

Summary
Induced pluripotent stem cells (iPSCs) acquire embryonic stem cell (ESC)-like epigenetic states, including the X chromosome. Previous studies reported that human iPSCs retain the inactive X chromosome of parental cells, or acquire two active X chromosomes through reprogramming. Most studies investigated the X chromosome states in established human iPSC clones after completion of reprogramming. Thus, it is still not fully understood when and how the X chromosome reactivation occurs during reprogramming. Here, we report a dynamic change in the X chromosome state throughout reprogramming, with an initial robust reactivation of the inactive X chromosome followed by an inactivation upon generation of nascent iPSC clones. iPSCs with two active X chromosomes or an eroded X chromosome arise in passaging iPSCs. These data provide important insights into the plasticity of the X chromosome of human female iPSCs and will be crucial for the future application of such cells in cell therapy and X-linked disease modeling.
Graphical Abstract
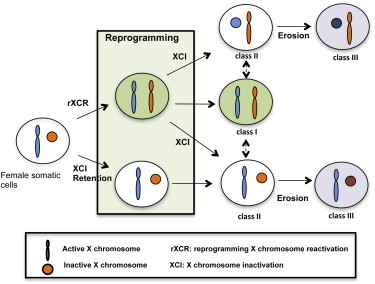
Highlights
-
•
The X chromosome state changes dynamically during human somatic cell reprogramming
-
•
Ectopic reprogramming factors transiently activate the inactive X chromosome
-
•
Nascent iPSC colonies carry an inactive X chromosome
-
•
Class I and class III iPSCs arise from nascent iPSCs
Determining the X chromosome state in human iPSCs is crucial for modeling X-linked diseases. Park and colleagues trace the X chromosome state in reprogramming and show that reprogramming reactivates the inactive X chromosome in female cells. Activated X chromosomes are randomly inactivated in nascent iPSCs. In early passaging, iPSC clones arise that contain two active X chromosomes or an eroded X chromosome.
Introduction
Expression of a defined set of transcription factors (OCT4, SOX2, KLF4, and c-MYC) reprograms human somatic cells to a pluripotent state, generating induced pluripotent stem cells (iPSCs) (Park et al., 2008b; Takahashi et al., 2007; Yu et al., 2007). iPSCs are similar to embryonic stem cells (ESCs) and are capable of indefinite self-renewal and differentiation into cells of all three germ layers. iPSCs also maintain the genomic composition of parental somatic cells and thus are considered as autologous cellular resources that are critical for cell therapy and in vitro disease modeling (Park et al., 2008a; Wu and Hochedlinger, 2011). Detailed genetic and epigenetic comparisons between iPSCs and ESCs, however, have shown that they are close but not identical (Chin et al., 2009). Reprogramming leaves reprogramming-specific epigenetic marks and produces copy number variation (Hussein et al., 2011; Lister et al., 2011). In addition, de novo mutations seem to accompany reprogramming and cause genetic alterations in iPSCs, although more in-depth analyses are needed before we can draw definite conclusions regarding the genetic changes in reprogramming (Abyzov et al., 2012; Gore et al., 2011).
Reprogramming affects the X chromosome status in female cells. During early development, one of the active X chromosomes in the inner cell mass (ICM) cells of the blastocyst undergoes random X chromosome inactivation (XCI) when ICM cells differentiate into epiblast cells (Mak et al., 2004). Only cells that are committed to developing as primordial germ cells (PGCs) start to reactivate the inactive X chromosome during migration to the genital ridge. In contrast, somatic cells maintain the inactive X chromosome throughout their life (de Napoles et al., 2007). Murine ESCs derived from ICM cells are considered to be in a naive state, and there are two active X chromosomes in female ESCs (Hanna et al., 2010). The X chromosome status in murine female iPSCs is indistinguishable from that in murine ESCs. Reprogramming activates the inactive X chromosome to produce iPSCs with two active X chromosomes (Maherali et al., 2007). Human ESCs are presumed to be derived from the epiblast cells of the embryo and have one inactive X chromosome. However, successful derivation of human ESCs with two active X chromosomes suggested that human ESCs are counterparts of ICM cells as well, but are prone to undergo XCI unless they are maintained in a pristine physiological condition, including a hypoxic oxygen concentration and no oxidative stress (Diaz Perez et al., 2012; Lengner et al., 2010). Thus, most human ESCs were reported to carry only one active X chromosome. In-depth studies on female human ESCs categorized them into three classes according to their X chromosome status (Kim et al., 2011; Lessing et al., 2013). Class I female human ESCs have two active X chromosomes, like murine ESCs, and show neither H3K27me3 nor XIST foci. When differentiated, class I ESCs undergo random XCI and form H3K27me3 foci and a XIST cloud. Spontaneous inactivation of one of the two X chromosomes in class I ESCs results in the formation of H3K27me3 and XIST foci, leading to the conversion of class I to class II cells. Class II ESCs maintain the inactive X chromosome after differentiation. However, the inactive X chromosome in class II ESCs is reversible and becomes reactivated with treatment of HDAC inhibitors (Diaz Perez et al., 2012). Continuous long-term passaging of H3K27me3 foci-positive class II ESCs triggers them to become H3K27me3 foci-negative class III ESCs. Although they are negative for H3K27me3 foci and XIST expression, class III ESCs carry one inactive X chromosome whose status seems to be permanent, and do not show H3K27me3 foci upon differentiation (Diaz Perez et al., 2012). As in the case of human ESCs, female iPSCs seem to have only one active X chromosome because they retain the inactive X chromosome (Tchieu et al., 2010). However, some groups, including ours, have found that iPSCs with two active X chromosomes can be generated via reprogramming (Kim et al., 2011; Marchetto et al., 2010; Tomoda et al., 2012). Others found that reprogramming does not reactivate the inactive X chromosome, and instead the unstable inactive X chromosome undergoes epigenetic erosion, producing class III iPSCs (Mekhoubad et al., 2012). The female iPSCs that have lost XIST expression seem to be less desirable cells for cell therapy or disease modeling because XIST loss is highly correlated with upregulation of X-linked oncogenes, which leads to a high growth rate and poor differentiation (Anguera et al., 2012). A recent study showed that high expression of leukemia inhibitory factor (LIF) facilitates the derivation of iPSCs with two active X chromosomes (Tomoda et al., 2012). The difference in X chromosome status in iPSCs among different labs suggests that the X chromosome is not in a stable state in current culture conditions.
There are many X-linked diseases in females for which iPSC-based disease modeling and future cell therapies are readily applicable. Thus, information regarding X chromosome status during reprogramming and in established iPSCs is critical. Here, we set out to detail the change in X chromosome status that occurs during human female somatic cell reprogramming. Remarkably, we found that the change in the X chromosome is dynamic during reprogramming. Reprogramming at the early stage causes reactivation of the inactive X chromosome of the parental fibroblast, which does not seem to be permanent and rapidly becomes inactivated in the nascent iPSC colonies. The inactive X chromosome can be reactivated, eroded, or maintained during early passages in established iPSCs, producing class I, II, and III iPSCs that have different states of X chromosome. Our data suggest that the X chromosome status is not permanently fixed, but is plastic during human somatic cell reprogramming.
Results
Reprogramming Changes the X Chromosome Status
In order to monitor changes in the inactive X chromosome state during female somatic cell reprogramming, we took advantage of a monoallelic D551-iPSCK1 clone that is derived from normal female Detroit 551 fibroblasts and has only one active X chromosome. We monitored the X chromosome status of the monoallelic D551-iPSCK1 clone by H3K27me3 staining and by performing a SNP analysis of X-linked genes (e.g., the TT allele in GRPR; Figure 1A). Differentiation in the D551-iPSCK1 cells was induced to produce dfD551-K1 fibroblast-like cells that maintained the specificity of X chromosome allelic gene expression, and expressed only the TT SNP of the GRPR gene. We then reprogrammed dfD551-K1 cells and derived ten clones of dfD551K1-iPSC lines, and examined the X chromosome status (Figure 1A; Figure S1A available online; Table 1). Two clones of dfD551K1-iPSC showed expression of the TT SNP GRPR of the parental fibroblasts (Tchieu et al., 2010). We found that six dfD551K1-iPSC clones expressed both AA and TT alleles of GRPR that may have acquired two active X chromosomes via X chromosome reactivation (XCR) during reprogramming (Tomoda et al., 2012). However, unexpectedly, two clones of dfD551K1-iPSC expressed GRPR with the AA allele. If the iPSC retains the inactive X chromosome state or reactivates the inactive X chromosome, iPSC clones expressing X-linked genes with the opposite SNP of the parental fibroblasts are not expected to be produced. We confirmed the X chromosome status of the dfD551K1-iPSC clones by examining the presence of H3K27me3 foci and XIST clouds, which are present in monoallelic clones and absent in biallelic clones (Figures 1B and 1C). We then compared the transcription of XCI-related genes and X-linked genes between mono- and biallelic dfD551K1-iPSC clones. iPSC lines without H3K27me3 foci showed lower expression of XCI-related XIST, EZH2, and RNF12, but showed higher expression of X-linked MECP2 (methyl CpG-binding protein 2) and HPRT (hypoxanthine-guanine phosphoribosyl transferase), representing the absence of the inactive X chromosome (Figure S1B). These data suggest that the inactive X chromosome of fibroblasts may have undergone a reactivation and then a subsequent inactivation during reprogramming. Similarly, we differentiated monoallelic Rett syndrome (RTT) iPSC lines and generated fibroblast-like cells that expressed only one allele of the X-linked gene MECP2 (dfRTT3-46m, dfRTT4-24w, and dfRTT5-34m). We generated clones of iPSCs from these fibroblast-like cells and examined the allelic expression of MECP2. As with the iPSC clones derived from dfD551-K1 cells, we found iPSC clones with the same allelic expression of MECP2 as the parental fibroblasts, the opposite SNP, or both (Figures S1C–S1E; Table 1), further supporting our finding of X chromosome dynamics, including reactivation, during reprogramming.
Figure 1.
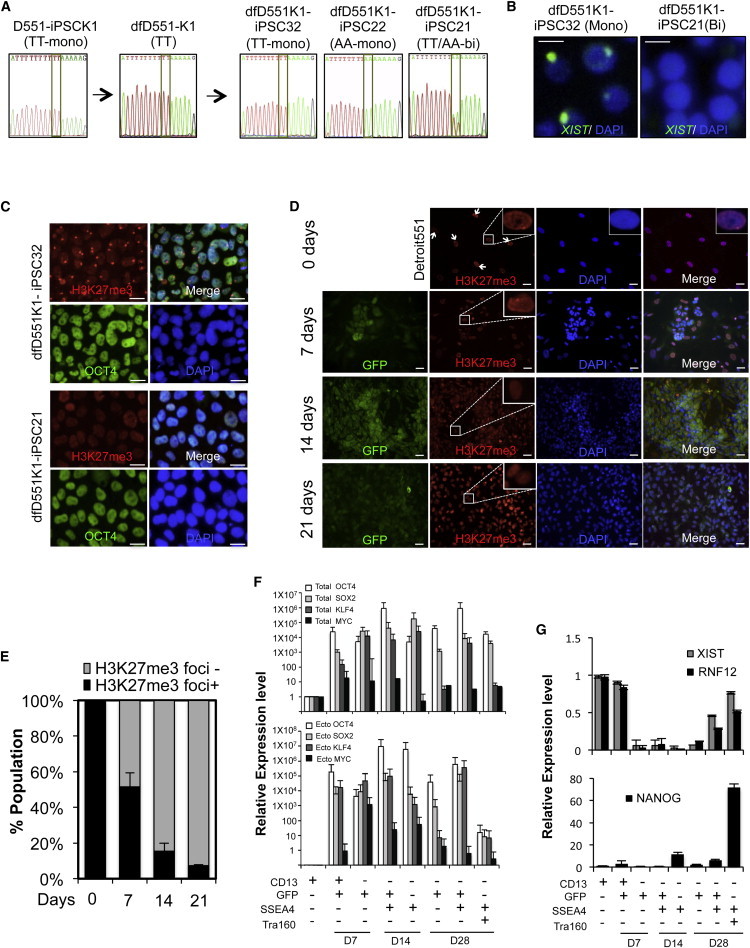
Analysis of the X Chromosome Status of Female iPSC Lines Derived from Secondary Reprogramming
(A) SNP analysis of allelic-specific expression of X-linked GRPR gene in iPSC (D551-iPSCK1) derived from Detroit 551 fibroblasts, differentiated fibroblasts (dfD551-K1), and secondary iPSC clones from dfD551-K1. When monoallelic dfD551-K1 cells having TT SNP in GRPR were reprogrammed, secondary iPSC clones that express TT SNP (dfD551K1-iPSC32), AA SNP (dfD551K1-iPSC22), or TT/AA (dfD551K1-iPSC21) were generated.
(B) RNA FISH for XIST in secondary iPSC lines at passage 12. Monoallelic dfD551-PSC32 shows XIST clouds, whereas biallelic dfD551K1-iPSC21 does not, confirming the X chromosome status. Scale bar, 20 μm.
(C) Immunostaining of H3K27me3 and OCT4 in secondary iPSC lines derived from dfD551-K1 fibroblasts. dfD551K1-iPSC32 shows H3K27me3 foci, representing the presence of inactive X chromosome, whereas dfD551K1-iPSC21 does not. Nuclei were counterstained with DAPI. Scale bar, 20 μm.
(D) Immunostaining of H3K27me3 (red) in Detroit 551 fibroblasts undergoing reprogramming on the indicated days. GFP (green) represents the expression of retrovirus-mediated reprogramming factors, and nuclei were counterstained with DAPI (blue). GFP+ cells under reprogramming lose H3K27me3 foci. Scale bar, 20 μm.
(E) Quantification of the H3K27me3 foci+ and H3K27me3 foci− cells in (A). Error bars represent mean ± SEM of three independent experiments.
(F) Relative expression of total (upper panel) and ectopic (lower panel) OCT4, SOX2, KLF4, and MYC.
(G) Relative mRNA expression of genes involved in XCI and pluripotency during reprogramming. Homogeneous populations of Detroit 551 fibroblasts undergoing reprogramming were sorted according to the expression of fibroblast marker CD13, pluripotency markers SSEA4 and TRA160, and retroviral GFP. Error bars represent mean ± SEM of three independent experiments.
Table 1.
Clonal Fibroblast Lines and the iPSC Clonal Lines Derived from Them
| Parental Cells | Clonal Fibroblasts | SNP Gene | SNP (A/B) and Location | Secondary iPSC with A Allele | Secondary iPSC with A/B Allele | Secondary iPSC with B Alleles |
|---|---|---|---|---|---|---|
| D551-iPSCK1 | dfD551-K1 | GRPR | TT/AA at 2,411–2,412 | 2 | 6 | 2 |
| 6TG-iPSC3 | df6TG-3 | GYG2 | C/T at 998 | 2 | 2 | 2 |
| GYG2 | A/G at 1,127 | |||||
| HAT-iPSC1 | dfHAT-1 | GYG2 | C/T at 998 | 2 | 3 | 1 |
| GYG2 | A/G at 1,127 | |||||
| MAOA | A/G at 4,100 | |||||
| RTT3-iPSC-46m | dfRTT3-46m | MECP2 | Del G at 705 | 4 | 3 | 2 |
| RTT4-iPSC-24w | dfRTT4-24w | MECP2 | C/T at 916 | 4 | 2 | 5 |
| RTT5-iPSC-34m | dfRTT5-34m | MECP2 | A/G at 1,461 | 2 | 4 | 3 |
| fLNS HPRT+/− | fLNS-6TG | GYG2 | C/T at 998 | 4 | 3 | 2 |
| GYG2 | A/G at 1,127 | |||||
| MAOA | A/G at 4,100 |
Fibroblast-like cells were generated from monoallelic iPSCs by differentiation. fLNS-6TG fibroblasts resistant to 6TG or fLNS-HAT fibroblasts resistant to HAT were selected from LNS fibroblasts with 6TG treatment or HAT medium. Differentiated fibroblasts, fLNS-6TG, and fLNS-HAT have monoallelic expression of genes on the X chromosome. When induced for reprogramming, iPSC clones with the same allelic expression of X-linked genes of the parental fibroblasts, iPSC clones with the opposite allele, or both are produced.
In order to elucidate the dynamics of X chromosome during reprogramming, we traced the X chromosome status in cells undergoing reprogramming. Following reprogramming of Detroit 551 fibroblast, cells were fixed at 7, 14, and 21 days for analysis in X chromosome state. First, we examined the change in H3K27me3 foci, which is the most reliable marker for the presence of an inactive X chromosome (Plath et al., 2003). As reprogramming proceeded, the percentage of cells with H3K27me3 foci gradually decreased and became 7% at day 21 (Figures 1D and 1E). These data further support the notion that reprogramming reactivates the inactive X chromosome. We also found a gradual decrease of H3K27me3 foci-positive cells in three other primary fibroblast cell lines (RTT3, WI38, and IMR90; Figures S2A–S2C).
Next, we examined whether the expression of genes that are critical for XCI changes during reprogramming. We isolated total RNA in cells undergoing reprogramming at 10, 14, 21, and 28 days after reprogramming, and analyzed the expression of XCI-related and pluripotent genes (Figure S2D). XIST is a noncoding RNA whose expression and spreading on the X chromosome is essential for XCI. Recently, the LIM-domain protein RNF12 was shown to be a positive transcription activator for XIST (Gontan et al., 2012; Navarro et al., 2011). The expression of XIST and RNF12 decreased during reprogramming, consistent with the decrease in the percentage of cells with H3K27me3-positive foci, whereas the pluripotency markers increased (Figure S2D). Previously, we found that reprogramming is a progressive process that can be defined by the change in cell-surface markers and the retroviral gene expression (Chan et al., 2009). Prior to reprogramming, fibroblasts express the cell-surface marker CD13, which is rapidly repressed by the expression of reprogramming factors marked by the expression of GFP contained in retroviral vector. The silencing of GFP marks the formation of bona fide iPSCs that express SSEA4 and TRA160 cell-surface markers (Chan et al., 2009). These cell-surface markers can be utilized to further dissect and isolate cells in progressive reprogramming stages: CD13+GFP-SSEA4-TRA160−, fibroblasts; CD13-GFP+SSEA4+TRA160−, partially reprogrammed cells; and CD13-GFP-SSEA4+TRA160+, fully reprogrammed cells (Chan et al., 2009). Using fluorescence-activated cell sorting (FACS), we isolated cells using a combination of markers and purified total RNA, and performed a gene-expression analysis (Figure 1F). CD13+ fibroblasts that had strong H3K27me3 foci showed high expression of XIST and RNF12, which is consistent with their role in XCI. CD13-GFP+ cells that were isolated at days 14 and 28 after reprogramming showed high expression of ectopic reprogramming factors and a dramatic reduction in expression of XIST and RNF12 (Figures 1F and 1G). When cells became bona fide iPSCs and expressed TRA160, the ectopic expression of four reprogramming factors was dramatically reduced (Figure 1F). These data suggest that the high expression of ectopic reprogramming factors induces X reactivation during reprogramming.
In order to rule out the possibility that the presence of contaminating cells with the opposite SNP of genes in parental fibroblasts is responsible for the formation of secondary iPSC clones with the opposite SNP, we used female fibroblasts from Lesch-Nyhan syndrome (LNS) cells with a mutation in HPRT. LNS fibroblasts display a mosaic pattern of X-linked HPRT expression. Fibroblasts that have mutant HPRT on an active X chromosome (XaHPRT−XiHPRT+), and therefore lack HPRT activity, are sensitive to hypoxanthine-aminopterin-thymidine (HAT) in medium. However, they do not metabolize 6-thio-guanine (6TG), whose toxic metabolites kill cells with an active HPRT, and thus they can grow in medium with 6TG. In contrast, fibroblasts with a wild-type HPRT allele on an active X chromosome (XaHPRT+XiHPRT−) are resistant to HAT but sensitive to 6TG (Figure 2A). Using this differential sensitivity to HAT and 6TG, we isolated two homogeneous cell populations: one with 6TG resistance (fLNS-6TG [XaHPRT−XiHPRT+]) and one with HAT resistance (fLNS-HAT [XaHPRT+XiHPRT−]). In order to test the stability of drug selectivity, cells that were selected with either drug were cultured without the drug for 2 weeks, and then drug sensitivity was tested. fLNS-HAT fibroblasts previously selected for HAT resistance showed no resistance to 6TG, and fLNS-6TG fibroblasts previously selected for 6TG did not survive in the HAT condition (Figure S3A). These data suggest that XCI states in the drug-selected cells were stably maintained. These two homogeneous populations of cell lines having only one active X chromosome were reprogrammed. We examined the X chromosome status of iPSC clones by testing the drug sensitivity and analyzing SNPs of genes on the X chromosome (Figure 2A). Like the iPSCs derived from clonally differentiated dfD551-K1, the iPSC clones that were generated from fLNS-6TG had different X chromosome states and drug sensitivities: 6TG-iPSC3, 6TG resistance (XaHPRT−XiHPRT+); 6TG-iPSC1, HAT resistance (XaHPRT+XiHPRT−); and 6TG-iPSC6, HAT resistance (XaHPRT+XaHPRT−; Figure 2B). In addition to drug resistance, we performed a SNP analysis in the X-linked GYG2 gene. Consistent with drug-resistance phenotypes, these iPSC clones showed allelic specificity (at nucleotide 1,127 of mRNA) of the GYG2 gene: 6TG-iPSC3, (G) SNP; 6TG-iPSC1, (A) SNP; and 6TG-iPSC6, (G/A) SNPs (Figure 2B). The generation of HAT-resistant clones (e.g., 6TG-iPSC1) from 6TG-selected fibroblasts further supports our finding that XCR occurs during reprogramming. In order to further confirm the conversion of the drug sensitivity of iPSCs, we differentiated 6TG-resistant monoallelic 6TG-iPSC3 and generated df6TG-3 fibroblast (XaHPRT−XiHPRT+). df6TG-3 cells were reprogrammed (Figure 2A). HPRT activity and SNP were again examined with secondary iPSC clones. As with the iPSC clones derived from drug-selected primary fibroblasts, the converted SNP and HPRT activities were observed in secondary iPSC lines (Figure 2C). When six secondary iPSC clones from df6TG-3 were treated with either 6TG or HAT, two clones showed resistance only to 6TG. The other four clones died under the 6TG culture condition, but survived in the presence of HAT in the medium (Figure 2C). In addition, SNP of the X-linked GYG2 gene was examined. Two of six df6TG3-iPSC clones had the same SNP (at 998 nt of mRNA) GYG2 (T) as the parental df6TG-3 cells (XaHPRT−XiHPRT+). Out of four df6TG3-iPSC clones that acquired HAT resistance, two showed monoallelic (C) SNP in GYG2 (XaHPRT+XiHPRT−), and the other two showed biallelic SNP patterns (XaHPRT−XaHPRT+; Figure S3B). The differential GYG2 SNPs were also confirmed via restriction-enzyme-sensitive digest (Figure 2D). GYG2 SNPs (either C or T) were differentially cleaved depending on the XCI status, as shown in Figure 2D, and strongly correlated with the status of H3K27me3 foci (Figure 2E). A similar conversion of X chromosome status or the reactivation of the inactive X chromosome was found in reprogramming fLNS-HAT (XaHPRT+ XiHPRT−) and secondary reprogramming dfHAT-1 (XaHPRT+ XiHPRT−). Not only HAT-resistant iPSCs but also 6TG-resistant iPSC clones were isolated from reprogramming of fLNS-HAT cells and dfHAT-1 cells (Figure S3C). The allelic specificity of the X chromosome of dfHAT1-iPSC was further supported by SNP analysis of MAOA and GYG2, and H3K27me3 staining (Figures 2E and S3D–S3F). These results support our finding that reactivation of the inactive parental X chromosome occurs, followed by inactivation.
Figure 2.
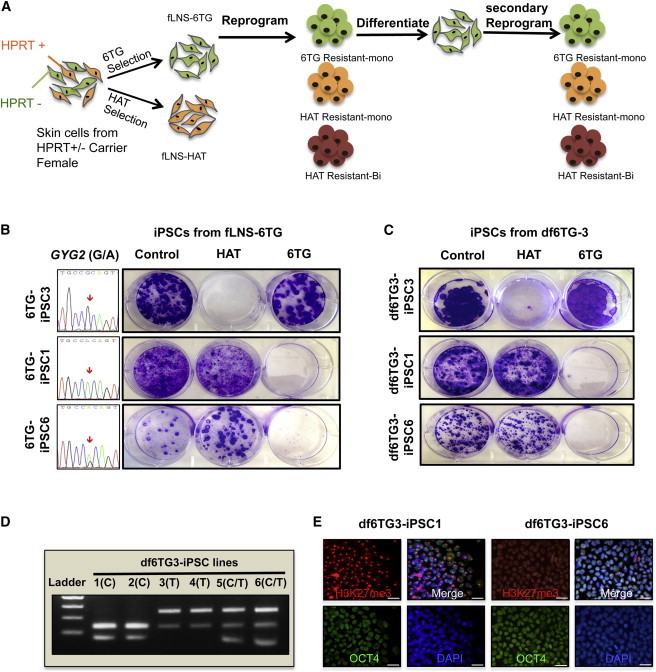
Formation of Female iPSCs with a Different X Chromosome State Compared with the Parental fLNS-6TG (XaHPRT−XiHPRT+) Fibroblast
(A) Schematic representation of strategy for generating homogeneous fibroblasts from LNS patients with a HPRT mutation. Culture with 6TG or HAT produces two homogeneous populations of fibroblasts with X chromosome in either the (XaHPRT−XiHPRT+, fLNS-6TG) or (XaHPRT+XiHPRT−, fLNS-HAT) state.
(B) Generation of HAT-resistant clones from 6TG-selected fLNS-6TG fibroblasts. Allelic-specific expression of GYG2 is shown in the left column, and resistance to HAT or 6TG is shown by crystal violet staining of iPSC clones after treatment with HAT or 6TG for 10 days.
(C) Generation of HAT-resistant secondary iPSC clones from 6TG-resistant fibroblasts (df6TG-3) of the 6TG-resistant iPSC clone (6TG-iPSC3). Crystal violet staining of secondary iPSC lines after selection with HAT or 6TG is shown.
(D) Analysis of allele-specific expression of GYG2 in secondary iPSC lines via restriction-enzyme-sensitive SNP. Allele-specific GYG2 SNPs were amplified by PCR and digested with BstIMutI restriction enzyme.
(E) Representative images of H3K27me3/OCT4 immunostaining with secondary df6TG-iPSC lines. Scale bar, 20 μm.
Reprogramming Activates the Inactive X Chromosome in Female Fibroblasts
We reasoned that if XCR occurs, cells with converted drug resistance might arise from cells undergoing active reprogramming. In order to test this hypothesis, we induced the reprogramming of fLNS-6TG cells (XaHPRT− XiHPRT+) and tested the 6TG and HAT resistance during reprogramming. At 10 days after reprogramming, we added HAT or 6TG into the reprogramming medium and continued reprogramming for an additional 2 weeks. Remarkably, iPSC colonies that were resistant to HAT arose (Figure 3A), suggesting that a reactivation of the inactive X chromosome occurs during reprogramming. Previous studies (Mekhoubad et al., 2012; Tchieu et al., 2010) reported that the X chromosome status is maintained during reprogramming. The Plath group used a lentiviral vector system that expresses four reprogramming factors in one backbone (STEMCCA vector), whereas we used a retroviral vector, which may explain the difference in results. Thus, we tried to reprogram the fLNS-6TG with STEMCCA vector. However, we could isolate iPSC colonies with an opposite X chromosome state (HAT-resistant clones; Figure 3A). Thus, the reprogramming vectors do not seem to be responsible for the reactivation of the X chromosome. The relatively high expression of reprogramming factors in our reprogramming condition may have resulted in reactivation of the X chromosome during reprogramming. We performed Southern blot analysis in 12 iPSC clones that were characterized for their X chromosome status (Figure S3G). No clones showed the same provirus integration patterns. A total of >30 provirus integrations and eight integrations of OCT4 or SOX2 were found in each clone (Figure S3G). No previous studies have directly addressed the relationship between the X chromosome status of female iPSCs and the total number of provirus integrations. However, the female iPSC clones derived from three independent individuals via the reprogramming protocol of the Eggan group showed two to six integrations in OCT4 provirus and two to four integrations in SOX2 provirus (see Figure S6 in Boulting et al., 2011), which is fewer than observed in the iPSC clones generated in our protocol. Thus, the reactivation of the inactive X chromosome in iPSC clones in our protocol may be due to the higher expression of the four factors during reprogramming. Since human ESCs with two active X chromosomes were successfully derived under the hypoxic condition, we tested whether the hypoxic reprogramming condition prevents the formation of an inactive X chromosome (Lengner et al., 2010). As in the normoxic condition, we obtained iPSC colonies from fLNS-6TG that showed HAT resistance as well as 6TG resistance (Figure 3A). The production of 6TG-resistant clones with only one active X chromosome suggests that hypoxic reprogramming conditions do not maintain the two active X chromosomes, consistent with a previous report (Pomp et al., 2011). We then asked whether the HAT resistance acquired by fLNS-6TG during the reprogramming accompanied the reactivation of XCI. At 14 days of reprogramming, the GYG2 and MAOA SNPs showed a biallelic pattern: GYG2 (C and T at 998 nt), GYG2 (A and G at 1,127 nt), and MAOA (A and G) (Figure 3B). Overall, our data showed that reprogramming reactivates the inactive X chromosome of fibroblasts.
Figure 3.
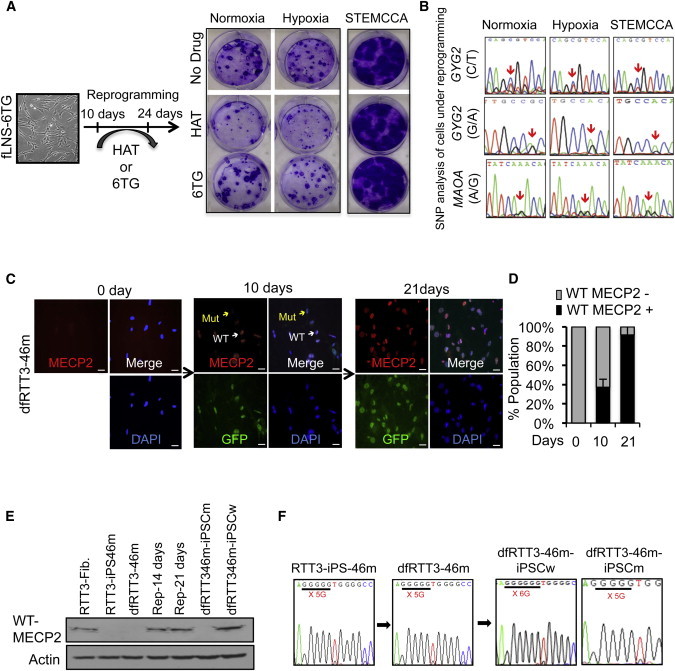
XCR during Active Reprogramming
(A) Crystal violet staining of a reprogrammed whole cell in a six-well plate. 6TG-resistant fLNS-6TG (XaHPRT−XiHPRT+) cells were induced for reprogramming in a normoxic or hypoxic condition using pMIG retroviral vector, or in a normoxic condition using STEMCCA lentiviral vector. After 10 days, reprogramming was continued in medium with HAT or 6TG for 2 weeks. Seven days after HAT was withdrawn, the plate was stained for crystal violet. In all three conditions, iPSCs with resistance to HAT were formed.
(B) Allelic-specific sequencing of GYG2 and MAOA in cells under 14 days of reprogramming.
(C) Immunostaining of wild-type (WT) MECP2 in dfRTT3-46m cells undergoing reprogramming at the indicated times with antibody recognizing the C terminus of MECP2 (red) and DAPI (blue). White arrows indicate WT of MECP2 and yellow arrows indicate the mutant type of MECP2. Scale bar, 20 μm.
(D) Quantification of WT MECP2+ cells in dfRTT3-46m cells undergoing reprogramming in (C). Four randomly chosen fields were used to count the number of MECP2+ cells and to calculate the percentage. Error bars represent mean ± SEM.
(E) Protein expression of WT of MECP2 in dfRTT3-46m fibroblasts undergoing reprogramming. At 10 and 21 days after reprogramming, whole-cell extracts were immunoblotted for MECP2 antibody recognizing the C terminus of MECP2.
(F) Representative SNP of MECP2 in secondary iPSCs.
We also took advantage of a monoallelic RTT3-iPSC-46m clone isolated from RTT3 fibroblasts to analyze the transcriptional activation of inactive X chromosome during reprogramming (Kim et al., 2011). RTT3 fibroblasts originate from a female RTT patient who had a nucleotide deletion (705 delG) in the middle of MECP2. This deletion causes a frameshift in the codon and ultimately produces C-terminal deletion MECP2 protein. The RTT3-iPSC-46m clone is monoallelic and expresses only mutant MECP2 allele (Figure 3F). Thus, the antibody against the C terminus of MECP2 does not recognize it. We differentiated RTT3-iPSC-46m iPSCs into fibroblasts to produce dfRTT3-46m cells. The inactive X chromosome status was maintained in dfRTT3-46m, and an antibody for C-terminal MECP2 did not detect mutant MECP2. We initiated reprogramming of dfRTT3-46m and analyzed the production of MECP2 at days 14 and 21 by performing western blotting and immunostaining. Remarkably, we found that cells undergoing reprogramming produced wild-type MECP2 from days 14 and 21 (Figures 3C–3E). These results provide definitive evidence that the XCR that resulted in loss of H3K27m3 foci and low expression of XIST and RNF12 upon reprogramming had become a state in which transcription was active.
Nascent iPSCs Contain the Inactivated X Chromosome
If the inactive X chromosome becomes activated during reprogramming, how are the monoallelic iPSCs that have one active X chromosome produced? In order to answer this question, we performed an extensive analysis of change in the X chromosome state in established iPSC clones. Our previous analysis of cellular marks during programming found that the silencing of retrovirus-mediated GFP expression occurs when cells become fully reprogrammed and make faithful iPSCs (Chan et al., 2009; Kim et al., 2012). At 28 days after reprogramming, when iPSC colonies arose, we examined X chromosome status by staining cells for H3K27me3. Unexpectedly, we found that all of the nascent iPSC colonies with no GFP expression displayed H3K27me3 foci, suggesting that all of them had an inactive X chromosome (Figure 4A). In order to examine the XIST and RNF12 expression in GFP− colonies, we performed quantitative PCR in cells isolated using surface markers and GFP expression. Consistent with the change in H3K27me3 staining, the expression of XIST and RNF12 together with pluripotency markers was highly upregulated in the SSEA4+/TRA160+/GFP− population isolated at day 28, whereas cells that showed GFP and thus had high expression of reprogramming factors showed low expression of RNF12 and XIST (Figures 1G and S2E). Thus, it seems that before cells become fully reprogrammed, the X chromosome is in an active state, perhaps due to the suppression of XIST and RNF12. The silencing of ectopic reprogramming factors activates XIST and RNF12, and the X chromosome becomes inactivated and marked by precipitous H3K27me3 foci formation in nascent iPSC clones (Shin et al., 2010). These results suggest that the active X chromosome during reprogramming is transient and the X chromosome becomes inactivated following completion of reprogramming, leading to the formation of a monoallelic X chromosome state in newly formed nascent iPSC clones.
Figure 4.
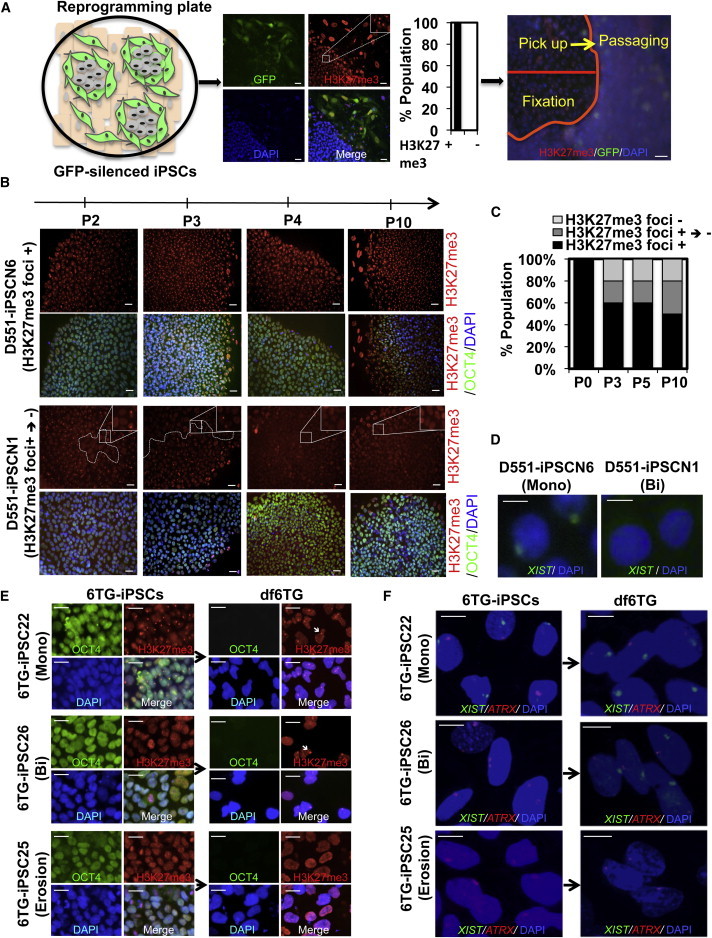
Formation of Class I and Class III iPSC Clones in Early Passaging
(A) Schematic of tracing the X chromosome after reprogramming. iPSCs undergo rapid XCI upon completion of reprogramming, and all GFP-silenced nascent iPSC colonies display H3K27me3 foci. In order to trace the X chromosome status after reprogramming, half of the colonies were fixed and half were used for passaging. Scale bar, 20 μm.
(B) Appearance of H3K27me3 foci-negative cells in iPSC clones during passaging. H3K27me3 foci-positive D551-iPSCN1 iPSCs became H3K27me3 foci-negative cells upon passaging, whereas D551-iPSCN6 remained an H3K27me3 foci-positive clone. Scale bar, 20 μm.
(C) Percentage of iPSC clones that underwent the H3K27me3 foci-positive to -negative transition during passaging in (B). A total of ten H3K27me3 foci-positive Detroit 551-derived iPSC clones were picked and expanded to trace the change of H3K27me3 status.
(D) FISH for XIST RNA in iPSCs at passage 10 to determine the X chromosome status. Scale bar, 20 μm.
(E) Representative images of H3K27me3 and OCT4 staining in 6TG-iPSCs at passage 10 and differentiated cells. 6TG-iPSC22 is a class II iPSC and displays H3K27me3 foci before and after differentiation. 6TG-iPSC26 is a class I iPSC and shows formation of H3K27me3 foci only after differentiation. 6TG-iPSC25 is a class III iPSC, and no H3K37me3 foci exist before or after differentiation. Arrow indicates H3K27me3 foci. Scale bar, 20 μm.
(F) Representative images of XIST and ATRX FISH in 6TG-iPSCs at passage 10 and differentiated cells. 6TG-iPSC22 is a class II iPSC and displays one XIST and one ATRX focus before and after differentiation. 6TG-iPSC26 is a class I iPSC and shows formation of XIST after differentiation. Two ATRX foci become one after differentiation. 6TG-iPSC25 is a class III iPSC, and no XIST foci exist before or after differentiation. Scale bar, 20 μm.
XCR in iPSCs
Although the initial female iPSC colonies displayed the marker for an inactive X chromosome in the current study (Figure 4A), other groups and we have previously reported the isolation of biallelic iPSCs with two active X chromosomes from female somatic cell reprogramming (Kim et al., 2011; Marchetto et al., 2010; Tomoda et al., 2012). It seems that biallelic iPSCs arise during picking and expansion. In order to test this, we closely examined X chromosome status in iPSC clones at each passage after the initial picking. First, the nascent iPSC clones picked from the original reprograming plate were denoted as “passage 0.” We picked ten iPSC colonies without GFP expression from a plate of reprogrammed Detroit 551 or RTT3 fibroblasts. During passaging, half of the colonies were picked up for the next passage and the other half were fixed for H3K27me3 staining (Figures 4A and S4A). We repeated the picking and passaging up to passage 10. At each passage, we examined the H3K27me3 status in iPSC colonies. Interestingly, four out of ten D551-iPSC colonies started to show an area without H3K27me3 foci following passage 1 or 2 (Figures 4B, 4C, and S4B). All cells in the clones that had begun to lose H3K27me3 foci eventually became H3K27me3 foci- and XIST-negative. The remaining six colonies maintained the H3K27me3 and XIST foci (Figures 4B–4D, S4C, and S4D). This gradual gaining of H3K27me3 foci-negative cells and the loss of XIST expression were also observed in iPSC clones from fLNS-HAT and fLNS-6TG fibroblasts (Figures S4E and S4F). These data suggest that either monoallelic iPSCs become biallelic iPSCs after picking and expansion or a few biallelic iPSCs exist in the colonies that have a growth advantage, and they become dominant cells during picking and expansion. Our current data cannot exclude either possibility.
The absence of H3K27me3 foci and XIST expression in iPSCs suggests that these cells are class I iPSCs that have two active X chromosomes. However, the continuous culture of class II iPSCs can result in a partial reactivation of the inactive X chromosome due to an epigenetic change of the X chromosome (so-called “erosion”), resulting in class III iPSCs (Mekhoubad et al., 2012). Erosion and full reactivation can be distinguished by the formation of H3K27me3 foci and XIST expression in cells differentiated from iPSCs. In order to determine whether iPSC clones that have no H3K27me3 foci or XIST are class I or class III iPSCs, we differentiated 12 iPSC clones derived from fLNS-6TG fibroblasts by treating them with retinoic acid for 14 days. The formation of H3K27me3 foci, XIST, and ATRX cloud was examined by fluorescence in situ hybridization (FISH). Six of 12 clones showed an X chromosome status of class II type, having XIST and H3K27me3 foci in iPSCs and differentiated cells (e.g., 6TG-iPSC22; Figures 4E and 4F). The other six iPSC clones showed no H3K27me3 foci or XIST cloud before differentiation, suggesting that they were either class I or class III iPSC clones. In three clones, H3K27me3 foci and XIST cloud arose after differentiation, confirming that they were class I iPSC clones (e.g., 6TG-iPSC26; Figures 4E and 4F). Meanwhile, the other three clones did not show H3K27me3 foci or XIST cloud after differentiation, representing the “eroded” state of the X chromosome (6TG-iPSC25; Figure 4F). SNPs in GYG and MAOA genes of 12 iPSCs further confirmed the allelic expression of X chromosome genes (Table 1). Our results indicate that either class I or class III iPSCs can arise from nascent iPSC colonies with inactive X chromosome marks.
Discussion
Here, we demonstrate that the status of the X chromosome is dynamic during human female somatic cell reprogramming. Extending previous studies that examined X chromosome status in iPSCs after completion of reprogramming, we determined the change of X chromosome status in cells at different stages of reprogramming and after they were established as iPSC clones. We found that strong ectopic expression of reprogramming factors markedly suppresses XIST and RNF12, and mediates the reactivation of the inactive X chromosome (reprogramming XCR [rXCR] in Figure S5D). Although it is transient, the reactivated X chromosome at rXCR is transcriptionally active (Figure 3). When reprogramming was completed, the nascent iPSC clones were shown to be composed mostly of cells possessing the inactive X chromosome marker H3K27me3 foci. These results suggest that the reactivated X chromosome state in cells under active reprogramming is transient, and the timing of inactivation of the X chromosome is well correlated with the silencing of ectopic reprogramming factors (Chan et al., 2009). In murine ESCs, the reprogramming factors Oct4 and Nanog bind to intron 1 of Xist and suppress its transcription, whereas Myc and Klf4 bind to DXPas34 of Tsix to activate the transcription, resulting in two active X chromosomes (Deuve and Avner, 2011). Likewise, the high expression of the reprogramming cocktail in the current study may suppress XIST in cells undergoing reprogramming and reactivate the inactive X chromosome. When cells become bona fide iPSCs, the retroviral silencing machinery becomes activated (Chan et al., 2009; Matsui et al., 2010) and reduces the ectopic expression of reprogramming factors and thus the suppression of XIST. Remarkably, the formation of monoallelic iPSC clones composed of the same inactive X chromosome indicates that out of many cells undergoing reprogramming, only one cell becomes an iPSC clone and thus has one allele of the inactive X chromosome.
Tracing of the nascent iPSC clones that were composed mostly of H3K27me3 foci-positive cells showed that cells with no H3K27me3 markers became dominant in some clones during very early passages. There are two possible explanations for this: either H3K27me3 foci-positive class II iPSCs become H3K27me3 foci-negative class I or class III cells, or some existing H3K27me3 foci-negative iPSCs that have a growth advantage become dominant during passaging. In a detailed analysis of the X chromosome state, we found that class I iPSCs with two active X chromosomes, as well as class III iPSCs with one active and one eroded X chromosome, arose in early passages (Figure 4F). Although a previous report by the Eggan lab suggested that the long-term culture of iPSCs results in X chromosome erosion (Mekhoubad et al., 2012), our data show that X chromosome erosion occurs even in very early passages. Currently, it is unknown how class I iPSC clones arise from class II nascent iPSCs. Perhaps the neighboring nonreprogrammed cells suppress the XCR in nascent iPSCs via paracrine factors (Bendall et al., 2007; Xu et al., 2005), and when the iPSCs are picked and placed in a new culture plate without the influence of these factors, XCR may occur. During development, the X chromosome shows dynamic changes in state. The X chromosome becomes activated in the ICM in mouse (Lessing et al., 2013). Preimplantation human embryos also show two X chromosomes in the pre-XCI state (Lengner et al., 2010; Okamoto et al., 2011). Following random inactivation in the epiblast stage, the X chromosome becomes reactivated in PGC development (Sugimoto and Abe, 2007). The reactivation of the X chromosome during reprogramming shown by our results suggests that reprogramming mimics either preimplantation embryo development or PGC formation where XCR occurs.
iPSC clones with different X chromosome status have been isolated by several groups (Ananiev et al., 2011; Cheung et al., 2011; Pomp et al., 2011; Tchieu et al., 2010). Some groups isolated iPSCs with one active X chromosome and others isolated two active X chromosomes. The medium used for reprogramming does not seem to be responsible for the different results, because all of these groups used a standard medium composed of knockout serum replacement and basic fibroblast growth factor (Amit et al., 2000). The reprogramming methods used by each group may not result in iPSCs with different X chromosome status. The Plath group used retro- or lentiviral polycistronic vectors that express four reprogramming factors in one backbone (Tchieu et al., 2010). The Ellis, Colman, Chang, and Eggan groups all used retrovirus for reprogramming (Ananiev et al., 2011; Mekhoubad et al., 2012; Pomp et al., 2011). We used a lentiviral STEMCCA vector that was used by the Plath group (Figure 3A). Although the Plath group did not report XCR, we found that the STEMCCA vector gives rise to iPSCs with XCR. Thus, the vectors used for reprogramming do not unambiguously explain the differential X chromosome status in iPSCs. Although isogenic iPSC clones can be isolated from reprogramming of female fibroblasts, some of the above-cited papers reported that monoallelic iPSC clones with only one inactive X chromosome, but no other X chromosomes, were isolated from some lines (Cheung et al., 2011; Pomp et al., 2011), whereas we readily isolated iPSC clones with two different X chromosome states. OCT4, SOX2, MYC, and KLF4 were shown to play critical roles in reactivation of the inactive X chromosome (Deuve and Avner, 2011; Lessing et al., 2013; Navarro et al., 2010). Differences in viral infectivity or the amount of virus added may have influenced the expression of reprogramming factors and thus X chromosome state during reprogramming. Indeed, the analysis of provirus integration in our iPSC clones showed many more integrations compared with those derived by the Eggan group (Figure S3G; Boulting et al., 2011; Mekhoubad et al., 2012). Another possibility is the difference in the fibroblast line of resistance to reprogramming, for which the epigenetic barriers may prevent the XCR during reprogramming. The different feeder conditions used cannot be ruled out as a possible cause, since the Yamanaka group showed that feeder cells that produce high LIF support the derivation of iPSCs with two active X chromosomes (Tomoda et al., 2012). However, the feeder we used does not express high LIF and is less likely to be a cause of X reactivation.
Considering the importance of female iPSCs for disease modeling and future cellular therapeutics, it is critical to acquire concrete information about the X chromosome state in given iPSC clones. X-linked monogenic diseases show different penetrance in males and females depending on the recessive or dominant role of the mutated genes (Dobyns et al., 2004). Maintaining one of the inactive X chromosomes in an inactive state in female iPSCs will be essential, especially for studying diseases such as Duchenne muscular dystrophy, hemophilia A and B, and α-thalassemia, where there is a low penetrance in the female and the mutated genes are recessive (Dobyns et al., 2004). When applying the in vitro differentiated derivatives of female iPSCs as therapeutics, it will be crucial to maintain the XCI because epigenetically unstable XIST-negative cells express oncogenes (Anguera et al., 2012) and could lead to tumor, as reported in XIST-depleted leukemia in a murine model (Yildirim et al., 2013). Thus, our current study delineating the X chromosome status of cells during and after reprogramming provides an important foundation for the use of female iPSCs in disease modeling and cell therapeutics.
Experimental Procedures
Cell Culture and Reprogramming
Normal primary fibroblasts Detroit 551, WI38, and IMR90 were purchased from the American Type Culture Collection (CCL-110, CCL-75, and CCL-186, respectively). Fibroblast cell lines from patients with RTT (RTT3, GM07982; RTT4, GM11270; and RTT5, GM17567) and fLNS-HPRT+/− (GM02226) were obtained from the Coriell Institute for Medical Research. iPSCs were reprogrammed and maintained as described in Supplemental Experimental Procedures. dfD551-K1, dfRTT1-13w, dfRTT3-46m, dfRTT4-24w, and dfRTT5-34m were generated by differentiating monoallelic D551-iPSCK1, RTT1-iPSC-13w, RTT3-iPSC-46m, RTT4-iPSC-24w, and RTT5-iPSC-34m iPSC lines, respectively, into fibroblast-like cells. To induce differentiation, iPSCs were dissociated using Accutase (Millipore) with the addition of rock inhibitor (Y27632; Sigma), and cells were plated on gelatin-coated plates. Cells were cultured in Dulbecco’s modified Eagle’s medium with 15% fetal bovine serum and nonessential amino acid over 4 weeks, and used for reprogramming.
HAT and 6TG Selections
In order to isolate two homogeneous subpopulations of fLNS-HAT (XaHPRT+XiHPRT−) and fLNS-6TG (XaHPRT−XiHPRT+) from HPRT+/− fibroblasts, cells were incubated with culture medium containing either 60 μM of 6TG or 1× HAT for 14 days. Each selected subpopulation was used for reprogramming to generate iPSCs. In order to determine the allelic specificity of HPRT in iPSC clones derived from fLNS-HAT and fLNS-6TG subpopulations, iPSCs were treated with collagenase and plated as small colony clumps in six-well plates coated with mouse embryonic fibroblasts. Three days after plating, cells were cultured with human ESC culture medium containing HAT or 6TG for 10 days. Cells were fixed with 4% formaldehyde/PBS and stained with crystal violet.
Gene-Expression and SNP Analyses
RNA was isolated from iPSCs using an RNeasy minikit (QIAGEN), and 1 μg of RNA was used for reverse transcription with iScript (BioRad) according to the manufacturer’s protocol. Gene-expression and SNP analyses were performed as described in Supplemental Experimental Procedures.
Immunostaining
iPSCs or cells undergoing reprogramming were fixed for 10 min at room temperature with 4% paraformaldehyde in PBS and stained as described in Supplemental Experimental Procedures.
FISH for XIST/ATRX, Western Blot, and Southern Blot
RNA FISH was carried out as described previously (Tchieu et al., 2010). Detailed methods for FISH, western blot, and Southern blot are described in Supplemental Experimental Procedures.
Acknowledgments
We thank Dr. Andrew Xiao for use of Olympus IX81 and radioisotope protective equipment. I.-H.P. was partly supported by the NIH (GM0099130-01); the CSCRF (12-SCB-YALE-11); the KRIBB/KRCF Research Initiative Program (NAP-09-3); CTSA grant UL1 RR025750 from the National Center for Advancing Translational Science (NCATS), a component of the NIH; and the NIH Roadmap for Medical Research.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Abyzov A., Mariani J., Palejev D., Zhang Y., Haney M.S., Tomasini L., Ferrandino A.F., Rosenberg Belmaker L.A., Szekely A., Wilson M. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit M., Carpenter M.K., Inokuma M.S., Chiu C.P., Harris C.P., Waknitz M.A., Itskovitz-Eldor J., Thomson J.A. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev. Biol. 2000;227:271–278. doi: 10.1006/dbio.2000.9912. [DOI] [PubMed] [Google Scholar]
- Ananiev G., Williams E.C., Li H., Chang Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS ONE. 2011;6:e25255. doi: 10.1371/journal.pone.0025255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anguera M.C., Sadreyev R., Zhang Z., Szanto A., Payer B., Sheridan S.D., Kwok S., Haggarty S.J., Sur M., Alvarez J. Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell. 2012;11:75–90. doi: 10.1016/j.stem.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall S.C., Stewart M.H., Menendez P., George D., Vijayaragavan K., Werbowetski-Ogilvie T., Ramos-Mejia V., Rouleau A., Yang J., Bossé M. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature. 2007;448:1015–1021. doi: 10.1038/nature06027. [DOI] [PubMed] [Google Scholar]
- Boulting G.L., Kiskinis E., Croft G.F., Amoroso M.W., Oakley D.H., Wainger B.J., Williams D.J., Kahler D.J., Yamaki M., Davidow L. A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol. 2011;29:279–286. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.M., Ratanasirintrawoot S., Park I.H., Manos P.D., Loh Y.H., Huo H., Miller J.D., Hartung O., Rho J., Ince T.A. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat. Biotechnol. 2009;27:1033–1037. doi: 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]
- Cheung A.Y., Horvath L.M., Grafodatskaya D., Pasceri P., Weksberg R., Hotta A., Carrel L., Ellis J. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin M.H., Mason M.J., Xie W., Volinia S., Singer M., Peterson C., Ambartsumyan G., Aimiuwu O., Richter L., Zhang J. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Napoles M., Nesterova T., Brockdorff N. Early loss of Xist RNA expression and inactive X chromosome associated chromatin modification in developing primordial germ cells. PLoS ONE. 2007;2:e860. doi: 10.1371/journal.pone.0000860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuve J.L., Avner P. The coupling of X-chromosome inactivation to pluripotency. Annu. Rev. Cell Dev. Biol. 2011;27:611–629. doi: 10.1146/annurev-cellbio-092910-154020. [DOI] [PubMed] [Google Scholar]
- Diaz Perez S.V., Kim R., Li Z., Marquez V.E., Patel S., Plath K., Clark A.T. Derivation of new human embryonic stem cell lines reveals rapid epigenetic progression in vitro that can be prevented by chemical modification of chromatin. Hum. Mol. Genet. 2012;21:751–764. doi: 10.1093/hmg/ddr506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobyns W.B., Filauro A., Tomson B.N., Chan A.S., Ho A.W., Ting N.T., Oosterwijk J.C., Ober C. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am. J. Med. Genet. A. 2004;129A:136–143. doi: 10.1002/ajmg.a.30123. [DOI] [PubMed] [Google Scholar]
- Gontan C., Achame E.M., Demmers J., Barakat T.S., Rentmeester E., van IJcken W., Grootegoed J.A., Gribnau J. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature. 2012;485:386–390. doi: 10.1038/nature11070. [DOI] [PubMed] [Google Scholar]
- Gore A., Li Z., Fung H.L., Young J.E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M.A., Kiskinis E. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J.H., Saha K., Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–525. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein S.M., Batada N.N., Vuoristo S., Ching R.W., Autio R., Närvä E., Ng S., Sourour M., Hämäläinen R., Olsson C. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- Kim K.Y., Hysolli E., Park I.H. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:14169–14174. doi: 10.1073/pnas.1018979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.Y., Hysolli E., Park I.H. Reprogramming human somatic cells into induced pluripotent stem cells (iPSCs) using retroviral vector with GFP. J. Vis. Exp. 2012 doi: 10.3791/3804. () [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner C.J., Gimelbrant A.A., Erwin J.A., Cheng A.W., Guenther M.G., Welstead G.G., Alagappan R., Frampton G.M., Xu P., Muffat J. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 2010;141:872–883. doi: 10.1016/j.cell.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Lessing D., Anguera M.C., Lee J.T. X chromosome inactivation and epigenetic responses to cellular reprogramming. Annu. Rev. Genomics Hum. Genet. 2013;14:85–110. doi: 10.1146/annurev-genom-091212-153530. [DOI] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Kida Y.S., Hawkins R.D., Nery J.R., Hon G., Antosiewicz-Bourget J., O’Malley R., Castanon R., Klugman S. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N., Sridharan R., Xie W., Utikal J., Eminli S., Arnold K., Stadtfeld M., Yachechko R., Tchieu J., Jaenisch R. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Mak W., Nesterova T.B., de Napoles M., Appanah R., Yamanaka S., Otte A.P., Brockdorff N. Reactivation of the paternal X chromosome in early mouse embryos. Science. 2004;303:666–669. doi: 10.1126/science.1092674. [DOI] [PubMed] [Google Scholar]
- Marchetto M.C., Carromeu C., Acab A., Yu D., Yeo G.W., Mu Y., Chen G., Gage F.H., Muotri A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T., Leung D., Miyashita H., Maksakova I.A., Miyachi H., Kimura H., Tachibana M., Lorincz M.C., Shinkai Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464:927–931. doi: 10.1038/nature08858. [DOI] [PubMed] [Google Scholar]
- Mekhoubad S., Bock C., de Boer A.S., Kiskinis E., Meissner A., Eggan K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell. 2012;10:595–609. doi: 10.1016/j.stem.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P., Oldfield A., Legoupi J., Festuccia N., Dubois A., Attia M., Schoorlemmer J., Rougeulle C., Chambers I., Avner P. Molecular coupling of Tsix regulation and pluripotency. Nature. 2010;468:457–460. doi: 10.1038/nature09496. [DOI] [PubMed] [Google Scholar]
- Navarro P., Moffat M., Mullin N.P., Chambers I. The X-inactivation trans-activator Rnf12 is negatively regulated by pluripotency factors in embryonic stem cells. Hum. Genet. 2011;130:255–264. doi: 10.1007/s00439-011-0998-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I., Patrat C., Thépot D., Peynot N., Fauque P., Daniel N., Diabangouaya P., Wolf J.P., Renard J.P., Duranthon V., Heard E. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature. 2011;472:370–374. doi: 10.1038/nature09872. [DOI] [PubMed] [Google Scholar]
- Park I.H., Arora N., Huo H., Maherali N., Ahfeldt T., Shimamura A., Lensch M.W., Cowan C., Hochedlinger K., Daley G.Q. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.H., Zhao R., West J.A., Yabuuchi A., Huo H., Ince T.A., Lerou P.H., Lensch M.W., Daley G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Plath K., Fang J., Mlynarczyk-Evans S.K., Cao R., Worringer K.A., Wang H., de la Cruz C.C., Otte A.P., Panning B., Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- Pomp O., Dreesen O., Leong D.F., Meller-Pomp O., Tan T.T., Zhou F., Colman A. Unexpected X chromosome skewing during culture and reprogramming of human somatic cells can be alleviated by exogenous telomerase. Cell Stem Cell. 2011;9:156–165. doi: 10.1016/j.stem.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Shin J., Bossenz M., Chung Y., Ma H., Byron M., Taniguchi-Ishigaki N., Zhu X., Jiao B., Hall L.L., Green M.R. Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature. 2010;467:977–981. doi: 10.1038/nature09457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M., Abe K. X chromosome reactivation initiates in nascent primordial germ cells in mice. PLoS Genet. 2007;3:e116. doi: 10.1371/journal.pgen.0030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tchieu J., Kuoy E., Chin M.H., Trinh H., Patterson M., Sherman S.P., Aimiuwu O., Lindgren A., Hakimian S., Zack J.A. Female human iPSCs retain an inactive X chromosome. Cell Stem Cell. 2010;7:329–342. doi: 10.1016/j.stem.2010.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda K., Takahashi K., Leung K., Okada A., Narita M., Yamada N.A., Eilertson K.E., Tsang P., Baba S., White M.P. Derivation conditions impact X-inactivation status in female human induced pluripotent stem cells. Cell Stem Cell. 2012;11:91–99. doi: 10.1016/j.stem.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.M., Hochedlinger K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat. Cell Biol. 2011;13:497–505. doi: 10.1038/ncb0511-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R.H., Peck R.M., Li D.S., Feng X., Ludwig T., Thomson J.A. Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat. Methods. 2005;2:185–190. doi: 10.1038/nmeth744. [DOI] [PubMed] [Google Scholar]
- Yildirim E., Kirby J.E., Brown D.E., Mercier F.E., Sadreyev R.I., Scadden D.T., Lee J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. 2013;152:727–742. doi: 10.1016/j.cell.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.