

Abstract
In Western countries, invasive infections caused by M1T1 serotype group A Streptococcus (GAS) are epidemiologically linked to mutations in the control of virulence regulatory 2-component operon (covRS). In indigenous communities and developing countries, severe GAS disease is associated with genetically diverse non-M1T1 GAS serotypes. Hypervirulent M1T1 covRS mutant strains arise through selection by human polymorphonuclear cells for increased expression of GAS virulence factors such as the DNase Sda1, which promotes neutrophil resistance. The GAS bacteremia isolate NS88.2 (emm 98.1) is a covS mutant that exhibits a hypervirulent phenotype and neutrophil resistance yet lacks the phage-encoded Sda1. Here, we have employed a comprehensive systems biology (genomic, transcriptomic, and proteomic) approach to identify NS88.2 virulence determinants that enhance neutrophil resistance in the non-M1T1 GAS genetic background. Using this approach, we have identified streptococcal collagen-like protein A and general stress protein 24 proteins as NS88.2 determinants that contribute to survival in whole blood and neutrophil resistance in non-M1T1 GAS. This study has revealed new factors that contribute to GAS pathogenicity that may play important roles in resisting innate immune defenses and the development of human invasive infections—Tsatsaronis, J. A., Hollands, A., Cole, J. N., Maamary, P. G., Gillen, C. M., Ben Zakour, N. L., Kotb, M., Nizet, V., Beatson, S. A. Walker, M. J., Sanderson-Smith, M. L. Streptococcal collagen-like protein A and general stress protein 24 are immunomodulating virulence factors of group A Streptococcus.
Keywords: systems biology, next-generation sequencing, innate immunity
Streptococcus pyogenes [group A Streptococcus (GAS)] infection is responsible for human mortality and morbidity on a global scale. Severe disease pathologies caused by GAS include acute invasive conditions, such as bacteremia, streptococcal toxic shock-like syndrome (STSS), and necrotizing fasciitis, as well as postinfectious immune-mediated sequelae in the form of acute rheumatic fever and glomerulonephritis (1). Epidemiologically, severe GAS infections in developed countries are dominated by a handful of serotypes (2). Notably, the well-characterized M1T1 GAS clone is frequently isolated from invasive infection (3). A resurgence in the rates of severe GAS disease over the past 3 decades has been paralleled by the global emergence of clonal hypervirulent M1T1 and M3 isolates (4, 5). In contrast, diverse GAS serotypes are endemic in indigenous and developing communities, where no serotype predominates (6). Necrotizing fasciitis isolates from tropical northern Australia exhibit emm diversity, and serotypes that monopolise disease epidemiology in the Western hemisphere are seldom encountered (7). Thus isolates from indigenous and developing areas make ideal model organisms for the study of emergent, invasive GAS strains.
Multiple factors appear to underlie the pandemic spread of M1T1, including the recent acquisition of phage-encoded virulence factors that dampen innate immune responses (8, 9). Bacteriophage-encoded deoxyribonuclease Sda1 enables M1T1 GAS to degrade neutrophil extracellular traps (NETs; ref. 10), and the transfer of Sda1 to M1T1 provides a selective trigger for acquiring mutations in the control of virulence covRS regulator (11). Certain mutations of covRS result in the up-regulation of many GAS virulence factor genes including sda1 and those encoding streptolysin O and hyaluronic acid capsule (12–14). Furthermore, expression of the broad spectrum cysteine protease SpeB is also abrogated as a result of covRS mutation, preserving the integrity of many GAS virulence proteins (15) and allowing the pathogen to acquire cell-surface plasmin activity capable of degrading fibrin clots that impede bacterial dissemination (16). Thus, the in vivo selection for GAS covRS mutants by the host innate immune system inadvertently initiates a permanent genetic switch to a hypervirulent phenotype, capable of resisting neutrophil-mediated killing and subverting the host plasminogen activation system for systemic infection (1).
The mechanisms coordinating the virulence of non-M1T1 GAS are less defined. It has recently been shown that mutation of covRS in a range of GAS M-types results in a genetic switch analogous to that seen in M1T1 (17) and that GAS of divergent M types with mutations in covRS and/or the regulatory ropB gene are frequently isolated from patients with STSS (18). However, the mechanisms underlying the hypervirulent phenotype in the absence of the bacteriophage encoded DNase Sda1 are yet to be characterized. The clinical isolate NS88.2 (emm 98.1) encodes a mutated covS gene, is highly encapsulated, acquires cell-surface plasmin activity, is resistant to killing by human neutrophils, and is hypervirulent in a humanized plasminogen mouse model (17, 19), while repair of the covS mutation renders NS88.2 highly sensitive to neutrophil killing (17). Here, we apply a comprehensive systems biology approach to analyze this representative non-M1 isolate to identify virulence determinants contributing to GAS neutrophil resistance, leading to invasive infection in the absence of sda1.
MATERIALS AND METHODS
Ethics statement
Permission to collect human blood under informed consent was approved by the Univeristy of California–San Diego (UCSD) Human Research Protections Program and the University of Wollongong Human Ethics Committee. All animal use and procedures were approved by the UCSD Institutional Animal Care and Use Committee and the University of Wollongong Animal Ethics Committee.
Bacterial strains and growth conditions
The widely disseminated GAS isolate 5448 (M1T1) and the animal-passaged covS mutant derivative 5448AP have been previously characterized (11). Clinical GAS isolate NS88.2 (emm 98.1) was obtained from a patient with severe bacteremia in Australia's Northern Territory and contains a covS mutation (20). Isogenic NS88.2 derivative strains with the covS mutation repaired (NS88.2rep) and the covS regulator returned to nonfunctionality via reverse complementation (NS88.2covS) have been previously described (17). GAS isolates were routinely cultured at 37°C on horse blood agar (HBA), Todd-Hewitt yeast agar (THYA) or in static cultures of yeast-supplemented (1%, w/v) Todd-Hewitt broth (THBY). GAS strains containing the pDCerm derivative constructs (pDC-gls24 or pDC-sclA) were cultured in the presence of 2 μg/ml erythromycin. GAS cultures for use in microarray experiments were cultured in THBY supplemented with 1.5% (w/v) yeast extract. GAS cultures for biofilm, keratinocyte assays, and in vivo adherence were grown in THB without yeast supplementation.
Biofilm formation
Measurement of biofilm formation on polystyrene was performed essentially as described previously (21). Briefly, 12 individual wells of a tissue culture-treated 96-well microtiter plate were inoculated with 150 μl of overnight GAS culture diluted 1:100 in THB and incubated for 24 h at 37°C. Plates were washed with sterile phosphate-buffered saline (PBS), and cells were fixed with 4% paraformaldehyde. Wells were stained with 0.2% crystal violet, extracted in ethanol/acetone (80:20), and assayed for crystal violet absorbance at 595 nm for biofilm quantification.
Epithelial cell adherence and invasion assay
Assays measuring the adherence of GAS to human keratinocyte cells (HaCaT line) were performed as described previously (21). Midlogarithmic phase GAS [2×106 colony-forming units (CFU)] were added to HaCaT cells (2×105 cells) in the wells of a 24-well plate, centrifuged for 10 min at 500 g, and incubated at 37°C in 5% CO2. For adherence assays, plates were incubated for 30 min before being washed with PBS, with subsequent release and lysing of cells. Bacteria were serially diluted and plated on THYA for enumeration. For invasion assays, plates were incubated for 2 h. Plates were then washed as described previously and treated with gentamicin and penicillin G. Plates were then incubated for a further 2 h before being washed and treated as described above. Bacterial adherence and invasion were calculated as a percentage of the original inoculum. Statistical significance was determined using 1-way ANOVA with the Tukey post hoc test.
Murine skin adherence assay
Adherence of GAS to mouse flanks was assessed in vivo as previously conducted (21). Midlogarithmic phase GAS culture (2×105 CFU) was spotted onto THYA plates and air dried, and agar disks containing the bacteria were excised using a biopsy punch. Bacterial agar disks were affixed to a total of 10 CD1 mice, each with 3 disks (1 each of the wild-type NS88.2, NS88.2rep, and NS88.2covS). After 1 h, the mice were euthanized, and skin under the bacterial disks was excised. Tissue was thoroughly washed with sterile PBS to remove nonadherent bacteria before homogenization. Homogenate was serially diluted in sterile PBS and plated on THYA for enumeration. Bacterial adherence was calculated as a percentage of the original inoculum. Statistical significance was determined using 1-way ANOVA with the Tukey post hoc test.
Construction of NS88.2 isogenic deletion mutants and complemented strains
The sclA and gls24 genes were isogenically removed from the NS88.2 genome via precise, allelic replacement, essentially as described previously (22). Briefly, 250-bp upstream and downstream regions of sclA and gls24 and the chloramphenicol acetyltransferase (cat) gene were amplified via PCR using sclA upstream sense (5′-TTCTTCCTGTCTGTTTACATTTCAAAAAAGAGTGACC-3′) and antisense (5′-GTGGCTTTTTTCTCCATATGTTGTTCTCTCTTTCTCT-3′) primers; sclA downstream sense (5′-GTGGCTGGGCGGGGCGTAATCCTCTAAATTGAGAGGCCT-3′) and antisense (5′-TTGTCCTCTTCTGTTTTCGTTCCTCATCTAGCATTT-3′) primers; gls24 upstream sense (5′-TTCTTCCTGTCTGTTTAAGGTATTATTGATCAATTTC-3′) and antisense (5′-GTGGCTTTTTTCTCCATGTTCACGCCATCACGTACAG-3′) primers; gls24 downstream sense (5′-GTGGCTGGGCGGGGCGTAATCATATGTCGTCAATTAGGT-3′) and antisense (5′-TTGTCCTCTTCTGTTTATGTCCCCTTTATTTATCTT-3′) primers; cat sense (5′-GGAGAAAAAAGCCACTGGATATACCACC-3′) and antisense (5′-ACGCCCCGCCCAGCCACTCATCGCAATACTGTT-3′) primers. PCR amplicons were subsequently combined with PmeI-digested pHY304-LIC, and recombinant vectors were constructed using ligation-independent cloning. Vectors were introduced into NS88.2 via electroporation and double crossover mutants encoding in-frame allelic exchanges of sclA or gls24 with cat generated via growth of transformants at 30°C before shifting to the nonpermissive temperature for plasmid replication (37°C). The absence of erythromycin, gene target, and the presence of cat were confirmed by PCR of each mutant strain. Complementation of the NS88.2ΔsclA and NS88.2Δgls24 isogenic deletion mutants was conducted via cloning of the sclA and gls24 protein coding sequences into the gram-positive constitutive expression vector pDCerm (23). Primers annealing to sclA (sense primer 5′-CCGGTCTAGATAAAGGAGGACTCTTCATGTTGACATCAAAACACCACAA-3′, antisense primer 5′-ATATGAATTCTTAGTTGTTTTCTTTGCGTTTTGT-3′) and gls24 (sense primer 5′-CCGGTCTAGATAAAGGAGGACTCTTCGTGGAAGTTGGAAAAAAACAAGTTGCCG-3′, antisense primer 5′-ATATGAATTCTTATTTTACACGTGGCTCAGCTTTTTGATC-3′) were designed with 5′ extensions containing XbaI and EcoRI restriction sites and amplified via PCR. Purified gls24 and sclA amplicons were subcloned into pCR2.1 (Invitrogen, Carlsbad, CA, USA) before directional cloning into pDCerm, generating pDC-gls24 and pDC-sclA. Recombinant plasmids (pDC-gls24 and pDC-sclA) were transformed into electrocompetent NS88.2ΔsclA and NS88.2Δgls24 via electroporation with the presence of erythromycin and reintroduction of gls24 or sclA for NS88.2Δgls24 (pDC-gls24) and NS88.2ΔsclA (pDC-sclA), respectively, confirmed via PCR screening.
Genome sequencing, de novo assembly, and annotation
We extracted RNA-free genomic DNA from an NS88.2 liquid culture as described previously (17). Genomic DNA extract was read using an Illumina GAII (Illumina, San Diego, CA, USA) sequencer at the Australian Genome Research Facility (Brisbane, QLD, Australia). Raw 75mer sequence reads were assembled de novo into 298 contigs with an average coverage of > 25 using Velvet 1.0.13 (24), with iterative refinement of assembly parameters to obtain optimal assembly outcomes. Genomic distances between the NS88.2 scaffold and publicly available GAS genomes were given based on DNA maximal unique match indexes (MUMis) calculated using MUMmer3 (25). Contigs in the scaffold were then reordered according to the closest MUMi neighbor (MGAS315, accession no. NC_004070) using the Mauve Contig Mover (26). Concatenated contigs were submitted to the RAST server (ref. 27; http://rast.nmpdr.org), where open reading frames were called using Glimmer3 (28), and putative protein function was assigned from FIGfam subsystem families. Secreted proteins were predicted using SignalP 3.0 (29). Cell-wall-associated proteins were predicted using the hmmer3 package (http://hmmer.org). Basic Local Alignment Search Tool (BLAST) searches and image generation were carried out using BLAST Ring Image Generator (BRIG; ref. 30). Manual curation and analysis of the genome draft were conducted using Artemis 12.0 (31) and read coverage mapped using BAMview (32). These sequence data have been submitted to the European Molecular Biology Laboratory (EMBL; Heidelburg, Germany) under accession number CAHN1000000 (project PRJEA84331).
Two-dimensional gel electrophoresis
Two-dimensional gel electrophoresis and peptide mass fingerprinting was conducted as described previously (15). Early stationary phase GAS cultures were grown in THBY containing 28 μM of the cysteine protease inhibitor N-[N-(L-3-trans-carboxyirane-2-carbonyl)-l-leucyl]-agmatine (E64; Sigma, St. Louis, MO, USA). GAS supernatant proteins were precipitated using trichloroacetic acid, and equal quantities of protein were loaded onto ReadyStrip IPG strips (Bio-Rad, Hercules, CA, USA) before rehydration for ≥15 h. Rehydrated IPG strips were isoelectrically focused using a Protean IEF Cell (Bio-Rad). Second-dimension separation of IPG strips was conducted using a Protean Dodeca Cell (Bio-Rad) with visualization of protein spots using Coomassie stain. Protein spots of interest were excised, tryptically digested before release of the digested peptides into 50 mM NH4HCO3 at 37°C. Digested peptides were loaded onto target plates, and mass spectra were generated using an Axima Confidence matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) mass spectrometer (Shimadzu, Kyoto, Japan). Spectra were analyzed using Shimadzu Biotech Launchpad 2.8.3 (Shimadzu). Peptide mass fingerprinting was conducted using exported protein coding sequence data from the NS88.2 genome draft and interrogated using the Mascot search algorithm (available through the Australian Proteomics Computational Facility, Parkville, VIC, Australia; http://www.apcf.edu.au).
Transcriptional microarray
The oligonucleotide microarray used in this study and the method for in vitro transcriptional microarray have been described previously (17). Midlogarithmic phase GAS cultures were grown in THBY, and RNA was extracted using the RNeasy Mini kit (Qiagen, Hilden, Germany). RNA was DNase treated and converted to dendrimer-labeled cDNA using the Genisphere 3DNA Array 900MPX kit (Genisphere, Hatfield, PA, USA) according the manufacturer's guidelines. Dendrimer-labeled cDNA was hybridized to the array and labeled with Alexa Fluor 546 or Alexa Fluor 647. Sliders were scanned with a GenePix 4000B scanner (Molecular Devices, Sunnyvale, CA, USA), and images were processed using GenePixPro 4.0 software (Molecular Devices). Transcriptional analyses were performed with GeneSpring GX 10 (Agilent, Santa Clara, CA, USA). All transcriptional microarray data were submitted to the U.S. National Center for Biotechnology Information (NCBI; Bethesda, MD, USA) Gene Expression Omnibus (GEO) according to the Minimum Information about a Microarray Experiment (MIAME) standards (GEO accession no. GSE23825).
Quantitative real-time PCR
RNA isolation from midlogarithmic phase GAS cultured in THBY was conducted as described previously for microarray experiments. RNA from midlogarithmic phase GAS cultured in whole blood was isolated following midlogarithmic growth of GAS in THBY and washed once in sterile PBS, and 0.2 ml of GAS resuspension was added to 1.8 ml of anticoagulated blood. GAS blood cultures were incubated at 37°C for 1 h with gentle agitation before hypotonic lysis of blood cultures using 40 ml of sterile dH2O and GAS recovery via centrifugation (7000 g, 5 min). RNA was extracted from blood cultured GAS as previously described. Purified GAS RNA was double DNase digested using an RNase-free DNase set (Qiagen) before cDNA synthesis using a Tetro cDNA Synthesis kit (Bioline, London, UK). NS88.2 transcripts were quantified using a SensiFAST SYBR No-Rox kit (Bioline). The absence of contaminating chromosomal DNA in RNA samples was confirmed via PCR analysis of purified RNA samples. Fold changes in expression were normalized according to amplification efficiency for each gene as described previously (33) and to the housekeeping gene proS, the expression of which does not vary with covS mutation or growth cycle (34).
Whole-blood growth kinetics
Estimation of GAS ability to survive and replicate in whole blood was conducted via the Lancefield method as described previously (35). Venous blood from healthy donors was collected and inoculated with 0.1 vol of midlogarithmic phase GAS culture and incubated at 37°C for 3 h with gentle agitation on a rotating mixer. Fold growth was calculated as resultant CFU per milliliter after 3 h over initial CFU per milliliter. All individual assays were conducted in triplicate, with a minimum of 3 donors for calculation of growth kinetics for each strain assayed. Statistical significance was determined using 1-way ANOVA with the Tukey post hoc test.
Polymorphonuclear leukocyte (PMN) bactericidal activity assay
Measurement of GAS resistance to PMN-mediated killing in vitro was conducted essentially as previously (36). Human neutrophils were purified from venous blood using a PolyMorphPrep kit (Axis-Shield, Oslo, Norway) as per the manufacturer's instructions. Midlogarithmic phase GAS (2×106 CFU) were added to 2 × 105 neutrophils seeded into 96-well plates in RPMI supplemented with 2% heat-inactivated plasma and brought into close proximity via centrifugation (200 g, 10 min, 4°C) before incubation for 30 min at 37°C. Postincubation, PMNs were hypotonically lysed and surviving GAS enumerated via serial dilution and overnight incubation on THYA. Growth controls consisted of GAS grown under identical conditions in the absence of PMNs. GAS survival was calculated as a percentage of GAS surviving following PMN incubation compared with growth controls. All individual assays were conducted in triplicate, with 3 independent assays using different blood donors. Statistical significance was determined using 1-way ANOVA with the Tukey post hoc test.
NET degradation
Visualization of NET degradation was conducted as previously (10). Human neutrophils were purified from venous blood using a PolyMorphPrep kit (Axis-Shield) as per the manufacturer's instructions and seeded at 2 × 105 cells/well in 96-well plates. GAS was added to the wells at a multiplicity of infection of 1:100 (GAS:neutrophils), and Sytox Orange (Invitrogen) was added at a final concentration of 0.1 μM. Cells were visualized without fixation or washing using a Zeiss Axiovert 100 inverted microscope with appropriate fluorescent filters, and images were captured with a CCD camera. For quantification, NETs were enumerated for each treatment by counting 1 transect after staining from 3 independent wells; a NET was defined as a discrete area of bright orange fluorescence larger than the size of a neutrophil. Statistical significance was determined using 1-way ANOVA with the Tukey post hoc test.
Hyaluronic acid capsule determination
Measurement of hyaluronic capsule production by the NS88.2 strains was conducted as described previously by Schrager et al. (37). Chloroform extractions of capsule from midlogarithmic phase GAS cultures were quantified using Stains-all (Sigma). Biosynthesis of hyaluronic acid was calculated per CFU of GAS from the original culture, determined via serial dilution and enumeration of viable CFUs.
SpeB degradation of purified general stress protein 24 (Gls24) protein
Recombinant Gls24 protein was expressed with an incorporated N-terminal histidine hexamer and isolated using nickel-affinity chromatography. The coding sequence of gls24 from NS88.2 was amplified via PCR using primers with 5′ extensions containing SmaI and PaeI restriction sites (sense primer 5′-CGCGCATGCCTGGTTCCGCGTGGCTCTGTGGAAGTTGGAAAAAAACAAGTTGCCGTTGATCTTG-3′, antisense primer 5′-GGCCCCGGGTTATTTTACACGTGGCTCAGCTTTTTGATC-3′) and subcloned into pCRScript (Agilent) before directional cloning into pQE30. Recombinant Gls24 was expressed in Escherichia coli and purified via binding to Ni-NTA resin. SpeB protease activity was assayed as described by Cole et al. (16). Purified mature SpeB protease (5 μg; Toxin Technologies, Sydney, NSW, Australia) was mixed with 5 μg of purified Gls24 or 25 μg of casein (Sigma), adjusted to a final volume of 25 μl with PBS, and incubated at 37°C for 3 h. Proteolysis of casein and Gls24 was determined by SDS-PAGE analysis. Negative control assays containing casein, Gls24, or SpeB only were also included.
RESULTS
Reduction of NS88.2 colonization potential due to covS mutation
Similar to the globally disseminated M1T1 GAS, mutation of covS in non-M1T1 GAS results in a hypervirulent, neutrophil-resistant phenotype (17). Such mutations in the M1T1 background have come with a potential fitness cost, as the covS mutant displays reduced capacity for biofilm formation and epithelial adherence and invasion (21). To determine whether this phenomenon occurred in the non-M1T1 background, clinical covS mutant GAS isolate NS88.2 (emm 98.1) from a case of severe bacteremia in Australia's Northern Territory (20) was compared with isogenic derivative strains with the covS mutation repaired (NS88.2rep) and the covS regulator returned to nonfunctionality via reverse complementation (NS88.2covS) (17). Restoration of functional covS in NS88.2rep resulted in a significant increase in biofilm formation compared with the wild-type NS88.2 or the reverse complemented mutant NS88.2covS, which each express truncated CovS (P<0.001; Fig. 1A). A similar result was observed with respect to the ability of the NS88.2 strains to adhere to and invade epithelial cells. The intact covS NS88.2rep strain showed a significant increase in adherence to and invasion of the HaCaT keratinocyte cell line over the covS mutant NS88.2 and NS88.2covS strains (P<0.001; Fig. 1B). These in vitro data were further corroborated by in vivo assays, as adhesion to live mouse skin was also compromised by covS mutation in NS88.2 and NS88.2covS (P<0.01; Fig. 1C).
Figure 1.

Reduction of colonization propensity due to covS inactivation in NS88.2. A) Biofilm formation of covS inactive strains NS88.2 and NS88.2covS and covS intact strain NS88.2rep. B) Adherence and invasion of HaCaT human keratinocytes by NS88.2, NS88.2rep, and NS88.2covS. C) Adherence of NS88.2, NS88.2rep, and NS88.2covS to live mouse flanks. Values for panels B and C are expressed as a percentage of adherent/invasive bacteria of the original inoculum. Values shown for panels A and B are means ± sd. **P < 0.01; ***P < 0.001.
NS88.2 neutrophil resistance does not require neutrophil extracellular trap degradation
NS88.2 is highly resistant to neutrophil killing (17); however, PCR screening indicates that NS88.2 does not contain the sda1 gene (data not shown). The ability of NS88.2 and the isogenic covS derivative mutants NS88.2rep and complemented mutant NS88.2covS to degrade NETs was examined. We observed no significant differences in NET degradation between NS88.2, the NS88.2 derivative strains, or 5448Δsda1, an M1T1 GAS strain with sda1 deleted via precise allelic exchange (Fig. 2). In contrast, the covRS mutant animal-passaged 5448AP strain exhibited significantly higher NET degradation in comparison to all of the NS88.2 strains and 5448Δsda1 (P<0.001). We concluded that in the absence of Sda1, other virulence factors play a role in NS88.2 neutrophil resistance. To test this hypothesis, we undertook a systems biology approach to identify virulence determinants contributing to neutrophil resistance in this genetic background.
Figure 2.

GAS-mediated degradation of extracellular neutrophil DNA NETs. NET degradation by GAS strains NS88.2, NS88.2rep, and NS88.2covS and M1T1 GAS strains 5448AP (animal passaged 5448 encoding a truncated covS protein) and 5448Δsda1 (a 5448 derivative with sda1 isogenically deleted). Values shown are means ± sd. ***P < 0.001.
NS88.2 genome sequence
The NS88.2 genome draft consists of 298 contigs concatenated in a single circular chromosome of an estimated size of 1.7 Mbp with a G + C content of 39.35% and a multilocus sequence type of 205. From Glimmer prediction, we found 1659 protein coding DNA sequences (CDSs) that account for 86.3% of the genome. The NS88.2 genome encodes many previously identified virulence factors, including streptolysin O (slo), Mac-1-like protein (mac), SmeZ, cysteine protease SpeB, and C5a peptidase (scpA), although not streptococcal inhibitor of complement nor serum opacity factor. As an M-pattern D isolate (20), NS88.2 also encodes the plasminogen-binding M-protein-like protein Prp, and 2 M-family proteins, Enn and Mrp. Comparison of the genomic content of NS88.2 to 13 previously sequenced GAS genomes was undertaken via whole-genome BLAST analysis (ref. 30 and Fig. 3). The majority of genetic diversity was confined to prophage-like content. NS88.2 contains at least 1 lysogenized prophage element, which encodes the streptococcal superantigen speL. Recombinational hotspots were also identified in the Mga regulon, wherein M family proteins are encoded, and the fibronectin-, collagen-, and T-antigen (FCT) locus (38). The FCT region is a streptococcal pathogenicity island, containing genes responsible for adherence to the host, including the GAS pilus components (39). Classification of NS88.2 by the FCT typing scheme corresponds to FCT-3, which also encodes a collagen-binding protein Cpa and fibronectin-binding protein PrtF2 (40). Identification of NS88.2 proteins that may interact with the host during infection was conducted via prediction of putatively surface-exposed proteins. A total of 190 genes encoding N-terminal secretion signal peptides and 7 genes containing gram-positive cell-wall anchor motifs were identified using hidden Markov models.
Figure 3.

Genome-wide BLAST comparison of the NS88.2 draft genome to publicly available fully sequenced GAS genomes. Rings are annotated from outermost to innermost. Location of the variable fibronectin-binding, collagen-binding, T antigen (FCT) locus, and MGA regulatory region (ring 1). Colored blocks denoting BLAST matches of 80–100% nucleotide identity between NS88.2 and query genomes (rings 2–11; GenBank accession numbers are indicated in parentheses): NZ131 (NC_011375), MGAS6180 (NC_007296), MGAS8232 (NC_003485), MGAS9469 (NC_008021) and MGAS2096 (NC_008023), MGAS10394 (NC_006086), Manfredo (NC_009332), MGAS10750 (NC_008024), SSI-1 (NC_004606), MGAS315 (NC_004070), MGAS10270 (NC_008022), MGAS5005 (NC_007297), and SF370 (NC_002737). Mapping coverage: red bars denote <200 fold coverage, and blue bars denote >200 fold coverage (ring 12); guanine and cytosine deviation (ring 13); percentage guanine and cytosine content (ring 14); and NS88.2 genome draft concatenated backbone (kbp, ring 15).
Screening of the NS88.2 secretome
GAS secrete many proteins that play immune-modulating roles during infection. Screening of the NS88.2, NS88.2rep, and NS88.2covS supernatant protein fractions was conducted via 2-dimensional electrophoresis to experimentally confirm the presence of putatively secreted proteins (Fig. 4). To analyze peptide mass fingerprinting data, we used CDS data from the NS88.2 draft genome and formatted this information as a database that could be interrogated by the Mascot search engine (41). Multiple putatively cytoplasmic proteins were detected in the supernatant, as has been noted previously (42). Notably, of the 42 supernatant proteins identified (Supplemental Table S1), SclA, which binds the alternative complement factor H (FH) and FH-related protein 1 (43), and Gls24 were both found in the supernatants of NS88.2 and NS88.2covS but not in NS88.2rep. As with M1T1 GAS (15), expression of cysteine protease SpeB resulted in the degradation of much of the NS88.2rep secretome when grown in the absence of cysteine protease inhibitor E64 (data not shown).
Figure 4.
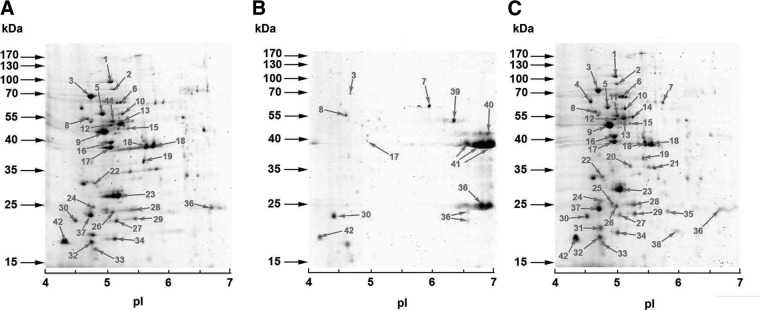
Screening of the NS88.2 secretome. Secreted proteomic profiles of NS88.2 (A), NS88.2rep (B), and NS88.2covS (C).Proteins identified via MALDI-TOF MS are indicated with numbered arrows (Supplemental Table S1). Molecular masses (kDA) of marker proteins and approximate isoelectric point (pI) values of each gel are indicated. Results shown are representative of duplicate gels from 2 independent protein isolations.
Expression of the NS88.2 transcriptome
The covRS operon has been shown to control ∼10–15% of the GAS genome, including many genes that have proven or putative roles in host-pathogen interactions (12). We subjected the parental NS88.2 strain bearing a mutated covS gene and the derivative NS88.2rep with intact covS to transcriptional microarray analysis during midlogarithmic phase growth. Comparison of the transcriptional profile of NS88.2 to NS88.2rep showed significant derepression of many virulence genes associated with resistance to innate immune responses, including emm, scpA, slo and the hyaluronic acid capsule biosynthesis genes hasA and hasB (Fig. 5). The sclA gene was also found to be highly up-regulated in NS88.2 compared with NS88.2rep. A putative antibiotic resistance gene norA and speB were found to be repressed in NS88.2 as a result of covS mutation, in accordance with previous studies (12, 17).
Figure 5.

Differential regulation of genes between the parental NS88.2 wild-type strain, which contains a covS-inactivating mutation, and derivative NS88.2rep, which encodes a functional covS gene. Selected genes are significantly differentially expressed (P<0.05).
To validate the microarray data, a panel of 5 genes was chosen for interrogation using quantitative real-time PCR analysis (Fig. 6A). The strong up-regulation of the emm, sclA, and hasA genes and down-regulation of speB in wild-type NS88.2 relative to the NS88.2rep strain were found to be consistent with the microarray analysis. In addition, expression of the gls24 gene was analyzed and did not show significant differences in regulation between the 2 covS variants. However, in vitro assays utilizing purified Gls24 protein suggest that expression of Gls24 may be regulated at the protein level via SpeB-mediated degradation (Supplemental Fig. S1A). Exposure of GAS to whole blood is likely to play an important role in the regulation of GAS genes during infection, and gene expression levels were also investigated following 1 h incubation in sanguis (Fig. 6B). Transcripts were quantified relative to the proS housekeeping gene and the relative transcript abundance between NS88.2 and NS88.2rep, both strains were grown in THY, and blood was estimated. Growth of NS88.2 and NS88.2rep in blood resulted in a significant (P<0.001) up-regulation of gls24 expression relative to growth of the same strains in THY. Other NS88.2 genes (sclA, emm) also displayed increased expression in response to exposure to blood, while speB showed consistent down-regulation in response to growth in whole blood and covS mutation.
Figure 6.

SclA and Gls24 are up-regulated in response to growth in whole blood. A) Quantitative real-time PCR analysis of NS88.2 genes during midlogarithmic phase growth. Fold change in expression between NS88.2 relative to NS88.2rep is indicated. B) Quantitative real-time PCR analysis of NS88.2 genes during midlogarithmic phase growth in THY or after 1 h incubation in whole blood. Transcript abundance is expressed relative to the house-keeping gene proS. ***P < 0.001.
SclA and Gls24 are necessary for NS88.2 survival in whole-blood and neutrophil resistance
Taken collectively, the presence of SclA and Gls24 in the NS88.2 secretome; up-regulation of sclA as a result of covS mutation; and up-regulation of Gls24 after incubation in whole blood and the putative roles of these proteins in virulence indicated these factors may function alone or in synergy as virulence determinants of NS88.2. SclA exhibits a variety of binding propensities including alternative complement FH and FH-related protein 1 (43), thrombin-activable fibrinolysis inhibitor (44), low-density lipoproteins (45), and α2β1-integrins (35). It has also been shown that SclA is expressed during human infection and that SclA elicits a humoral immune response against the collagen-like region of this protein (46). Gls24 contributes to the virulence of Enterococcus faecalis in an endocarditis model and is involved in stress tolerance of that pathogen (47, 48). To assess the effect of these proteins on NS88.2 innate immune resistance, the sclA and gls24 genes were isogenically deleted from the NS88.2 genome via precise allelic replacement with the cat gene. Complementation of the sclA- and gls24-deficient strains was conducted via transformation with sclA- or gls24-expressing pDCerm variants.
The ability of NS88.2 and the sclA and gls24 isogenic deletion mutants to replicate in whole blood was assessed via Lancefield bactericidal assays (ref. 35 and Fig. 7A, B). Deletion of either sclA or gls24 significantly impaired the survival of the isogenic mutants in comparison with NS88.2 (NS88.2ΔsclA, P<0.001; NS88.2Δgls24, P<0.01). Complementation of the sclA or gls24 deletion via heterologous expression restored survival in whole blood to both isogenic mutants (P>0.05; Fig. 7A, B). These changes were resultant of the activity of SclA and Gls24 in whole blood, as the growth kinetics of the isogenic mutants and complemented strains in THY were unchanged relative to the wild-type NS88.2 (Supplemental Fig. S1B). Further examination of the role of SclA and Gls24 in innate immune responses was studied via measurement of the NS88.2 strains to neutrophil-mediated killing (Fig. 7C, D). In comparison with the NS88.2 wild type, both NS88.2ΔsclA and NS88.2Δgls24 displayed significantly reduced neutrophil resistance (P<0.05) while the complemented isogenic mutants exhibited neutrophil resistance equivalent to the wild type (P>0.05; Fig. 7C, D). This difference in resistance to neutrophil killing was independent of capsule expression as the isogenic deletion mutations and complemented strains express equivalent amounts of hyaluronic acid (Supplemental Fig. S1C).
Figure 7.

Deletion of sclA or gls24 impairs NS88.2 growth in whole blood and resistance to neutrophil-mediated killing. A, B) Fold growth of NS88.2, the sclA-deficient mutant NS88.2ΔsclA, and complemented derivative NS88.2ΔsclA (pDC-sclA; A); the gls24-deficient mutant NS88.2Δgls24 and complemented derivative NS88.2Δgls24 (pDC-gls24; B) after 3 h incubation in human whole blood. C, D) Ability of NS88.2, NS88.2ΔsclA, NS88.2ΔsclA (pDC-sclA; C), NS88.2Δgls24, and NS88.2Δgls24 (pDC-gls24; D) to survive neutrophil-mediated killing following coculture with purified human PMNs. All assays were performed with a minimum of 3 donors; all assays were conducted in triplicate. Values shown are means ± sd of triplicate assays. *P < 0.05; **P < 0.01; ***P < 0.01,
DISCUSSION
Recent increases in the incidences of life-threatening invasive GAS infections highlight the need to identify bacterial factors that explain epidemic behavior. Despite extensive research focusing on Western centralized serotypes (8, 13), comparatively fewer studies have focused on disparate GAS strains originating from less-developed areas. In many cases, the rates of severe GAS diseases from these areas far exceed those observed in urban centers (49). The use of genomic sequencing and comparative bioinformatics has enabled high-throughput screening of other M2, M4, M6, M12, and M28 GAS strains, which determine distinguishing genetic features (50, 51). Currently, genomic research has focused exclusively on GAS serotypes that are more prevalent in developed countries such as M1, M3, M12, and M28 (52). While these serotypes are frequently isolated from severe infections in the Western hemisphere, and/or are associated with distinct disease pathologies, the majority of GAS infections nonetheless occur in developing areas (53). Here, we have utilized genomic sequencing coupled with microarray and proteomic screening to identify a cohort of virulence factors that distinguish the hypervirulent, neutrophil-resistant GAS isolate NS88.2.
Genetic integrity of the 2-component gene regulator covRS is a major determining factor of GAS virulence and colonization. M1T1 GAS that bear the intact form of this operon are more highly suited to adherence and colonization of host tissues (21). In this study, we have characterized the emm 98.1 invasive isolate NS88.2. Phenotypically, this isolate is hypervirulent and neutrophil resistant (17), while the covS intact derivative (NS88.2rep) is avirulent and exhibits increased biofilm formation and adhesion to epithelial cells. However, NS88.2 does not contain sda1 nor does it degrade neutrophil NETs, and so the mechanism of neutrophil resistance exhibited by NS88.2 is unaccounted for. Transcriptomic and proteomic data generated here demonstrate a cohort of immunomodulating virulence factors is up-regulated as a result of covS mutation in NS88.2, including the antiphagocytic M protein and SclA.
Isogenic deletion of sclA and gls24 from the NS88.2 genome was conducted to investigate their role in GAS pathogenesis in resisting innate immune responses in particular. Multiple attempts to generate a double-knockout mutant were unsuccessful. The finding that loss of sclA expression results in attenuated growth in whole blood and increased neutrophil sensitivity is consistent with previous work describing the binding of complement regulatory factors to SclA (43, 44, 54). SclA is ubiquitously distributed in GAS strains; however, sclA shows evidence of recombination and immune-selection analogous to the gene encoding M protein (46, 55), which gives credence to an antiphagocytic role in pathogenesis. GAS binding of mammalian integrins with SclA may also facilitate escape from phagolysosomal killing via uptake into the cytoplasm (35). Previous work implicates Gls24 in the survival of E. faecalis during culture in human blood and in urine (56, 57). Gls24 was also found to be up-regulated at a protein level in response to growth of GAS in hyaluronic acid-enriched medium simulating an infection scenario (58) and during microarray analysis of murine soft tissue GAS infection (59). To date, this is the first work demonstrating the direct effect of gls24 mutation on GAS survival and work is currently ongoing to determine the precise mechanism of action of Gls24 in systemic GAS infection.
Mutations in covRS have been linked to the hypervirulence of GAS in animal models of infection (16, 17). Moreover, the recently published recovery of an emm 81.0 covS mutant GAS isolate following 13 d of human carriage supports a model that predicts that such mutations occur clinically in a range of serotypes and in doing so escalate the severity of infection (17, 18, 60). Recent work has shown that functional emm and hasA genes are also essential for the acquisition of covRS mutations (61). Data from this study support a model in which GAS utilize a cohort of immune modulating virulence factors to attain a highly neutrophil-resistant phenotype, leading to more severe infection.
Despite ongoing research effort, the burden of GAS infection in developing areas and indigenous populations remains high and is likely to be underestimated. Here, we demonstrate the utility of systems biology approaches to identify novel bacterial virulence factors. Such virulence factors may prove valuable targets for future therapeutic or vaccine interventions to treat this globally important pathogen.
Supplementary Material
Acknowledgments
M.L.S-S., M.J.W., A.H., J.N.C., and N.L.B Z are recipients of Australian National Health and Medical Research Council (NHMRC) fellowships. J.A.T. and P.G.M. are recipients of Australian Postgraduate Awards.
Proteomic data analysis described in this work was supported by the use of the Australian Proteomics Computational Facility, funded by Australian NHMRC grant 381413. All other work described was funded by the Australian NHMRC.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- BLAST
- Basic Local Alignment Search Tool
- CDS
- coding DNA sequence
- CFU
- colony-forming unit
- FCT
- fibronectin-, collagen-, and T-antigen
- FH
- factor H
- GAS
- group A Streptococcus
- Gls24
- general stress protein 24
- MALDI-TOF
- matrix-assisted laser desorption/ionization–time of flight
- NET
- neutrophil extracellular trap
- PBS
- phosphate-buffered saline
- PMN
- polymorphonuclear leukocyte
- SLO
- streptolysin O
- STSS
- streptococcal toxic shock-like syndrome
- THB
- Todd-Hewitt broth
- THBY
- yeast-supplemented Todd-Hewitt broth
- THYA
- Todd-Hewitt yeast agar
REFERENCES
- 1. Cole J. N., Barnett T. C., Nizet V., Walker M. J. (2011) Molecular insight into invasive group A streptococcal disease. Nat. Rev. Microbiol. 9, 724–736 [DOI] [PubMed] [Google Scholar]
- 2. Schwartz B., Facklam R. R., Breiman R. F. (1990) Changing epidemiology of group A streptococcal infection in the U. S. A. Lancet 336, 1167–1171 [DOI] [PubMed] [Google Scholar]
- 3. Chatellier S., Ihendyane N., Kansal R. G., Khambaty F., Basma H., Norrby-Teglund A., Low D. E., McGeer A., Kotb M. (2000) Genetic relatedness and superantigen expression in group A Streptococcus serotype M1 isolates from patients with severe and nonsevere invasive diseases. Infect. Immun. 68, 3523–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ikebe T., Wada A., Inagaki Y., Sugama K., Suzuki R., Tanaka D., Tamaru A., Fujinaga Y., Abe Y., Shimizu Y., Watanabe H., and Working Grp Grp, A. S. J. (2002) Dissemination of the phage-associated novel superantigen gene speL in recent invasive and noninvasive Streptococcus pyogenes M3/T3 isolates in Japan. Infect. Immun. 70, 3227–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharkawy A., Low D. E., Saginur R., Gregson D., Schwartz B., Jessamine P., Green K., McGeer A., and Ontario Group A Streptococcal Study Group (2002) Severe group a streptococcal soft-tissue infections in Ontario: 1992–1996. Clin. Infect. Dis. 34, 454–460 [DOI] [PubMed] [Google Scholar]
- 6. Carapetis J. R., Walker A. M., Hibble M., Sriprakash K. S., Currie B. J. (1999) Clinical and epidemiological features of group A streptococcal bacteraemia in a region with hyperendemic superficial streptococcal infection. Epidemiol. Infect. 122, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hassell M., Fagan P., Carson P., Currie B. J. (2004) Streptococcal necrotising fasciitis from diverse strains of Streptococcus pyogenes in tropical northern Australia: case series and comparison with the literature. BMC Infect Dis. 4, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sumby P., Porcella S. F., Madrigal A. G., Barbian K. D., Virtaneva K., Ricklefs S. M., Sturdevant D. E., Graham M. R., Vuopio-Varkila J., Hoe N. P., Musser J. M. (2005) Evolutionary origin and emergence of a highly successful clone of serotype M1 group a Streptococcus involved multiple horizontal gene transfer events. J. Infect. Dis. 192, 771–782 [DOI] [PubMed] [Google Scholar]
- 9. Aziz R. K., Kotb M. (2008) Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg. Infect. Dis. 14, 1511–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buchanan J. T., Simpson A. J., Aziz R. K., Liu G. Y., Kristian S. A., Kotb M., Feramisco J., Nizet V. (2006) DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 16, 396–400 [DOI] [PubMed] [Google Scholar]
- 11. Walker M. J., Hollands A., Sanderson-Smith M. L., Cole J. N., Kirk J. K., Henningham A., McArthur J. D., Dinkla K., Aziz R. K., Kansal R. G., Simpson A. J., Buchanan J. T., Chhatwal G. S., Kotb M., Nizet V. (2007) DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 13, 981–985 [DOI] [PubMed] [Google Scholar]
- 12. Sumby P., Whitney A. R., Graviss E. A., DeLeo F. R., Musser J. M. (2006) Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kansal R. G., Datta V., Aziz R. K., Abdeltawab N. F., Rowe S., Kotb M. (2010) Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J. Infect. Dis. 201, 855–865 [DOI] [PubMed] [Google Scholar]
- 14. Aziz R. K., Kansal R., Aronow B. J., Taylor W. L., Rowe S. L., Kubal M., Chhatwal G. S., Walker M. J., Kotb M. (2010) Microevolution of group A streptococci in vivo: capturing regulatory networks engaged in sociomicrobiology, niche adaptation, and hypervirulence. PLoS One 5, e9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aziz R. K., Pabst M. J., Jeng A., Kansal R., Low D. E., Nizet V., Kotb M. (2004) Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol. Microbiol. 51, 123–134 [DOI] [PubMed] [Google Scholar]
- 16. Cole J. N., McArthur J. D., McKay F. C., Sanderson-Smith M. L., Cork A. J., Ranson M., Rohde M., Itzek A., Sun H., Ginsburg D., Kotb M., Nizet V., Chhatwal G. S., Walker M. J. (2006) Trigger for group A streptococcal M1T1 invasive disease. FASEB J. 20, 1745–1747 [DOI] [PubMed] [Google Scholar]
- 17. Maamary P. G., Sanderson-Smith M. L., Aziz R. K., Hollands A., Cole J. N., McKay F. C., McArthur J. D., Kirk J. K., Cork A. J., Keefe R. J., Kansal R. G., Sun H., Taylor W. L., Chhatwal G. S., Ginsburg D., Nizet V., Kotb M., Walker M. J. (2010) Parameters governing invasive disease propensity of non-M1 serotype group A streptococci. J. Innate Immun. 2, 596–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ikebe T., Ato M., Matsumura T., Hasegawa H., Sata T., Kobayashi K., Watanabe H. (2010) Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog. 6, e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanderson-Smith M. L., Dinkla K., Cole J. N., Cork A. J., Maamary P. G., McArthur J. D., Chhatwal G. S., Walker M. J. (2008) M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB J. 22, 2715–2722 [DOI] [PubMed] [Google Scholar]
- 20. McKay F. C., McArthur J. D., Sanderson-Smith M. L., Gardam S., Currie B. J., Sriprakash K. S., Fagan P. K., Towers R. J., Batzloff M. R., Chhatwal G. S., Ranson M., Walker M. J. (2004) Plasminogen binding by group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infect. Immun. 72, 364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hollands A., Pence M. A., Timmer A. M., Osvath S. R., Turnbull L., Whitchurch C. B., Walker M. J., Nizet V. (2010) Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group A Streptococcus M1T1 clone. J. Infect. Dis. 202, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cook S. M., Skora A., Gillen C. M., Walker M. J., McArthur J. D. (2012) Streptokinase variants from Streptococcus pyogenes isolates display altered plasminogen activation characteristics–implications for pathogenesis. Mol Microbiol. 86, 1052–1062 [DOI] [PubMed] [Google Scholar]
- 23. Jeng A., Sakota V., Li Z., Datta V., Beall B., Nizet V. (2003) Molecular genetic analysis of a group A Streptococcus operon encoding serum opacity factor and a novel fibronectin-binding protein, SfbX. J. Bacteriol. 185, 1208–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zerbino D. R., Birney E. (2008) Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurtz S., Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., Salzberg S. L. (2004) Versatile and open software for comparing large genomes. Genome Biol. 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Darling A. C. E., Mau B., Blattner F. R., Perna N. T. (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aziz R. K., Bartels D., Best A. A., DeJongh M., Disz T., Edwards R. A., Formsma K., Gerdes S., Glass E. M., Kubal M., Meyer F., Olsen G. J., Olson R., Osterman A. L., Overbeek R. A., McNeil L. K., Paarmann D., Paczian T., Parrello B., Pusch G. D., Reich C., Stevens R., Vassieva O., Vonstein V., Wilke A., Zagnitko O. (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Delcher A. L., Bratke K. A., Powers E. C., Salzberg S. L. (2007) Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bendtsen J. D., Nielsen H., Heijne G. V., Brunak S. (2004) Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795 [DOI] [PubMed] [Google Scholar]
- 30. Alikhan N. F., Petty N. K., Ben Zakour N. L., Beatson S. A. (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rutherford K., Parkhill J., Crook J., Horsnell T., Rice P., Rajandream M.-A., Barrell B. (2000) Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 [DOI] [PubMed] [Google Scholar]
- 32. Carver T., Bohme U., Otto T. D., Parkhill J., Berriman M. (2010) BamView: viewing mapped read alignment data in the context of the reference sequence. Bioinformatics 26, 676–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Graham M. R., Smoot L. M., Migliaccio C. A., Virtaneva K., Sturdevant D. E., Porcella S. F., Federle M. J., Adams G. J., Scott J. R., Musser J. M. (2002) Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc. Natl. Acad. Sci. U. S. A. 99, 13855–13860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Humtsoe J. O., Kim J. K., Xu Y., Keene D. R., Hook M., Lukomski S., Wary K. K. (2005) A streptococcal collagen-like protein interacts with the alpha2beta1 integrin and induces intracellular signaling. J. Biol. Chem. 280, 13848–13857 [DOI] [PubMed] [Google Scholar]
- 36. Hollands A., Aziz R. K., Kansal R., Kotb M., Nizet V., Walker M. J. (2008) A naturally occurring mutation in ropB suppresses SpeB expression and reduces M1T1 group A streptococcal systemic virulence. PLoS One 3, e4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schrager H. M., Rheinwald J. G., Wessels M. R. (1996) Hyaluronic acid capsule and the role of streptococcal entry into keratinocytes in invasive skin infection. J. Clin. Invest. 98, 1954–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bessen D. E., Kalia A. (2002) Genomic localization of a T serotype locus to a recombinatorial zone encoding extracellular matrix-binding proteins in Streptococcus pyogenes. Infect. Immun. 70, 1159–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mora M., Bensi G., Capo S., Falugi F., Zingaretti C., Manetti A. G., Maggi T., Taddei A. R., Grandi G., Telford J. L. (2005) Group A Streptococcus produce pilus-like structures containing protective antigens and Lancefield T antigens. Proc. Natl. Acad. Sci. U. S. A. 102, 15641–15646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kratovac Z., Manoharan A., Luo F., Lizano S., Bessen D. E. (2007) Population genetics and linkage analysis of loci within the FCT region of Streptococcus pyogenes. J. Bacteriol. 189, 1299–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 42. Lei B., Mackie S., Lukomski S., Musser J. M. (2000) Identification and immunogenicity of group A Streptococcus culture supernatant proteins. Infect. Immun. 68, 6807–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Caswell C. C., Han R., Hovis K. M., Ciborowski P., Keene D. R., Marconi R. T., Lukomski S. (2008) The Scl1 protein of M6-type group A Streptococcus binds the human complement regulatory protein, factor H, and inhibits the alternative pathway of complement. Mol. Microbiol. 67, 584–596 [DOI] [PubMed] [Google Scholar]
- 44. Pahlman L. I., Marx P. F., Morgelin M., Lukomski S., Meijers J. C., Herwald H. (2007) Thrombin-activatable fibrinolysis inhibitor binds to Streptococcus pyogenes by interacting with collagen-like proteins A and B. J. Biol. Chem. 282, 24873–24881 [DOI] [PubMed] [Google Scholar]
- 45. Han R., Caswell C. C., Lukomska E., Keene D. R., Pawlowski M., Bujnicki J. M., Kim J. K., Lukomski S. (2006) Binding of the low-density lipoprotein by streptococcal collagen-like protein Scl1 of Streptococcus pyogenes. Mol. Microbiol. 61, 351–367 [DOI] [PubMed] [Google Scholar]
- 46. Hoe N. P., Lukomska E., Musser J. M., Lukomski S. (2007) Characterization of the immune response to collagen-like proteins Scl1 and Scl2 of serotype M1 and M28 group A Streptococcus. FEMS Microbiol. Lett. 277, 142–149 [DOI] [PubMed] [Google Scholar]
- 47. Nannini E. C., Teng F., Singh K. V., Murray B. E. (2005) Decreased virulence of a gls24 mutant of Enterococcus faecalis OG1RF in an experimental endocarditis model. Infect. Immun. 73, 7772–7774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Teng F., Nannini E. C., Murray B. E. (2005) Importance of gls24 in virulence and stress response of Enterococcus faecalis and use of the Gls24 protein as a possible immunotherapy target. J. Infect. Dis. 191, 472–480 [DOI] [PubMed] [Google Scholar]
- 49. Carapetis J. R., Wolff D. R., Currie B. J. (1996) Acute rheumatic fever and rheumatic heart disease in the top end of Australia's Northern Territory. Med. J. Aust. 164, 146–149 [DOI] [PubMed] [Google Scholar]
- 50. Beres S. B., Richter E. W., Nagiec M. J., Sumby P., Porcella S. F., DeLeo F. R., Musser J. M. (2006) Molecular genetic anatomy of inter- and intraserotype variation in the human bacterial pathogen group A Streptococcus. Proc. Natl. Acad. Sci. U. S. A. 103, 7059–7064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Green N. M., Zhang S., Porcella S. F., Nagiec M. J., Barbian K. D., Beres S. B., LeFebvre R. B., Musser J. M. (2005) Genome sequence of a serotype M28 strain of group A Streptococcus: potential new insights into puerperal sepsis and bacterial disease specificity. J. Infect. Dis. 192, 760–770 [DOI] [PubMed] [Google Scholar]
- 52. Musser J. M., Shelburne S. A., 3rd (2009) A decade of molecular pathogenomic analysis of group A Streptococcus. J. Clin. Invest. 119, 2455–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Carapetis J. R., Steer A. C., Mulholland E. K., Weber M. (2005) The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 [DOI] [PubMed] [Google Scholar]
- 54. Reuter M., Caswell C. C., Lukomski S., Zipfel P. F. (2010) Binding of the human complement regulators CFHR1 and factor H by streptococcal collagen-like protein 1 (Scl1) via their conserved C termini allows control of the complement cascade at multiple levels. J. Biol. Chem. 285, 38473–38485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rasmussen M., Eden A., Bjorck L. (2000) SclA, a novel collagen-like surface protein of Streptococcus pyogenes. Infect. Immun. 68, 6370–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vebo H. C., Snipen L., Nes I. F., Brede D. A. (2009) The transcriptome of the nosocomial pathogen Enterococcus faecalis V583 reveals adaptive responses to growth in blood. PLoS One 4, e7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vebo H. C., Solheim M., Snipen L., Nes I. F., Brede D. A. (2010) Comparative genomic analysis of pathogenic and probiotic Enterococcus faecalis isolates, and their transcriptional responses to growth in human urine. PLoS One 5, e12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang M., McDonald F. M., Sturrock S. S., Charnock S. J., Humphery-Smith I., Black G. W. (2007) Group A Streptococcus cell-associated pathogenic proteins as revealed by growth in hyaluronic acid-enriched media. Proteomics 7, 1379–1390 [DOI] [PubMed] [Google Scholar]
- 59. Graham M. R., Virtaneva K., Porcella S. F., Gardner D. J., Long R. D., Welty D. M., Barry W. T., Johnson C. A., Parkins L. D., Wright F. A., Musser J. M. (2006) Analysis of the transcriptome of group A Streptococcus in mouse soft tissue infection. Am. J. Pathol. 169, 927–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garcia A. F., Abe L. M., Erdem G., Cortez C. L., Kurahara D., Yamaga K. (2010) An insert in the covS gene distinguishes a pharyngeal and a blood isolate of Streptococcus pyogenes found in the same individual. Microbiology 156, 3085–3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cole J. N., Pence M. A., von Kockritz-Blickwede M., Hollands A., Gallo R. L., Walker M. J., Nizet V. (2010) M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. MBio 1, pii: e00191-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.