but NPC1 deficiency in human disease-affected cell types has not been analyzed because of a lack of availability. We detected cholesterol accumulation in the LE/L compartments by Filipin staining in NPC1-deficient hepatic-like and neuronal cells (Figures 3A and 3B) similar to that observed in NPC1 iPSC-derived fibroblasts (Figures S3A and S3B). Sequestration of cholesterol in the LE/L compartments consequently affects downstream pathways, such as cholesterol ester (CE) synthesis (Abi-Mosleh et al., 2009; Brown and Goldstein, 1997; Goldstein et al., 1983), prompting us to analyze cholesterol esterification and the cholesterol sensing machinery in mutant hepatic cells. Defects in acetyl-CoA acetyltransferase-mediated synthesis of CE were detected by measuring the incorporation of [14C]oleate (Brown and Goldstein, 1997; Goldstein et al., 1983). Cholesteryl [14C]oleate synthesis was markedly increased in control hepatic-like cells after LDL treatment, whereas no increase was detected in NPC1-deficient hepatic-like cells (Figure 3C), suggesting an impairment in CE synthesis. Hydroxypropyl-β-cyclodextrin (HP-β-cyclodextrin), which releases cholesterol from the LE/L compartments (Abi-Mosleh et al., 2009), allowed CE synthesis in NPC1-deficient hepatic-like cells but not in the control cells (Figure 3D). HP-β-cyclodextrin (0.2%) was sufficient to induce maximal formation of cholesteryl [14C]oleate in NPC1-deficient hepatic-like cells. We did not detect any general effect on triglyceride formation in the treated hepatic cells (Figure S3C). NPC1-deficient and control cells treated with 25-hydroxycholesterol (25-HC), which stimulates cholesterol esterification by causing cholesterol to translocate directly from the plasma membrane to the ER, responded in a similar range (Figure S3D) (Abi-Mosleh et al., 2009), indicating that components of the cholesterol pathway downstream of LE/L compartments were intact. Our data show that sequestration of LE/L-resident cholesterol impairs cholesterol esterification in NPC1-deficient hepatic-like cells.
Figure 3.
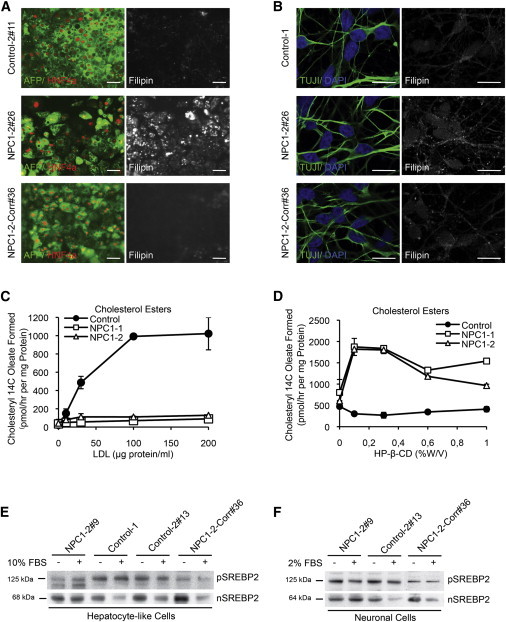
Analysis of Cholesterol Metabolism in Isogenic NPC1 iPSC-Derived Cell Types
(A and B) Immunofluorescence staining of representative control, mutant, and corrected NPC1 iPSC-derived hepatic (A) and neuronal (B) cells with lineage markers [AFP, green; HNF4a, red (A); TUJI, green (B)], respectively. Endogenous cholesterol was detected by Filipin staining (white) in the same samples as the lineage marker staining (A) or in duplicate samples of the same experiment (B). Scale bar, (A) 100 μm and (B) 10 μm. Results shown are representative of at least three independent experiments using two different clones of each line.
(C) Cholesterol ester formation in NPC1-deficient and control hepatic-like cultures after exposure to different concentrations of low-density lipoprotein (LDL). Mean variation for each of the duplicate incubations for control, NPC1-1, and NPC1-2 were in a range between 9% and 20% for the untreated samples and between 1%–31% for the data points at the different LDL concentrations, respectively.
(D) Cholesterol ester formation in NPC1 iPSC-derived hepatic cultures after exposure to different concentrations of HP-β-cyclodextrin (HP-β-CD [%w/v]). Mean variations for each of the duplicate incubations for control, NPC1-1, and NPC1-2 were in a range between 1% and 37% for the untreated samples and between 0.2% and 24% for the data points at the different HP-β-CD concentrations, respectively.
(C and D) Results shown are the mean of duplicates of each cell line and representative for three independent experiments using different clones of indicated cell lines.
(E and F) Immunoblot analysis detecting the activation of sterol regulatory element-binding protein 2 (SREBP2) cleavage in NPC1 iPSC-derived hepatic (E) and neuronal (F) cells after incubation with indicated serum concentrations. pSREBP2 (precursor SREBP2) in the cytoplasmic and nSREBP2 in the nuclear fraction of cell lysates were detected. Protein sizes are indicated. Results shown are representative of at least three independent experiments using two different clones of each line.
Rescue of Cholesterol Defects by Treatment with HP-β-Cyclodextrin or by Genetic Correction of the NPC1I1061T Mutation
A block in cholesterol transport to the ER impairs transcriptional regulation caused by constant activation of proteolytic cleavage of the sterol regulatory element-binding proteins (SREBPs) (Horton et al., 2002). Because cleaved SREBP translocates to the nucleus and activates transcription, we analyzed the formation of nuclear SREBP-2 (nSREBP-2) in NPC1-deficient hepatic cells. Although in control cells nSREBP-2 was decreased in the nuclear fraction in response to treatment with 10% serum, we did not detect any changes in NPC1-deficient cells (Figure 3E). Similar observations were made in NPC1 iPSC-derived neurons after treatment with 2% serum (Figure 3F). In addition, increased nSREBP-2 levels in NPC1-deficient hepatic cells lead to upregulation of transcription of specific target genes, such as SREBPs themselves. We detected an increase in SREBP RNA levels in NPC1-deficient hepatic cells after treatment with 10% serum, whereas RNA levels in controls were downregulated (Figure S3E). Upon combined treatment with 10% serum and 0.2% HP-β-cyclodextrin, transcript levels of SREBP and low-density lipoprotein receptor (LDL-R) decreased in NPC1-deficient hepatic cultures and were comparable to those in control cells after treatment with 10% serum only (Figures S3F and S3G). After correction of the NPC1I1061T mutation, NPC iPSC-derived hepatic and neuronal cells showed normal cholesterol distribution (Figures 3A and 3B) and responded to serum treatment by suppressing SREBP2 cleavage, as evident by a reduction in nSREBP2 protein and SREBP transcript levels (Figures 3E, 3F, and S3E). These results confirmed defects in cholesterol sensing in NPC1 iPSC-derived hepatic and neuronal cells that could be rescued by genetic correction of NPC1 I1061T.
Impairment of Autophagic Flux in NPC1 iPSC-Derived Human Disease-Affected Cell Types
Emerging evidence indicates that impaired autophagy contributes to neurodegenerative and liver disorders (Hara et al., 2006; Komatsu, 2012; Mizushima et al., 2008; Ravikumar et al., 2010) and is thought to be involved in NPC1 disease (Elrick et al., 2012; Ordonez et al., 2012; Sarkar et al., 2013). We assessed perturbation in autophagic flux in NPC1-deficient hepatic and neuronal cells. To monitor autophagic flux in NPC patient-specific iPSC-derived cells, we measured the levels of the specific autophagosome maker LC3-II (light chain 3 of microtubule-associated protein 1) and the pathway-specific substrate, p62. Levels of LC3-II directly correlate with the steady-state number of autophagosomes, whereas the levels of p62 reflect autophagic turnover (Klionsky et al. 2012, Bjørkøy et al., 2009). Both LC3-II and p62 levels were significantly increased in NPC1-deficient hepatic-like cells compared to the control cells (Figure 4A), suggesting impaired autophagic flux. However, p62 transcript levels in control and NPC1-deficient hepatic cells were comparable (Figure S4A), implying that accumulation of p62 protein is due to reduced clearance rather than diminished transcription. Similar observations were made in NPC1 iPSC-derived neuronal cultures (Figure 4B). We further analyzed autophagosome synthesis with a saturating concentration of bafilomycin A1 (BafA1), which inhibits lysosomal acidification and prevents LC3-II degradation, thus causing its accumulation (Klionsky et al., 2012). After BafA1 treatment, the accumulation of LC3-II in NPC1-deficient hepatic-like and neuronal cells was abrogated compared to controls (Figures 4C and 4D). These data suggest that accumulation of autophagosomes is not due to increased autophagosome synthesis but rather occurs because of a block in autophagic flux in NPC1-deficient cells. We did not find alterations in the upstream events regulating autophagosome synthesis, such as the levels of Beclin 1 and Atg5-Atg12 conjugation, in NPC1-deficient cells (Figures S4B and S4C). These data are consistent with our recent findings related to a block in autophagic flux seen in mouse NPC1 mutant cells that is attributed to impaired formation of amphisomes, resulting from defects in the SNARE machinery that regulates autophagosome maturation (Sarkar et al., 2013). Impaired SNARE functioning and autophagic flux are also reported in other lysosomal storage disorders, such as multiple sulphatase deficiency and mucopolysaccharidosis type IIIA, where LE/L-resident cholesterol accumulation is observed (Fraldi et al., 2010). We were not able to detect impaired autophagy in NPC1-deficient iPSCs, hepatic and neuronal progenitor cells, or early neurons (data not shown). Our data argue against previous findings that attributed the increase in steady-state LC3-II levels in human cells to an induction of autophagy (Ordonez et al., 2012; Pacheco et al., 2007).
Figure 4.
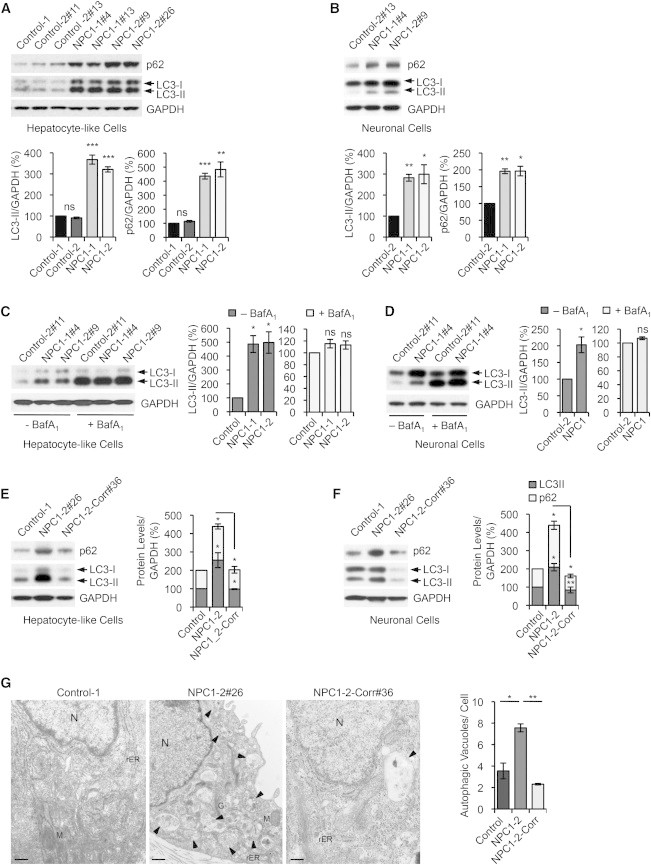
Genetic Correction of Autophagy Phenotype in NPC1 iPSC-Derived cells
(A and B) Immunoblot analysis and quantifications of p62 and LC3-II levels using anti-p62, anti-LC3, and anti-GAPDH antibodies in hepatic (A) and neuronal cultures (B) derived from indicated control and NPC1 iPSC lines. Graphical data represent mean ± SE (n = 4).
(C and D) Immunoblot analysis and quantification of LC3-II levels using anti-LC3 and anti-GAPDH antibodies in hepatic (C) and neuronal cultures (D) derived from control and NPC1 iPSC lines, treated with or without 400 nM bafilomycin A1 (BafA1) for 4 hr. Graphical data represent mean ± SE (n = 3).
(E and F) Immunoblot analysis and quantifications of p62 and LC3-II levels in hepatic (E) and neuronal cultures (F) derived from the NPC1-2 iPSC line after correction of the NPC1I1061T mutation. Graphical data represent mean ± SE (n = 3).
(G) Electron microscopy images of representative NPC1 iPSC-derived hepatic-like cells before and after genome editing. Arrows are indicating autophagic vacuoles. Graphical data represent mean ± SE (n = 3). Nucleus (N), rough endoplasmatic reticulum (rER), mitochondria (M), Golgi (G). Scale bar, 500 nm.
Results shown are representative for at least three independent experiments using two different clones of each line. ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, nonsignificant.
Defect in Autophagy Is Directly Linked to the NPC1I1061T Mutation
To assess whether the defect in autophagic flux is caused by NPC1 deficiency, we compared LC3-II and p62 levels in hepatic and neuronal cells carrying the NPC1I1061T mutation and their isogenic control. We found that basal autophagy was restored in the mutant cells after genetic correction (Figures 4E and 4F). In contrast, disruption of the NPC1 wild-type allele in control hepatic cells, which led to loss of NPC1 expression (Figure 2F), resulted in marked increase of LC3-II and p62 levels, which phenocopied the effects found in NPC1 iPSC-derived hepatic cells (Figure S4D). Electron microscopic analysis confirmed accumulation of autophagic vacuoles and lipid droplets (LDs) in NPC1 mutant hepatic cells, a phenotype that was abolished after correction of the NPC1I1061T mutation (Figures 4G and S4E). Thus, genetic correction of the NPC1 mutation rescued both the cholesterol and autophagy abnormalities.
Treatment with HP-β-cyclodextrin (Vance and Peake, 2011) did not rescue the perturbation in autophagic flux. Instead, we observed an increased block in autophagy, which was dose dependent as evident by elevation of LC3-II and p62 levels (Figure S5A). A lower dose of HP-β-cyclodextrin (0.2%), sufficient to restore defects in cholesterol metabolism as shown by induction of cholesterol esterification (Figure 3D), did not further impair autophagy in NPC1 iPSC-derived hepatic-like cells (Figure S5A). In summary, we show a block in autophagic flux in NPC1 disease-affected human cells. This defect was abrogated in hepatic-like and neuronal cells by genetic correction of the NPC1I1061T mutation but not by depleting cholesterol, thus implying that impaired autophagy is directly linked to loss of NPC1 protein function.
Induction of Autophagy Rescues the Defect in Basal Autophagy and Increases Cell Viability in NPC1-Deficient Hepatic-like and Neuronal Cells
Upregulation of autophagy is beneficial in transgenic models of several neurodegenerative and certain liver disorders (Rubinsztein et al., 2012; Sarkar, 2013). Using NPC iPSC-derived cells may allow the identification of potent compounds that can rescue the autophagy defects in human disease-affected cells. We found that induction of autophagy by inhibiting the mammalian target of rapamycin (mTOR) pathway with rapamycin (Rap) or starvation (Ravikumar et al., 2010) significantly reduced p62 levels in NPC1 iPSC-derived hepatic cells to amounts comparable to the basal levels in control cells (Figures 5A and S5B). Because long-term treatment with mTOR inhibitors may have unfavorable side effects due to critical functions of mTOR in growth and translation (Sarkar, 2013), we performed a small-scale candidate drug screen testing different concentrations of mTOR-independent autophagy enhancers, such as carbamazepine (CBZ), verapamil (Ver), trehalose (Tre), and SMER28 (Figures 5B and S5C; see Supplemental Information). These compounds are reported to be protective in Drosophila or mouse models of Alzheimer’s, Parkinson’s, and Huntington’s disease (Rubinsztein et al., 2012; Sarkar, 2013) or α-antitrypsin deficiency (Hidvegi et al., 2010; Sarkar, 2013). By measuring p62 clearance, we found CBZ to be the most potent drug in rescuing the defective autophagy phenotype in NPC1 iPSC-derived hepatic cells (Figure 5B). CBZ, which induces mTOR-independent autophagy by lowering inositol and IP3 levels (Sarkar et al., 2005), reduced p62 levels to a similar extent as observed with rapamycin treatment (Figure 5B). Our data suggest that CBZ-mediated restoration of autophagic flux was sufficient to overcome the block observed in NPC1-deficient hepatic cells, which may be due to a recently characterized bypass mechanism (Sarkar et al., 2013).
Figure 5.
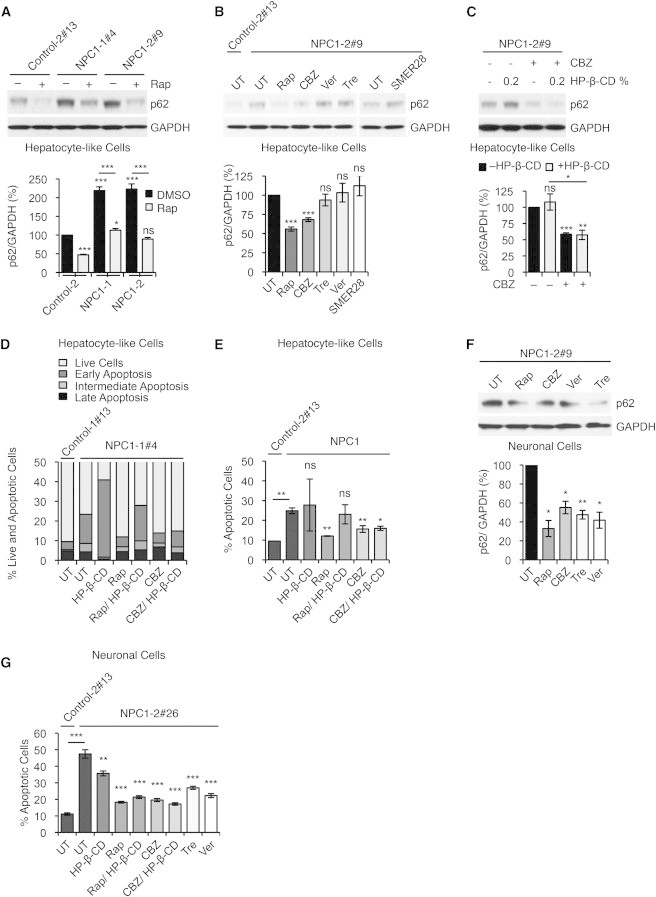
Chemical Correction of Autophagy Phenotype in NPC1 iPSC-Derived cells
(A) Immunoblot analysis and quantification of p62 levels using anti-p62 and anti-GAPDH antibodies in control and NPC1 iPSC-derived hepatic cultures, treated with either DMSO (vehicle control) or rapamycin (Rap). Graphical data represent mean ± SE (n = 3).
(B) Immunoblot analysis and quantification of p62 levels using anti-p62 and anti-GAPDH antibodies in control and NPC1 iPSC-derived hepatic cultures after treatment with autophagy inducing compounds: untreated (UT), rapamycin (Rap), carbamazepine (CBZ), verapamil (Ver), trehelose (Tre), and SMER28. Graphical data represent mean ± SE (n = 3).
(C) Immunoblot analysis and quantification of p62 levels using anti-p62 and anti-GAPDH antibodies in NPC1 iPSC-derived hepatic cultures, treated with CBZ and HP-β-CD as indicated. Graphical data represent mean ± SE (n = 3).
(D and E) FACS analysis of cell viability and apoptosis in NPC1 iPSC-derived hepatic cultures after treatment with indicated compounds measuring FITC-Annexin V and propidium iodide staining. (E) Graphical data represent mean ± SE (n = 3).
(F) Immunoblot analysis and quantification of p62 levels using anti-p62 and anti-GAPDH antibodies in NPC1 iPSC-derived neuronal cultures after treatment with autophagy inducing compounds: untreated (UT), Rap, CBZ, Ver, and Tre. Graphical data represent mean ± SE (n = 3).
(G) Analysis of cell death in NPC1 iPSC-derived 5-week-old neurons by assessing fragmented and TUNNEL positive nuclei. Graphical data represent mean ± SE (n = 6).
Results shown are representative for at least two independent experiments using two different clones of each line. ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, nonsignificant.
We assessed the possibility of using CBZ in combination with HP-β-cyclodextrin (0.2%, sufficient to induce cholesterol ester formation without perturbing autophagy; Figures 3D and S5A) to simultaneously restore both the cholesterol and autophagy defects. We found a significant reduction in p62 levels after dual treatment comparable to the effects of CBZ alone (Figure 5C), as well as a partial rescue of the defective cholesterol metabolism as observed by downregulation of SREBP protein, and SREBP and LDL-R transcript levels (Figures S5D and S5E). p62 transcript levels were not affected by the compound treatment (Figure S5F), suggesting that the clearance of p62 protein levels is due to increased autophagic degradation. Chemical induction of autophagy with Rap or CBZ, or CBZ in combination with a low dose of HP-β-cyclodextrin, significantly increased cell viability of NPC1-deficient hepatic cells (Figures 5D and 5E).
We further evaluated the effects of autophagy-inducing compounds using NPC1 iPSC-derived neuronal cultures. In addition to Rap and CBZ, Tre and Ver significantly rescued the defective autophagy phenotype in these cells, as assessed by p62 clearance (Figures 5F and S5G). Notably, cell viability in NPC1-deficient neuronal cultures was significantly increased with all the chemical inducers of autophagy alone, as well as in combination with 0.2% HP-β-cyclodextrin (Figure 5G). Although our data suggest that induction of autophagy by itself is cytoprotective in the context of NPC disease, combining this treatment with a partial release of the LE/L-resident cholesterol can restore the autophagy defects as well as improve cholesterol homeostasis in the liver and the brain.
Discussion
In summary, our data show that patient-specific NPC1 iPSC-derived hepatic and neuronal cells are less viable and develop disease-relevant defects, such as in cholesterol metabolism and autophagic flux (Figure 6). In contrast to previous reports, we do not observe similar defects in NPC1 iPSCs or in neuronal and liver progenitor cells (Trilck et al., 2013). Both these defects are abrogated after correction of the NPC1I1061T mutation, implying that the NPC1 protein has a functional role in both the processes. Recent studies provide evidence that regulation of lipolysis and autophagy are interrelated and controlled by similar regulatory mechanisms (Singh and Cuervo, 2012). We speculate that disturbance of this interrelation may trap NPC1-deficient cells in a vicious cycle where inhibition of autophagy will cause increased intracellular lipid content (Ouimet, 2013; Sarkar et al., 2013; Singh and Cuervo, 2012; Singh et al., 2009), a condition that may further deteriorate by the release of high levels of cholesterol (Peake and Vance, 2012) or by blocking autophagy (Meske et al., 2014; Sarkar et al., 2013). Brain and liver-specific deletion of essential autophagy genes (such as Atg5 or Atg7) in normal mice causes neurodegeneration and liver injury, respectively (Hara et al., 2006; Komatsu et al., 2006, 2007), similar to those observed in NPC1 patients. These observations further support our assumption that impairment in autophagy is a crucial contributing factor for NPC disease.
Figure 6.
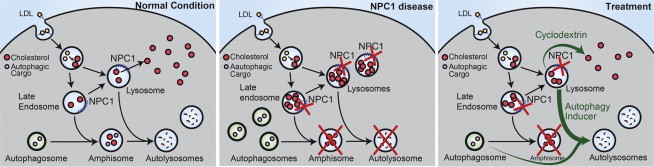
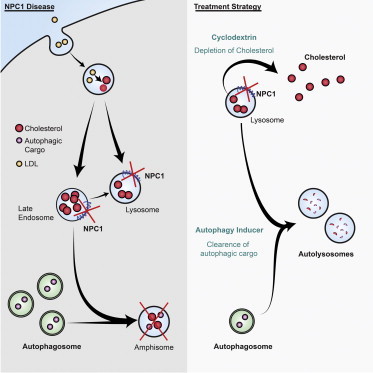
Schematic Overview of Correction of Functional Defects in NPC Disease-Affected Cell Types
Left panel shows cholesterol distribution and autophagic flux under normal conditions. Middle panel shows the effects due to loss of NPC1 protein function on cholesterol metabolism and autophagic flux. Mutations in the NPC1 gene on both alleles lead to accumulation of cholesterol in the LE/L compartments by inhibiting its efflux, and to a block in autophagic flux causing accumulation of autophagosomes and autophagy substrate arising due to impaired formation of amphisomes. Chemical correction of disease related phenotypes are achieved by HP-β-cyclodextrin-mediated cholesterol release and CBZ-mediated autophagy induction (green arrows, right panel). Restoration of autophagic flux by autophagy inducer is possibly through a bypass mechanism.
Our data argue that induction of autophagy might be a plausible treatment option for NPC disease. Such a strategy is supported by our recent study in NPC1 mutant mouse cells showing that a block in autophagic flux due to defects in amphisome formation can be bypassed by stimulating autophagy (Sarkar et al., 2013). Induction of autophagy has been shown to be protective in several models of neurodegenerative and liver diseases (Hidvegi et al., 2010; Ravikumar et al., 2010; Rubinsztein et al., 2012; Sarkar, 2013). Because mTOR governs critical cellular function such as translation and cell growth, small molecule autophagy enhancers acting independently of mTOR might be more suitable for long-term treatment of patients. Screening mTOR-independent autophagy inducers that are known to increase autophagic flux in various in vitro and in vivo transgenic disease models (Sarkar, 2013) revealed cell type specificity, because only CBZ could overcome the impairment in autophagy in both NPC1 iPSC-derived hepatic and neuronal cells. These findings underline the unique value of using human iPSC-derived disease-relevant cells for identifying potent compounds of biomedical relevance (Grskovic et al., 2011). We show that higher concentrations of HP-β-cyclodextrin treatment further perturb autophagic flux in NPC1 iPSC-derived hepatic cells with likely deleterious cellular consequences. These observations are consistent with earlier findings describing neurotoxic effects of HP-β-cyclodextrin treatment in neurons from NPC1 mutant mice (Peake and Vance, 2012).
Our data indicate that induction of autophagy itself is beneficial, and in combination with a cholesterol-depleting agent (such as low dose of HP-β-cyclodextrin) that does not impact on autophagy will rescue both the autophagy and cholesterol defects in NPC1 patients. Our study suggests that inducing autophagy with CBZ may have overall benefit for the treatment of NPC disease as it might be effective in liver and brain.
Experimental Procedures
HiPSC Derivation, Cultivation, and Differentiation
Transgene-free iPSCs were generated from fibroblasts of NPC patients (Table 1) using Cre-excisable lentiviruses encoding a polycistronic doxcycycline-inducible (DOX) expression cassette containing all four reprogramming factors Oct4, Klf4, Sox2, c-Myc (OKSM) (pHAGE2-TetOminiCMV-hSTEMCCA-loxP) (Sommer and Mostoslavsky, 2010) and a modified tetracycline-controlled trans-activator (FUW-M2rtTA-loxP) (Soldner et al., 2009). All pluripotent cell lines have been characterized using standard pluripotency assays (see Supplemental Experimental Procedures). All protocols were approved by the relevant Institutional Review Board (Massachusetts Institute of Technology) and the Embryonic Stem Cell Research Oversight Committees (Whitehead Institute). Differentiation of iPSCs into hepatic-like cells was induced after single-cell replating of iPSCs on matrigel following published protocols (Si-Tayeb et al., 2010). Neuronal progenitors (NPs) and neurons were derived using an embryoid body (EB)-based protocol (Marchetto et al., 2010).
TALEN and Donor Design and Generation
TALENs were designed according to previously published principles (Cermak et al., 2011; Miller et al., 2011). All TALENs used the +63 truncation point for fusion to the obligate heterodimeric FokI cleavage domain (Doyon et al., 2011; Miller et al., 2011). The tandem arrays of TALE repeats were assembled as described previously (Marchetto et al., 2010) (see Supplemental Information). Their editing activity was assayed using the Surveyor nuclease (Transgenomic) (Guschin et al., 2010). The targeted loci were PCR amplified using the following primer pairs: Cel-I-NPC1-F (5′-atgctgcctatagttctgcag-3′) and Cel-I-NPC1-R (5′-tcacagagactttagattctg-3′) for the NPC1 locus. PCR products were then denatured, rehybridized, digested with the Surveyor nuclease, and analyzed. NPC1I1061T-specific TALENS pairs introducing a DNA double-strand break in exon 21 (nt 3181C) were designed according to previously published principles (Cermak et al., 2011). To increase targeting efficiency, we used a donor construct containing a puromycin selection cassette (PGK-puroΔtk-pGH-pA cassette) flanked by piggyBac terminal repeats (Yusa et al., 2011). The 700 bp 3′ homology arm introduced the ACA (Thr) to ATA (Ile) switch correcting for the mutation. In addition, a silent CTT to TTA (Leu) codon switch 3 bp upstream of the mutation was introduced, generating the TTAA sequence necessary for the piggyBac excision (Figures 2A and 2B).
TALEN-Mediated Gene Targeting and Transposon Excision
Cells were prepared as described previously (Kondo et al., 2013; Soldner et al., 2009). Cells (1 × 107) were electroporated with 35 μg of donor plasmid (designed and assembled by H.W.) and 7.5 μg of each TALEN expression vector (Gene Pulser Xcell System, Bio-Rad: 250 V, 500 μF, 0.4 cm cuvettes) and subsequently plated on DR4 MEF feeder layers in hESC medium supplemented with ROCK inhibitor for the first 24 hr. Puromycin selection (0.5 μg/ml) was started 72 hr after electroporation. Individual colonies were picked 10–14 days after electroporation. Correctly targeted clones were confirmed by Southern blot (AseI, EcoRI digested) and used for transposon removal. Cells (1 × 107) were electroporated with 10 μg of hyperactive piggyBac transposase expression vector (pCMV-hypBase) as described previously (Soldner et al., 2009). On day 4, medium was changed to hESC medium containing 0.25 μM FIAU. Individual colonies were picked and expanded. Genotype and deletion of the piggyBac transposon were analyzed by Southern blot (AseI, EcoRI digest). Additionally PCR and sequencing mutation analysis was performed. Genomic DNA was amplified with primers NPC1-Wt-F (5′-cctttgattatacatgaaaccag-3′), NPC1-I1061T-Mut-F (5′-gaagaaagcccgacttac-3′), NPC1-I1061T-Corr-F (5′-gaagaaagcccgattaat-3′), respectively, and NPC1-R (5′-gagcattccacagcattctg-3′) under standard PCR conditions. PCR products were sequenced (Applied Biosystems Model 3730 capillary DNA sequencer with Big Dye Terminator Cycle Sequencing Kit).
Prediction of NPC1 TALENs Off-Target Sites
The position weight matrices (PWMs) of NPC1 TALEN pairs (see Supplemental Information) were constructed using model 3 as described previously (Moscou and Bogdanove, 2009) (see Supplemental Information).
Immunostaining
Immunostaining was performed according to standard protocols using primary antibodies listed in the Supplemental Information. Cholesterol was detected by Filipin staining (50 μg/ml Filipin complex, Streptomyces filipinensis; Sigma-Aldrich).
Acyl-CoA Acetyltransferase Assay
Defects in cholesterol metabolism were assessed using the Acyl-CoA Acetyltransferase Assay. The rate of incorporation of [14C]oleate into cholesterol [14C]oleate and [14C]triglycerides by intact cell monolayers was measured as previously described (Goldstein et al., 1983).
Immunoblot Analysis
Cell pellets were lysed on ice in Lysis Buffer (10 mM Tris-HCl [pH 7.4], 2% SDS, 1 mM DTT, 10% glycerol, and 120 mg/ml urea) for 30 min in presence of Complete EDTA-free Protease Inhibitor Cocktail (Roche Diagnostics) and subjected to SDS-PAGE and standard immunoblot analysis. Primary antibodies used are listed in the Supplemental Information. Amersham ECL Western Blotting Detection Reagent (GE Healthcare) was used for visualization.
Chemical Compounds
Autophagic flux in NPC1 iPSC-derived cells was assessed using a saturating concentration of bafilomycin A1 (BafA1) clamping LC3-II/autophagosome degradation (Klionsky et al., 2012). To test the possibility of inducing autophagy, we performed a candidate drug screen including described mTOR-independent autophagy inducers (Sarkar, 2013) (Figure S5G; Supplemental Information). Their effect was analyzed by measuring p62 levels and cell viability by FITC-Annexin V/ PI staining. Compounds used for treatment of hepatic-like cells were bafilomycin A1 (Enzo Life Sciences), rapamycin (LC Laboratories), carbamazepine, verapamil, trehalose, SMER28, and HP-β-cyclodextrin (all Sigma-Aldrich). Cells were treated with compounds as indicated.
Statistical Analyses
Densitometry analyses on the immunoblots were done by ImageJ software (NIH) by measuring levels of the protein of interest relative to the loading control, as previously described (Williams et al., 2008). The control condition was set to 100%, and the data were represented as mean ± SEM. The y axis values are shown in percentages (%). Experiments were performed in triplicates at least twice. The p values for densitometry analyses, vesicle number, and aggregate formation were determined by Student’s t test (unpaired) using Prism 6 software (GraphPad), as previously described (Korolchuk et al., 2009). ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, nonsignificant.
Author Contributions
D.M., S.S., H.W., and R.J. designed the experiments and wrote the paper. D.M. and H.W. designed and performed the TALEN-mediated gene correction experiments. D.M. and S.S. designed and performed the autophagy experiments. L.A.-M. performed the ACAT assay. M.M. and P.X. provided technical assistance. A.W.C. performed computational analysis for TALEN specificity and for TALEN off-target cutting sides. Q.G. analyzed teratomas. All other experiments were performed by D.M.
Acknowledgments
We thank J.L. Goldstein, M.S. Brown, and D.F. Voytas for their helpful advice; R. Alagappan, T. Lungjangwa, K. Ganz, R. Flannery, D. Fu, T. DiCesare, T. Volkert, N. Watson, and W. Salmon for technical assistance. We thank Keck Microscopy Facility; Whitehead Technology Core; G. Bell of Whitehead Bioinformatics & Research Computing. We thank all Jaenisch lab members for helpful discussion. D.M. was a Peter G. Pentchev Research Fellow of the NNPD Foundation. S.S. is a Former Fellow at Hughes Hall, University of Cambridge, UK. R.J. was supported by US NIH grant R01-CA084198 and Skoltech Center; J.L. Goldstein and M.S. Brown were supported by the US NIH grant HL20948. R.J. is an advisor to Stemgent and Fate Therapeutics.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Accession Numbers
The NCBI SRA accession number for the CMV analysis reported in this paper is SRP026624.
Supplemental Information
References
- Abi-Mosleh L., Infante R.E., Radhakrishnan A., Goldstein J.L., Brown M.S. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. USA. 2009;106:19316–19321. doi: 10.1073/pnas.0910916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T., Pankiv S., Øvervatn A., Brech A., Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- Brown M.S., Goldstein J.L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Carstea E.D., Morris J.A., Coleman K.G., Loftus S.K., Zhang D., Cummings C., Gu J., Rosenfeld M.A., Pavan W.J., Krizman D.B. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y., Vo T.D., Mendel M.C., Greenberg S.G., Wang J., Xia D.F., Miller J.C., Urnov F.D., Gregory P.D., Holmes M.C. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat. Methods. 2011;8:74–79. doi: 10.1038/nmeth.1539. [DOI] [PubMed] [Google Scholar]
- Elrick M.J., Yu T., Chung C., Lieberman A.P. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum. Mol. Genet. 2012;21:4876–4887. doi: 10.1093/hmg/dds324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraldi A., Annunziata F., Lombardi A., Kaiser H.J., Medina D.L., Spampanato C., Fedele A.O., Polishchuk R., Sorrentino N.C., Simons K., Ballabio A. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010;29:3607–3620. doi: 10.1038/emboj.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gelsthorpe M.E., Baumann N., Millard E., Gale S.E., Langmade S.J., Schaffer J.E., Ory D.S. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 2008;283:8229–8236. doi: 10.1074/jbc.M708735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein J.L., Basu S.K., Brown M.S. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- Grskovic M., Javaherian A., Strulovici B., Daley G.Q. Induced pluripotent stem cells—opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011;10:915–929. doi: 10.1038/nrd3577. [DOI] [PubMed] [Google Scholar]
- Guschin D.Y., Waite A.J., Katibah G.E., Miller J.C., Holmes M.C., Rebar E.J. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hidvegi T., Ewing M., Hale P., Dippold C., Beckett C., Kemp C., Maurice N., Mukherjee A., Goldbach C., Watkins S. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M. Liver autophagy: physiology and pathology. J. Biochem. 2012;152:5–15. doi: 10.1093/jb/mvs059. [DOI] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Koike M., Sou Y.S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Kondo T., Asai M., Tsukita K., Kutoku Y., Ohsawa Y., Sunada Y., Imamura K., Egawa N., Yahata N., Okita K. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Korolchuk V.I., Mansilla A., Menzies F.M., Rubinsztein D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulinski A., Vance J.E. Lipid homeostasis and lipoprotein secretion in Niemann-Pick C1-deficient hepatocytes. J. Biol. Chem. 2007;282:1627–1637. doi: 10.1074/jbc.M610001200. [DOI] [PubMed] [Google Scholar]
- Marchetto M.C., Carromeu C., Acab A., Yu D., Yeo G.W., Mu Y., Chen G., Gage F.H., Muotri A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meske V., Erz J., Priesnitz T., Ohm T.G. The autophagic defect in Niemann-Pick disease type C neurons differs from somatic cells and reduces neuronal viability. Neurobiol. Dis. 2014;64:88–97. doi: 10.1016/j.nbd.2013.12.018. [DOI] [PubMed] [Google Scholar]
- Millard E.E., Gale S.E., Dudley N., Zhang J., Schaffer J.E., Ory D.S. The sterol-sensing domain of the Niemann-Pick C1 (NPC1) protein regulates trafficking of low density lipoprotein cholesterol. J. Biol. Chem. 2005;280:28581–28590. doi: 10.1074/jbc.M414024200. [DOI] [PubMed] [Google Scholar]
- Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscou M.J., Bogdanove A.J. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Ordonez M.P., Roberts E.A., Kidwell C.U., Yuan S.H., Plaisted W.C., Goldstein L.S. Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum. Mol. Genet. 2012;21:2651–2662. doi: 10.1093/hmg/dds090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M. Autophagy in obesity and atherosclerosis: Interrelationships between cholesterol homeostasis, lipoprotein metabolism and autophagy in macrophages and other systems. Biochim. Biophys. Acta. 2013;1831:1124–1133. doi: 10.1016/j.bbalip.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Pacheco C.D., Kunkel R., Lieberman A.P. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007;16:1495–1503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- Peake K.B., Vance J.E. Normalization of cholesterol homeostasis by 2-hydroxypropyl-β-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 2012;287:9290–9298. doi: 10.1074/jbc.M111.326405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B., Sarkar S., Davies J.E., Futter M., Garcia-Arencibia M., Green-Thompson Z.W., Jimenez-Sanchez M., Korolchuk V.I., Lichtenberg M., Luo S. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Rosenbaum A.I., Maxfield F.R. Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J. Neurochem. 2011;116:789–795. doi: 10.1111/j.1471-4159.2010.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein D.C., Codogno P., Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha K., Jaenisch R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell. 2009;5:584–595. doi: 10.1016/j.stem.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013;41:1103–1130. doi: 10.1042/BST20130134. [DOI] [PubMed] [Google Scholar]
- Sarkar S., Floto R.A., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L.J., Rubinsztein D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005;170:1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S., Carroll B., Buganim Y., Maetzel D., Ng A.H., Cassady J.P., Cohen M.A., Chakraborty S., Wang H., Spooner E. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013;5:1302–1315. doi: 10.1016/j.celrep.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Tayeb K., Noto F.K., Nagaoka M., Li J., Battle M.A., Duris C., North P.E., Dalton S., Duncan S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Cuervo A.M. Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012;2012:282041. doi: 10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A.M., Czaja M.J. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F., Jaenisch R. Medicine. iPSC disease modeling. Science. 2012;338:1155–1156. doi: 10.1126/science.1227682. [DOI] [PubMed] [Google Scholar]
- Soldner F., Hockemeyer D., Beard C., Gao Q., Bell G.W., Cook E.G., Hargus G., Blak A., Cooper O., Mitalipova M. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F., Laganière J., Cheng A.W., Hockemeyer D., Gao Q., Alagappan R., Khurana V., Golbe L.I., Myers R.H., Lindquist S. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C.A., Mostoslavsky G. Experimental approaches for the generation of induced pluripotent stem cells. Stem Cell Res. Ther. 2010;1:26. doi: 10.1186/scrt26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trilck M., Hübner R., Seibler P., Klein C., Rolfs A., Frech M.J. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J. Rare Dis. 2013;8:144. doi: 10.1186/1750-1172-8-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J.E., Peake K.B. Function of the Niemann-Pick type C proteins and their bypass by cyclodextrin. Curr. Opin. Lipidol. 2011;22:204–209. doi: 10.1097/MOL.0b013e3283453e69. [DOI] [PubMed] [Google Scholar]
- Vanier M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A., Sarkar S., Cuddon P., Ttofi E.K., Saiki S., Siddiqi F.H., Jahreiss L., Fleming A., Pask D., Goldsmith P. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C., Turley S.D., Pentchev P.G., Dietschy J.M. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am. J. Physiol. 1999;276:E336–E344. doi: 10.1152/ajpendo.1999.276.2.E336. [DOI] [PubMed] [Google Scholar]
- Yusa K., Rashid S.T., Strick-Marchand H., Varela I., Liu P.Q., Paschon D.E., Miranda E., Ordóñez A., Hannan N.R., Rouhani F.J. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.