

Summary
The NAD-dependent deacetylase SIRT1 is involved in chromatin silencing and genome stability. Elevated SIRT1 levels in embryonic stem cells also suggest a role for SIRT1 in pluripotency. Murine SIRT1 attenuates telomere attrition in vivo and is recruited at telomeres in induced pluripotent stem cells (iPSCs). Because telomere elongation is an iPSC hallmark, we set out to study the role of SIRT1 in pluripotency in the setting of murine embryonic fibroblasts reprogramming into iPSCs. We find that SIRT1 is required for efficient postreprogramming telomere elongation, and that this effect is mediated by a c-MYC-dependent regulation of the mTert gene. We further demonstrate that SIRT1-deficient iPSCs accumulate chromosomal aberrations and show a derepression of telomeric heterochromatin. Finally, SIRT1-deficient iPSCs form larger teratomas that are poorly differentiated, highlighting a role for SIRT1 in exit from pluripotency. In summary, this work demonstrates a role for SIRT1 in the maintenance of pluripotency and modulation of differentiation.
Highlights
-
•
SIRT1 is required for mTERT-dependent telomere elongation after reprogramming
-
•
Sirt1−/− xenografts are larger than wild-type and display delayed differentiation
-
•
SIRT1 regulates TERRA transcription and contributes to maintain genomic stability
-
•
SIRT1 deficiency impairs telomeric chromatin remodeling upon reprogramming
SIRT1 is a multifunctional protein that also plays a role in pluripotency and telomere dynamics. Blasco and colleagues demonstrate that SIRT1, although not directly involved in the reprogramming of somatic cells, is required for telomere elongation taking place after reprogramming. Moreover, SIRT1 maintains genomic stability upon long-term cell culture and modulates the differentiation potential of iPSCs.
Introduction
SIRT1 belongs to the sirtuin family of NAD+-dependent lysine deacetylases, homologs of the yeast protein Sir2. In mammals, the sirtuin family includes seven members (SIRT1–SIRT7), with SIRT1 having the closest homology to Sir2. SIRT1 has been shown to regulate several key cellular processes, among them chromatin remodeling, transcriptional silencing, and genomic stability (Haigis and Sinclair, 2010), via the deacetylation of diverse substrates, including p53 (Cheng et al., 2003), FoxO transcription factors (Frescas et al., 2005), and NF-κB (Yeung et al., 2004).
Sir2 is part of the Sir silencing complex, which counteracts aging in yeast by suppressing recombination at the ribosomal DNA locus, and contributes to heterochromatin formation (Sinclair and Guarente, 1997). Importantly, the Sir silencing complex is also involved in the formation and maintenance of telomeric heterochromatin in budding yeast (Xu et al., 2007).
Telomeres are specialized nucleoprotein structures that protect the ends of eukaryotic chromosomes from unscheduled DNA repair (Chan and Blackburn, 2004). In vertebrates, telomeres are constituted by TTAGGG tandem repeats, bound by a specialized multiprotein complex, known as shelterin, which plays a pivotal role in the regulation of telomere length and protection of chromosome ends (de Lange, 2005). Telomeric repeats are synthesized by telomerase (Greider and Blackburn, 1985), a ribonucleoprotein reverse transcriptase expressed mainly during embryonic development, as well as in adult stem cell compartments (Flores et al., 2006a; Liu et al., 2007). Telomerase levels in adult somatic cells are not sufficient to prevent the telomere shortening that occurs with aging, and telomeres suffer a progressive attrition with cell divisions.
Chromatin at mammalian telomeres is underacetylated and shares the typical features of other repeat-containing constitutive heterochromatic elements, such as pericentric chromatin. Telomeric repeats are also enriched for H3K9me3, which represents a docking site for the binding of HP1 isoforms (Benetti et al., 2007).
Despite evidence that telomeres are heterochromatic structures, our group and others have reported that the C-rich strand of eukaryotic telomeres is transcribed by RNA polymerase II, giving rise to noncoding transcripts, heterogeneous in size, known as TelRNAs or TERRAs (Azzalin et al., 2007; Schoeftner and Blasco, 2008). TERRAs closely associate with telomeres and negatively regulate telomerase activity in vitro (Schoeftner and Blasco, 2008). Telomeric RNA abundance is modulated by several factors, including telomere length; consistently, TERRA transcription increases upon somatic cell reprogramming (Marion et al., 2009).
Murine and human somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) by the overexpression of exogenous transcription factors related to pluripotency, namely OCT4, KLF4, SOX2, and c-MYC (Takahashi and Yamanaka, 2006). During murine iPSC generation, telomeres are also “reprogrammed” to adopt features similar to those of embryonic stem cells (ESCs); iPSC telomere length increases in a telomerase-dependent manner, and this elongation continues with cell passages after reprogramming, until they reach a length comparable to that of ESCs (Marion et al., 2009). In addition, there is an increase in the binding of the shelterin TRF1 (Schneider et al., 2013), and the global epigenome is broadly rearranged to an ESC-like chromatin architecture (Meshorer et al., 2006). Therefore, besides stemness and pluripotency factors, cellular reprogramming also involves chromatin modifiers that contribute to the remodeling of chromatin, including telomere chromatin.
SIRT1 is highly expressed in mouse ESCs, and its expression progressively declines during in vitro differentiation (Calvanese et al., 2010). This downregulation is required to correctly establish differentiation programs, pointing to SIRT1 as a pluripotency and differentiation modulator. Given this, it is conceivable that SIRT1 may play a role in some of the events occurring during somatic cell reprogramming. We therefore set out to study the role of SIRT1 in this process and to analyze its role in the dynamic changes that telomeres undergo during reprogramming.
To address this, we employed both loss-of-function (Sirt1−/−) (Cheng et al., 2003) and gain-of-function (Sirt1Super) (Pfluger et al., 2008) mouse models. We generated iPSCs from Sirt1−/− and Sirt1Super murine embryonic fibroblasts (MEFs), together with their wild-type counterparts, and investigated the impact on reprogramming of SIRT1 deficiency or mild overexpression. Our results show that SIRT1, although not strictly required for reprogramming itself, is necessary for telomere elongation after reprogramming; this effect is mediated by the regulation of telomerase expression via c-MYC. In addition, SIRT1 is required to maintain iPSC genomic stability and is involved in telomeric transcription and in the remodeling of telomeric chromatin.
In conclusion, this work demonstrates a role for SIRT1 in regulating some of the hallmark features of iPSCs, namely genomic integrity and telomere expansion. Understanding the molecular mechanism by which such characteristics are maintained in established iPSC lines is critical to the advance of iPSC technology in regenerative medicine.
Results
Reprogramming Efficiency of Sirt1−/− and Sirt1Super Embryonic Fibroblasts
We first asked if SIRT1 plays a direct role in the conversion of fibroblasts to the pluripotent state. We infected wild-type (Sirt1+/+), SIRT1-deficient (Sirt1−/−), or SIRT1-overexpressing (Sirt1Super) MEFs with retroviral vectors expressing the three reprogramming factors (OCT4, KLF4, and SOX2) to generate iPSCs. We estimated reprogramming efficiency on the basis of the number of colonies positive for alkaline phosphatase staining, normalized to the percentage of infected cells (Figures 1A–1D). Sirt1−/−, as well as Sirt1Super MEFs, showed a reprogramming efficiency comparable to wild-type counterparts, indicating that SIRT1 does not impact the overall ability of the cells to be induced to a pluripotent state. The percentage of colonies staining positive for NANOG (Figures 1E and 1F), as well as NANOG and OCT3/4 protein levels in individual passage 5 iPSC clones (Figures 1G and 1H; Figures S1A and S1B available online), also revealed no differences in reprogramming efficiencies among genotypes.
Figure 1.
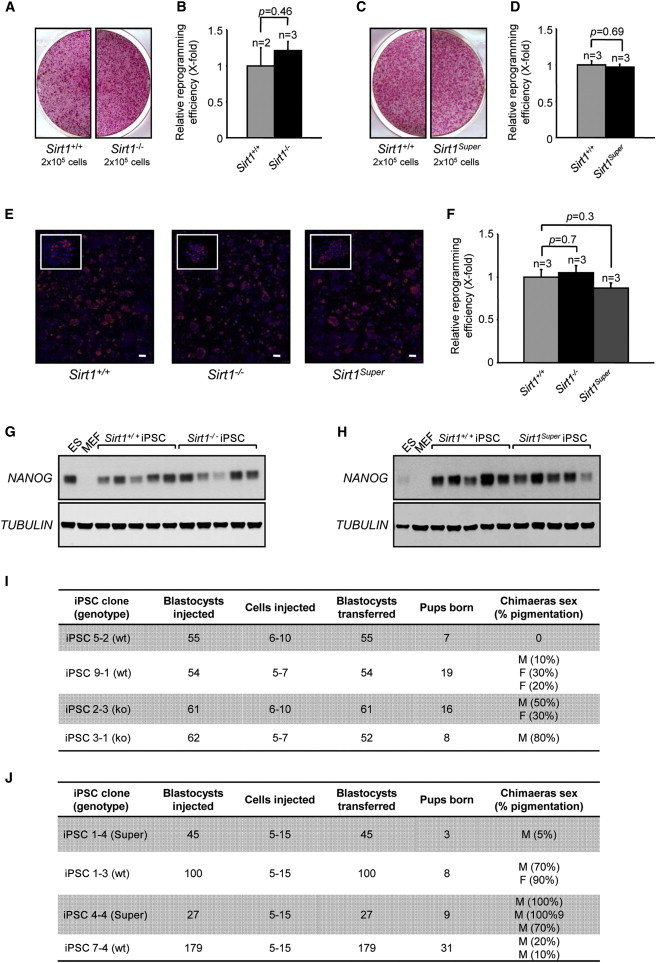
Reprogramming Efficiency and Chimera Generation
(A and C) Representative images of alkaline phosphatase-positive colonies derived from the reprogramming of Sirt1−/− (A) and Sirt1Super (C) MEFs, compared with the relevant wild-type.
(B and D) The relative reprogramming efficiency of Sirt1−/− (B) and Sirt1Super (D) MEFs, expressed as wild-type X-fold, has been calculated by counting the colonies and normalizing to viral transduction efficiency. n, number of independent clones. Error bars represent the SEM. Statistical analysis was performed using a two-tailed Student’s t test.
(E and F) Representative images of NANOG-positive colonies derived from the reprogramming of Sirt1+/+, Sirt1−/−, and Sirt1Super MEFs. (E) Scale bar represents 500 μm. The relative reprogramming efficiency, expressed as wild-type X-fold, has been calculated as in (B) and (D). Error bars represent the SEM. Statistical analysis was performed using a two-tailed Student’s t test.
(G and H) Western blot analysis showing the expression of NANOG in Sirt1−/− (G) and Sirt1Super (H) iPSCs. Tubulin was included as a loading control. Five individual iPSC clones per genotype were tested. An ESC and an MEF sample were included as positive and negative controls, respectively.
(I and J) Tables reporting the details of contribution to mouse chimerism obtained from microinjection of Sirt1−/− (I) and Sirt1Super (J) iPSCs.
See also Figure S1.
To more stringently test their pluripotency features, we injected Sirt1−/− and Sirt1Super iPSCs into C57BL/6-Tyrc (albino) blastocysts or aggregated them with CD1 (albino) morulae. For both Sirt1+/+ and Sirt1−/− iPSCs, we obtained black-haired chimeras with a variable extent of chimerism (Figure 1I). Sirt1Super iPSCs also generated chimeras similar to their wild-type counterparts (Figure 1J), suggesting that both SIRT1-deficient and SIRT1-overexpressing iPSCs can contribute to the formation of adult tissues.
Interestingly, previous results showed that SIRT1 is highly expressed in ESCs compared to differentiated cells (Calvanese et al., 2010). We therefore tested whether SIRT1 expression is also induced during reprogramming. Remarkably, we found that the expression of SIRT1, which is very low in parental MEFs, is increased in iPSCs comparable to levels seen in ESCs (Figure S1C).
SIRT1 Deficiency Correlates with a Delayed Differentiation In Vivo
To test the in vivo differentiation potential of SIRT1-deficient iPSCs, we injected Sirt1+/+ and Sirt1−/− iPSCs in athymic nude mice and screened the formation of subcutaneous teratomas. All the three germ layers could be found in teratomas derived from both Sirt1+/+ and Sirt1−/− iPSCs (Figure 2B). Interestingly, teratomas from Sirt1−/− iPSCs grew much faster, resulting in significantly bigger tumors, compared to those obtained from Sirt1+/+ iPSCs (Figure 2A and inset). This difference was most significant when comparing the final xenograft weights at the end of the experiment (Figure 2C) or the average xenograft volumes throughout the experiment (Figure 2D).
Figure 2.
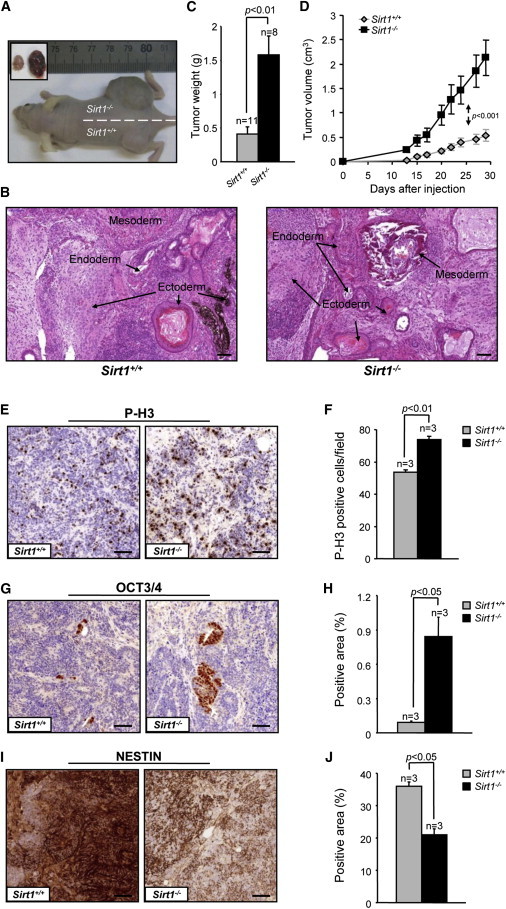
Sirt1−/− iPSC-Derived Teratomas Grow Faster than Wild-Type and Show Delayed Differentiation In Vivo
(A) Representative example of teratomas generated by Sirt1+/+ and Sirt1−/− iPSCs 29 days after injection.
(B) Hematoxylin and eosin-stained teratoma sections from both genotypes show tissues derived from the three germ layers for both genotypes. Scale bar, 100 μm.
(C and D) Average weight of Sirt1+/+ and Sirt1−/− tumors at the time of excision (C) and average volume of Sirt1+/+ and Sirt1−/− tumors, measured at the indicated day postinjection (D). n, number of different tumors per genotype. Error bars represent the SEM. Statistical analysis was performed using a two-tailed Student’s t test (C) or two-way ANOVA (D).
(E, G, and I) Representative areas of Sirt1+/+ (left) and Sirt1−/− (right) teratoma sections 14 days postinjection, stained with P-H3 (E), OCT3/4 (G), or NESTIN (I) antibodies. Scale bar represents 50 μm.
(F, H, and J) Quantification of P-H3 (F) was obtained scoring the number of positive cells in ten random fields. Quantification of OCT3/4 (H) and NESTIN (J) expression was obtained measuring throughout the whole slide the percentage of positive area with an intensity above an arbitrary threshold by means of the ImageJ software. n = 3 refers to three independent wild-type versus three independent knockout clones injected in parallel. Error bars refer to SEM. Statistical analysis was performed using a two-tailed Student’s t test.
See also Figure S2.
One explanation for the increased size of Sirt1−/− teratomas could be an enhanced intrinsic cell proliferation capacity in undifferentiated Sirt1−/− iPSCs, as reported in other cell types (Chua et al., 2005; Narala et al., 2008). To test this, we measured the proliferation rate of three independent clones per genotype over 5 days and found comparable values for all clones, ruling out this possibility (Figure S1D).
An alternative explanation for the enhanced growth of Sirt1−/− teratomas is that Sirt1−/− iPSCs may acquire a proliferative advantage while differentiating, because it has been previously reported that SIRT1 dually modulates differentiation and proliferation in mouse neurospheres (Hisahara et al., 2008) and human keratinocytes (Blander et al., 2009). Because teratomas are mainly composed of ectodermal tissues, we reasoned that SIRT1 deficiency could delay ectodermal differentiation, allowing Sirt1−/− iPSCs to maintain longer their stemness properties and proliferate more than the wild-type cells.
To verify this hypothesis, we microinjected Sirt1+/+ and Sirt1−/− iPSC clones (n = 3 each) in athymic nude mice and monitored early events by removing the tumors 14 days after injection. At this stage, Sirt1+/+ xenografts just started to become visible, whereas Sirt1−/− tumors were already appreciably larger. We stained a section from each tumor (n = 3 per genotype) for the proliferation marker phosphohistone 3 (P-H3; Figures 2E and 2F) and, as expected, we observed a higher number of proliferating cells in Sirt1−/− teratomas compared to wild-type. We next quantified the number of cells positive for the stemness marker OCT3/4 and for NESTIN, a neural lineage precursor marker. Interestingly, Sirt1−/− teratomas exhibited a higher number of cells expressing OCT3/4 (Figures 2G and 2H) and a consistently weaker NESTIN staining compared to wild-type xenografts (Figures 2I and 2J). No differences were observed in teratomas examined at later stages (data not shown).
To corroborate this observed effect in a different setting, we generated embryoid bodies (EBs) from four clones of each Sirt1+/+ and Sirt1−/− iPSCs in culture and assessed OCT4 and NESTIN levels at different time points (day 7, day 14, and day 21 after leukemia inhibitory factor [LIF] was withdrawn) through immunohistochemistry (Figures S2A and S2B, representative images). After 7 days, we could observe a higher number of OCT4-positive cells and a clear tendency for decreased NESTIN staining (though this was not statistically significant) in Sirt1−/− EBs. Consistent with protein levels, Oct4 mRNA levels decreased more slowly over time when assessed at 3 days of EB development compared to wild-type (Figure S2E), whereas Nestin expression was not significantly different, at every time point selected, between genotypes (Figure S2F).
Collectively, these results suggest that SIRT1 deficiency delays, both in vivo and in vitro, the ability of iPSCs to exit pluripotency and commit to ectodermal lineages.
SIRT1 Is Required for Efficient Telomere Elongation during Reprogramming
One of the critical events occurring during reprogramming is a telomerase-dependent telomere elongation, which gives rise to hyperlong telomeres comparable to those of ESCs. Because SIRT1 is able to positively regulate telomere length in MEFs, we asked whether it could also have a role in telomere elongation during reprogramming. To test this, we analyzed telomere length in four independent iPSC clones per genotype (Sirt1−/− versus Sirt1+/+), compared to the corresponding parental MEFs, using two experimental approaches.
First, we measured the length of telomeric TTAGGG track using Southern blot-based telomere restriction fragment (TRF) analysis comparing MEFs (passage 3) with iPSCs (passage 15). Nuclear reprogramming resulted in telomere elongation in both Sirt1+/+ and Sirt1−/− iPSCs compared to the corresponding parental MEFs; however, Sirt1−/− iPSCs displayed significantly shorter telomeres compared to wild-type iPSCs (Figure 3A). To validate these results, we performed a quantitative fluorescence in situ hybridization (Q-FISH) on metaphase spreads using a telomere-specific probe. The frequency distribution of telomere length confirmed a diminished telomere elongation in Sirt1−/− iPSCs compared with Sirt1+/+ controls (Figures 3B and 3E). This effect became even more apparent when comparing the absolute lengthening, in kilobases, in Sirt1+/+ versus Sirt1−/− iPSCs after 15 passages in culture (Figure 3F). These findings support a role for SIRT1 in telomere elongation during nuclear reprogramming of embryonic fibroblasts.
Figure 3.
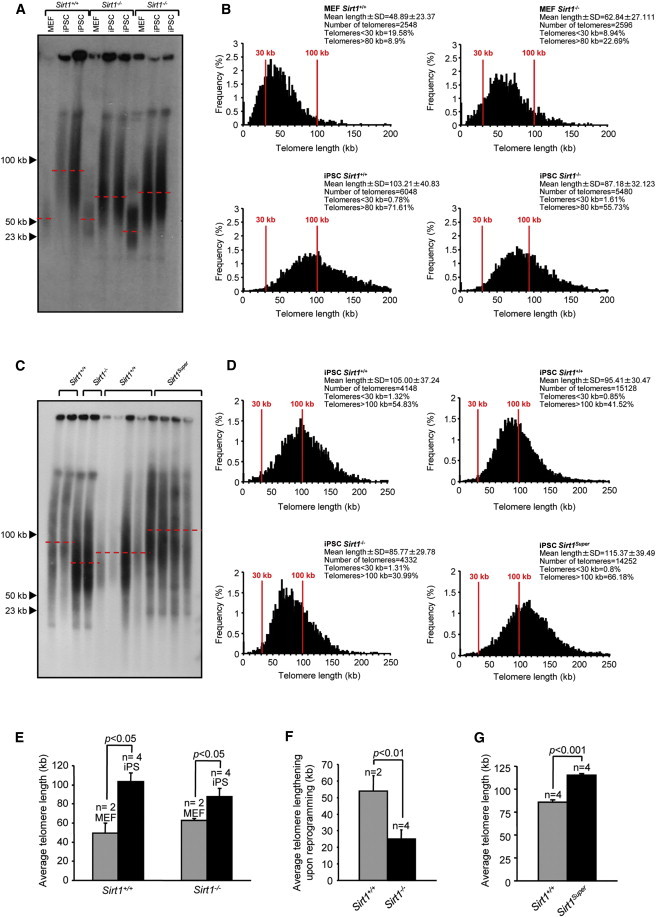
SIRT1 Is a Positive Regulator of Telomere Elongation Associated with Reprogramming
(A) TRF performed with MEFs (passage 3) derived from one embryo of Sirt1+/+ and two embryos of Sirt1−/− genotypes and two independent iPSC clones (passage 15) obtained from each MEF.
(B) Telomere length frequency distribution (in kilobases) as determined by Q-FISH on metaphase spreads from Sirt1+/+ and Sirt1−/−, two independent primary MEFs (passage 3) per genotype, and two independent clones of iPSCs (passage 15) per each parental MEFs. n indicates the number of independent MEF cultures or iPSCs clones. Error bars represent the SEM. Statistical analysis was performed using a two-tailed Student’s t test.
(C) TRF performed with Sirt1Super iPS (passage 15) and relative wild-type clones. Two samples of each Sirt1−/− and relative wild-type were included as a reference. Sirt1Super iPSCs were derived from two independent parental MEFs (two clones each; a total of four independent iPSC clones).
(D) Telomere length frequency distribution (in kilobases) as determined by Q-FISH on metaphase spreads from Sirt1+/+ and Sirt1Super, two independent clones of iPSCs (passage 15) per parental MEF, for a total of four independent clones.
(E) Mean telomere length in Sirt1+/+ and Sirt1−/− primary MEFs and iPSCs.
(F) Average telomere elongation upon reprogramming of Sirt1+/+ and Sirt1−/− MEFs.
(G) Mean telomere length in Sirt1+/+ and SirtSuper iPSCs. n indicates the number of independent MEF cultures or iPSCs clones. Error bars represent the SEM. Statistical analysis was performed using a two-tailed Student’s t test.
To further strengthen this conclusion, we also measured telomere length in Sirt1Super iPSCs (Figures 3C and 3D), and we observed the opposite trend: the presence of an extra copy of SIRT1 correlated with greater telomere elongation upon reprogramming; the quantitative analysis shows an average 20% higher telomere length in Sirt1Super compared to Sirt1+/+ iPSCs (Figure 3G). This result reinforces the view that SIRT1 plays a role in the elongation of telomere taking place during the generation of iPSCs by nuclear reprogramming.
iPSCs show a progressive elongation of telomeres postreprogramming; we thus measured telomere length at different passages of Sirt1+/+ and Sirt1−/− iPSCs. Although we observed the expected elongation with passages of Sirt1+/+ iPSCs, SIRT1-deficient cells showed only a mild elongation of telomeres upon the same passages (Figure 4A). We further confirmed these results by Q-FISH analysis in metaphase spreads from the same cells (Figures 4B and 4C). Interestingly, telomere length in Sirt1−/− iPSCs at early passages (passage 5) is comparable to that of wild-type iPSCs, as determined both by TRF and Q-FISH (Figures 4A and 4B). Collectively, these results demonstrate that although SIRT1 is not required for the initial telomere elongation during early reprogramming, it is required for the acquisition of the hyperlong telomeres during further passaging, an important feature of the pluripotent nature of iPSCs.
Figure 4.
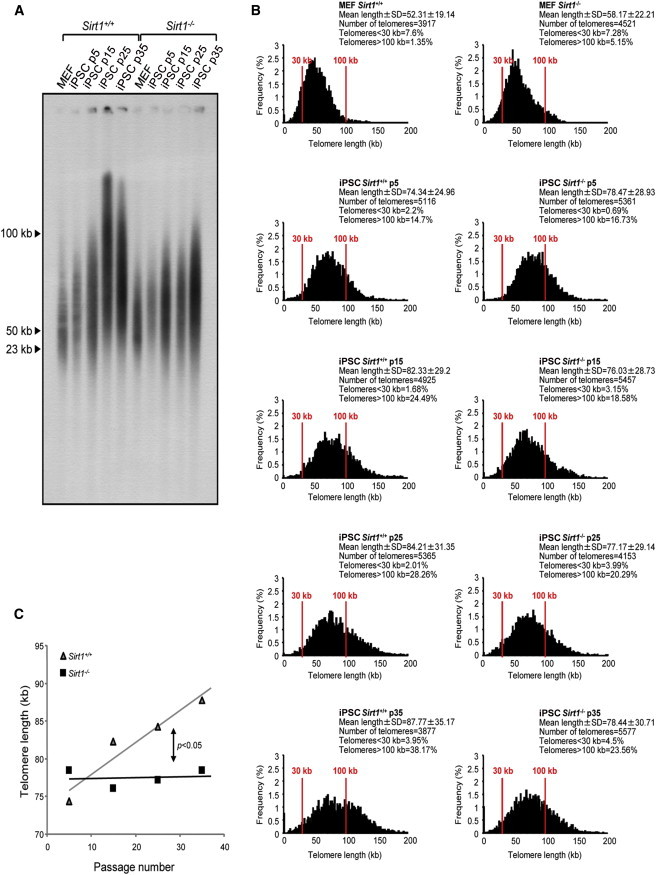
Telomere Elongation after Reprogramming Is Impaired in Sirt1−/− iPSCs
(A) TRF analysis of one Sirt1+/+ iPSC clone and one Sirt1−/− iPSC clone at increasing passages (p5, p15, p25, and p35). The progressive telomere lengthening is impaired in the absence of SIRT1. A sample from the parental primary MEF of each clone has been included as a reference.
(B) Telomere length frequency distribution, obtained from Q-FISH in metaphase spreads from one Sirt1+/+ iPSC clone and one Sirt1−/− iPSC clone at increasing passages (p5, p15, p25, and p35). The results obtained confirm the tendency observed by TRF. A sample from the parental primary MEF of each clone has been included as a reference.
(C) Linear regression analysis of telomere elongation with passaging in culture, comparing a Sirt1+/+ iPSC clone (gray line) with a Sirt1−/− iPSC clone (black line). The statistical comparison of the two slopes was performed using analysis of covariance.
c-MYC-Dependent mTERT Regulation Sustains Telomere Elongation after Reprogramming
We next asked if the effects of SIRT1 on telomere length during reprogramming could be due to a possible misregulation of mTERT expression. Despite significant variability among clones, western blot analysis of Sirt1+/+ versus Sirt1−/− iPSCs at passage 35, when the divergence in telomere length is more pronounced, showed a clear trend toward decreased mTERT expression in Sirt1−/− iPSCs compared to wild-type (Figures 5A and 5C, left panels). The correlation between SIRT1 and telomerase levels was further confirmed comparing Sirt1+/+ versus Sirt1Super iPSCs, with SIRT1-overexpressing cells showing an increase in telomerase expression compared to wild-type cells (Figures 5A and 5C, right panels). As expected, the decrease observed in telomerase abundance in Sirt1−/− iPSCs is paralleled by a consistent difference in mTert transcription (Figure S3A).
Figure 5.
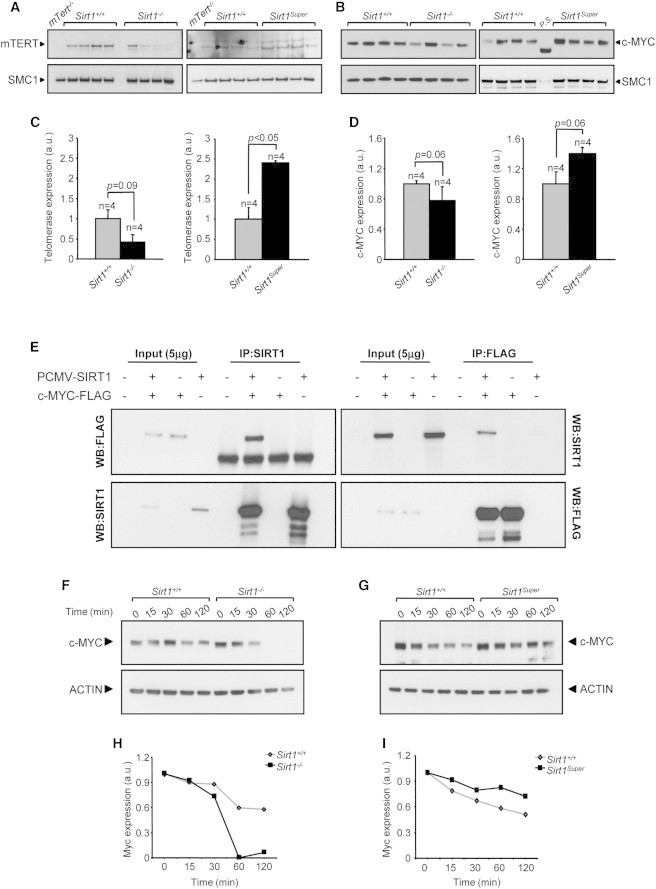
SIRT1 Regulates mTERT in a c-MYC-Dependent Manner
(A–D) Western blot showing the expression of mTERT (A) and c-MYC (B) in iPSCs with the indicated genotypes (passage 35). Cohesin SMC1 was used as loading control. The quantification of mTERT (C) or c-MYC expression (D) in Sirt1−/− (C and D, left panels) or SirtSuper (C and D, right panels) clones was performed through background subtraction and normalization based on SMC1 expression to correct for differences in loading. A nuclear extract sample obtained from mTert−/− MEFs was included as a negative control in (A). Four independent iPSC clones per genotype were tested. Error bars refer to SEM. Statistical analysis was performed using a two-tailed Student’s t test.
(E) Coimmunoprecipitation of SIRT1 and c-MYC in 293T cells transfected with the indicated constructs using a SIRT1-specific antibody (IP:SIRT1) or a FLAG-specific antibody (IP:FLAG). WB, western blot.
(F–I) c-MYC stability is modulated by SIRT1. Protein synthesis was inhibited with 20 μM cycloheximide. Cells were collected after the indicated periods of time and analyzed by western blot (F and G). The decline of c-MYC protein is accelerated in the absence of SIRT1 (H), whereas an extra copy of Sirt1 increases c-MYC stability (I).
See also Figure S3.
A positive-feedback loop involving SIRT1 and the c-MYC oncogene, which is known to regulate telomerase transcription in both human and mouse, has been previously described (Flores et al., 2006b; Wang et al., 1998). This prompted us to examine whether decreased telomerase expression in late-passage Sirt1−/− iPSCs correlates with diminished c-MYC expression. Western blot analysis revealed indeed a reduction in c-MYC expression in late-stage Sirt1−/− iPSCs that paralleled the decrease in telomerase expression (Figures 5B and 5D, left panels). However, we did not observe in this case any difference in c-Myc RNA, pointing to a posttranscriptional regulation of this protein (Figure S3B). Analogously, in cells overexpressing SIRT1, c-MYC expression correlated with increased telomerase expression compared to controls (Figures 5B and 5D, right panels).
To learn more about the mechanism through which SIRT1 regulates c-MYC expression, we first checked if SIRT1 and c-MYC physically interact. To do this, we transiently overexpressed murine SIRT1 and a FLAG-tagged version of murine c-MYC in 293T cells and assayed their coimmunoprecipitation. We were able to detect c-MYC upon SIRT1 immunoprecipitation and vice versa, supporting a direct interaction between the two proteins (Figure 5E). This result, together with the evidence that no significant difference when comparing c-Myc transcription in Sirt1+/+ and Sirt1−/− iPSCs was observed (Figure S3B), makes it unlikely that SIRT1 regulates c-Myc transcription through promoter deacetylation. We then set out to verify if the absence of SIRT1 could affect c-MYC stability. Upon treatment of Sirt1+/+, Sirt1−/−, or Sirt1Super iPSCs with the inhibitor of protein biosynthesis cycloheximide, we observed a faster decline of c-MYC protein in the absence of SIRT1 (Figures 5F and 5H), whereas the presence of an extra copy of Sirt1 significantly prolongs c-MYC half-life (Figures 5G and 5I).
Taken together, these results suggest that SIRT1 drives telomere elongation with passages of iPSCs by slowing down the degradation of c-MYC, which in turn promotes telomerase expression.
p53 Expression and Activity, as well as SIRT6 Expression, Are Not Affected in Sirt1−/− iPSCs
SIRT1 deacetylates and directly regulates p53, which has been linked to reprogramming, tumorigenesis, DNA damage repair, and the recruitment of histone modifiers (Han et al., 2008).
To exclude the possibility that the phenotype we observed in Sirt1−/− iPSCs might be p53 dependent, we analyzed the acetylation of p53 at Lys379, a known target of SIRT1 deacetylase activity, and observed increased levels in Sirt1−/− compared to Sirt1+/+ iPSCs (Figure S3E). However, no significant increase in total p53 protein levels (Figures S3C and S3D) or increased expression of the p53 target p21 was detected in Sirt1−/− iPSCs (Figure S5F).
SIRT6 is another member of the mammalian sirtuin family linked to telomere instability in human cells. SIRT1 is able to positively regulate SIRT6 in certain tissues (Kim et al., 2010), raising the possibility that the effect of SIRT1 deficiency on iPSC telomeres could actually result from SIRT6 downregulation. However, SIRT6 expression appears to be increased rather than diminished in Sirt1−/− cells (Figures S5G and S5H), ruling out this possibility.
SIRT1 Maintains Genomic Stability in iPSCs
Because SIRT1 has been shown to play important roles in genome integrity and DNA damage response (Oberdoerffer et al., 2008), we reasoned that SIRT1 deficiency during reprogramming, and the concomitant telomere elongation defects in the resulting iPSCs, could render these cells more prone to chromosomal and/or telomeric aberrations. To address this, we scored chromosomal aberrations in Sirt1+/+ and Sirt1−/− metaphases using a Cy-3-labeled peptide nucleic acid probe to stain telomeres, focusing on recurrent aberrations associated with telomere dysfunction, such as multitelomeric signals (MTSs), chromosome/chromatid breaks, and chromosome/chromatid fusions. Interestingly, we found that MTSs, which are associated with replication defects at telomeres (Martínez et al., 2009; Sfeir et al., 2009), were significantly reduced upon reprogramming in both Sirt1+/+ and Sirt1−/− cells (Figure 6A), suggesting that cells with this type of aberration are selected against during the reprogramming process. In agreement with a role for SIRT1 in DNA damage repair, we found that chromosome/chromatid breaks were significantly higher in Sirt1−/− MEFs compared to controls. Reprogramming significantly reduced chromosome/chromatid breaks in both genotypes, although the levels remained significantly higher in Sirt1−/− than in Sirt1+/+ iPSCs (Figure 6B).
Figure 6.
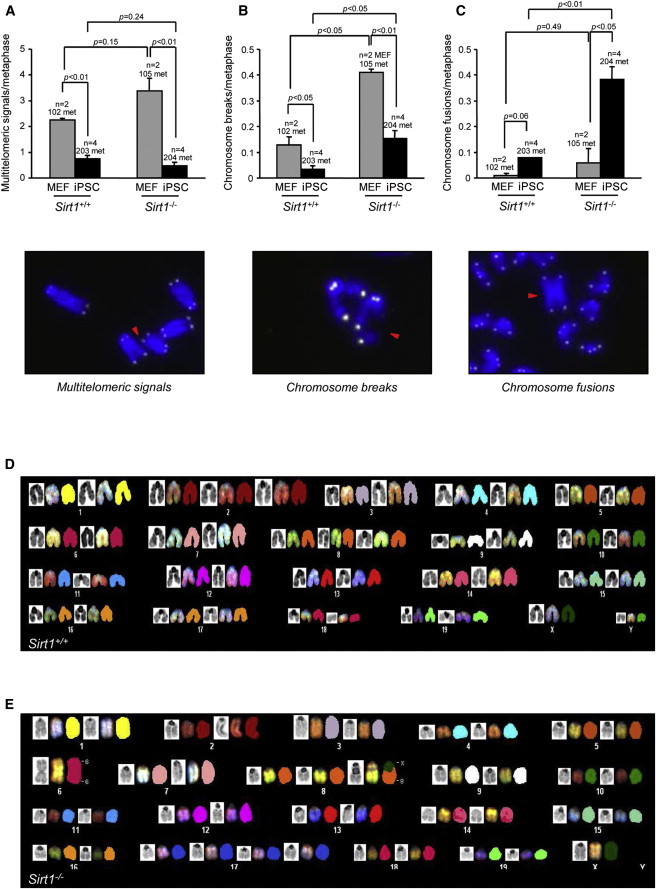
Sirt1−/− iPSCs Show Higher Chromosomal Instability
(A–C) Quantification of multitelomeric signals (MTS) (A), chromosome breaks (B), and chromosome fusions (C) in Sirt1+/+ and Sirt1−/− MEFs (passage 3; two independent cell lines per genotype) and iPSCs (passage 5; two independent clones per each parental MEF). A representative image of every chromosomal aberration is shown. Error bars represent the SEM. Statistical analysis was performed using a two-tailed Student’s t test.
(D and E) SKY analysis of metaphase spreads from Sirt1+/+ and Sirt1−/− iPSCs (passage 35). Each chromosome is represented from left to right by DAPI staining, the display color, and the classification pseudocolor. Karyotype formulas: 39−42.XY,+2,+8 (D); 35–41.X0,rob(6;6),+8,der(8)T(8;X),+17×2 (E).
See also Figure S4.
In contrast to MTSs, we observed a subtle increase in chromosome/chromatid fusions upon reprogramming of Sirt1+/+ MEFs, which was significantly greater in Sirt1−/− iPSCs even at early passages (Figure 6C) and became more evident with long-term culture. Spectral karyotyping (SKY) analysis of metaphase spreads from Sirt1+/+ and Sirt1−/− iPSCs confirmed that Sirt1−/− cells at passage 35 were more prone to accumulate chromosomal aberrations. In particular, the analysis of three independent clones per genotype showed that late-passage Sirt1+/+ iPSCs carried only a few recurrent aneuploidies (mainly chromosome 8), most likely due to adaptation to culture, and only a few structural aberrations (namely a duplication of part of chromosome 14 and a translocation involving chromosome 15 and chromosome 8). In marked contrast, the great majority of metaphase spreads from Sirt1−/− iPSC clones showed additional aneuploidies and aberrations, most notably Robertsonian translocation involving both homologs of chromosome 6, observed in all clones. Also of note, Sirt1−/− iPSC clones exhibited clone-specific structural aberrations involving chromosome 8. Figure 6 provides representative karyotyping by SKY analysis, of Sirt1+/+ (Figure 6D, karyotype formula: 39–42.XY,+2,+8) versus Sirt1−/− iPSCs [Figure 6E, karyotype formula: 35–41.X0,rob(6;6),+8,der(8)T(8;X),+17×2]. For the complete panel of karyotyping in all clones, see the table in Figure S4, as well as Figures S4A–S4K. All of these chromosomal aberrations arise specifically in Sirt1−/− cells during reprogramming, because the parental MEFs display a normal karyotype (Figures S4L and S4M).
SIRT1 Regulates TERRA Levels and Chromatin Remodeling Associated with iPSC Generation
Given its histone deacetylase activity, we next tested if SIRT1 deficiency or overexpression correlates with a deregulation in TERRA transcription associated with reprogramming. To assess this, we measured TERRA levels in Sirt1+/+ and Sirt1−/− iPSCs and their respective parental MEFs. Surprisingly, we observed an increase in baseline levels of TERRAs in Sirt1−/− MEFs, which did not increase upon reprogramming and compared to levels in Sirt1+/+ iPSCs (Figures 7A and 7B). On the other hand, Sirt1Super iPSCs exhibited a milder increase in telomeric transcription upon reprogramming, as compared to Sirt1+/+ iPSCs (Figures 7C and 7D). Thus, impaired TERRA transcription cannot explain decreased telomere elongation in Sirt1−/− iPSCs, and interestingly, our data are compatible with a role for SIRT1 in negatively regulating TERRA expression in differentiated and pluripotent cells.
Figure 7.
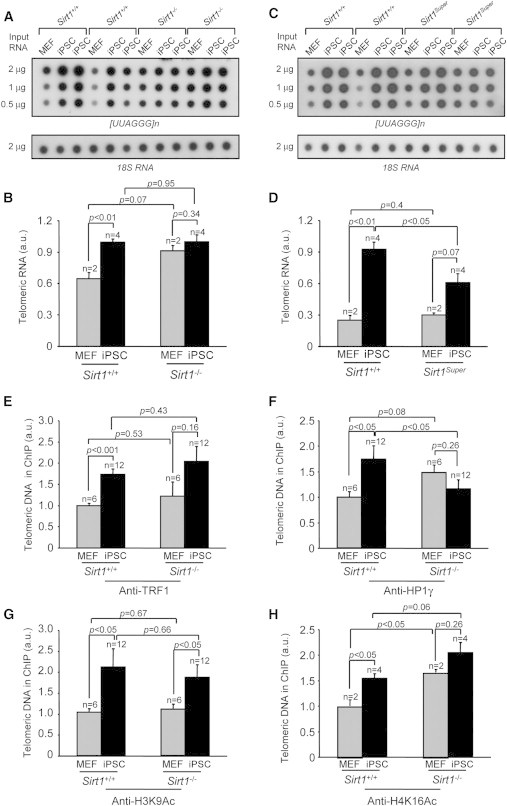
SIRT1 Is Involved in Regulation of TERRA Transcription and in the Reprogramming of Telomeric Chromatin
(A and C) Expression of TERRA in the indicated cells types as measured by dot blot hybridization with a [UUAGGG]n probe. 18S ribosomal subunit was used as a loading control.
(B and D) Quantification of TERRA levels reported in (A) and (C), respectively. Two independent MEFs per genotype (n = 2) and two independent iPSC clones per parental MEF (n = 4) were used. Error bars represent the SEM. Statistical analysis was performed using a two-tailed Student’s t test.
(E–H) Quantification of immunoprecipitated telomeric repeats with the antibodies indicated in each panel. Values were obtained after normalization to DNA input signal (anti-TRF1 and anti-HP1γ), to both DNA input signal and histone H3 abundance (anti-H3K9Ac), and to both DNA input signal and histone H4 abundance (anti-H4K16Ac). Error bars represent the SEM; n indicates the number of independent clones tested. Statistical analysis was performed using a two-tailed Student’s t test. MEF passage number = 3; iPSC passage number = 15.
To specifically address the possibility that SIRT1 may also play a role in the remodeling of telomere chromatin during reprogramming, we analyzed the abundance of different histone marks by chromatin immunoprecipitation. Dot blot analysis using a telomeric repeat-specific probe showed that TRF1 bound telomeres more abundantly in iPSCs compared to MEFs, independent of SIRT1 status (Figure 7E). We also observed a significant increase of HP1γ density at telomeres upon reprogramming, which did not occur in Sirt1−/− iPSCs (Figure 7F), consistent with our previous observations that SIRT1 deficiency in MEFs results in lower density of this mark (Palacios et al., 2010). Interestingly, two of the known targets of sirtuin deacetylase activity, namely H3K9Ac and H4K16Ac acetylation, were significantly increased in wild-type iPSCs compared to the parental MEFs (Figures 7G and 7H), consistent with the stronger expression of SIRT1 in iPSCs (Figure S1C). However, although H3K9Ac, which is mainly considered as a target of SIRT6 (Michishita et al., 2008), was similarly increased in Sirt1+/+ and Sirt1−/− iPSCs compared to the parental MEFs (Figure 7G), H4K16 acetylation was significantly more pronounced in Sirt1−/− MEFs and iPSCs than in the wild-type cells (Figure 7H). These results confirm H4K16Ac as a bona fide target of SIRT1; this mark is significantly elevated both in Sirt1−/− MEFs and iPSCs, suggesting a more “open” chromatin structure associated to SIRT1 deficiency, in line with increased TERRA transcription even in the absence of reprogramming. Of note, the more open chromatin landscape associated to SIRT1 deficiency is not accompanied by higher telomere elongation, supporting the idea that the chromatin conformation is a predisposing, but not determinant, factor for telomere elongation.
Discussion
SIRT1 is a multifunctional protein with roles in several key cellular processes (Haigis and Sinclair, 2010), and recent evidence has also implicated this protein in pluripotency. In particular, SIRT1 is highly expressed in ESCs and downregulated upon differentiation (Calvanese et al., 2010); furthermore, SIRT1 inhibits the p53-mediated repression of NANOG protein (Han et al., 2008). Here, we demonstrate that although SIRT1 upregulation occurs upon reprogramming (Figure S1C), it is not required for the process of reacquiring pluripotency itself. However, we have established a role for SIRT1 in postreprogramming events that are critical for the maintenance of iPSCs. In particular, we show here that Sirt1−/− iPSCs, although exhibiting pluripotency features (expression of stemness markers and the ability to form teratomas and contribute to the development of chimeras), fail to efficiently elongate telomeres after reprogramming and accumulate chromosomal aberrations upon long-term culture.
Telomere elongation is one of the hallmarks of the reprogramming process, and we previously showed that this is a two-step process: it starts during reprogramming and then continues while iPSCs have obtained full pluripotent status (Marion et al., 2009). In Sirt1−/− cells, the telomere elongation after reprogramming is significantly slower, which is consistent with our previous finding that SIRT1 is recruited to telomeres in iPSCs and positively regulates telomere length both in MEFs and tissues (Palacios et al., 2010). Although Sirt1−/− MEFs also exhibit a more pronounced telomere length attrition with cell generation, the role of SIRT1 in telomere elongation is more obvious during reprogramming, which involves a massive net telomere elongation; telomere length defect is indeed quite subtle in Sirt1−/− MEFs, and it is clearly appreciable only upon the screening of a high number of samples.
Importantly, we show here that the defective capacity of Sirt1−/− iPSCs to elongate telomeres after reprogramming is associated with decreased c-MYC protein and lower expression of telomerase, a known c-MYC target in both humans and mice (Flores et al., 2006b; Wang et al., 1998). These findings are consistent with published observations of the two effects independently in other cell lines. For example, a recent report showed that the small interfering RNA (siRNA)-mediated downregulation of Sirt1 in a human thyroid cancer cell line, though not affecting c-Myc transcription level, produces a robust reduction of c-MYC protein due to a decrease in its stability (Herranz et al., 2013).
The downregulation of c-MYC in SIRT1-deficient cells can also explain the apparent discrepancy between our results and a recent report showing that siRNA-mediated knockdown of SIRT1 affects reprogramming efficiency (Lee et al., 2012); the use of c-MYC as a reprogramming factor may add a further layer of variability in the expression of this protein, possibly impacting reprogramming efficiency.
Interestingly, although it has been described that Sirt1−/− MEFs show in certain conditions an increased proliferation capacity (Chua et al., 2005; Narala et al., 2008), Sirt1−/− iPSCs do not exhibit any proliferative advantage over their wild-type counterparts. However, when microinjected in athymic mice, they form teratomas that grow much faster than Sirt1+/+ xenografts, in accordance with the view of Sirt1 as a tumor suppressor gene. Consistently, Sirt1 knockdown in human colorectal carcinoma cells accelerates tumor xenograft formation (Kabra et al., 2009).
The higher proliferative rate of Sirt1−/− iPSCs in vivo correlates with a delay of differentiation: while forming teratomas, Sirt1−/− iPSCs retain longer their stemness properties, thus postponing ectodermal tissue specification. We could also confirm these findings in vitro, generating EBs from Sirt1+/+ and Sirt1−/− iPSCs. It has been previously described, in this respect, that SIRT1 is able to favor neuronal differentiation by repressing the Notch1 pathway (Hisahara et al., 2008) and promote keratinocyte differentiation in humans (Blander et al., 2009).
Furthermore, here we show that telomeric chromatin is not properly reprogrammed in SIRT1-deficient iPSCs, which show a higher than normal density of the H4K16Ac mark, characteristic of a more open chromatin structure. Of note, the evidence that Sirt1−/− iPSCs fail to elongate telomeres postreprogramming, despite the increased “accessibility” of telomeric chromatin, suggests that telomere elongation, at least in this case, depends primarily on telomerase availability and that a more open chromatin conformation does not compensate for a lower level of telomerase.
In agreement with the decreased deacetylation of telomeric chromatin, we also see increased TERRA transcription in Sirt1−/− parental MEFs. We could not detect a decrease of TERRA transcription in Sirt1−/− compared to Sirt1+/+ iPSCs, but we speculate here that we could probably detect it in Sirt1−/− iPSCs at late passages, when the defects induced by SIRT1 deficiency become more evident. These observations are particularly interesting, because they open the possibility that other mechanisms, besides the c-MYC-induced mTert downregulation, could contribute to slow down telomere lengthening in Sirt1−/− cells; mounting evidence, reviewed in Maicher et al. (2012), correlates increased transcription of TERRAs with telomere shortening. In Sirt1Super iPSCs, this correlation is already evident at passage 15.
Finally, it is also plausible that SIRT1 deficiency may impair proper telomere elongation in iPSCs through deregulation of telomere recombination in a telomerase-independent manner. Interestingly, recombination-mediated telomere elongation associated with the early stages of embryo development has been described (Liu et al., 2007).
In line with the role of SIRT1 in recombination, Sirt1−/− iPSCs show enhanced genome instability, as indicated by a significant increase in chromosome breaks and chromosomal/chromatid fusions in Sirt1−/− compared with Sirt1+/+ iPSCs. It is worth mentioning, in this respect, that siRNA depletion of TERRA has also been linked to an increase in aberrations in metaphase telomeres (Deng et al., 2009). Extensive genomic instability, leading to the appearance of chromosome breaks, unequal chromosome segregation, and aneuploidy, has also been reported both in SIRT1-deficient day 9.5 embryos and MEFs (Wang et al., 2008). Interestingly, we found that upon long-term culture, Sirt1−/− iPSCs, in addition to a few aneuploidies shared with the Sirt1+/+ iPSCs (mainly trisomy 8, described as one of the most recurrent numerical aberration in mouse ESCs/iPSCs; Chen et al., 2011; Liu et al., 1997), display a recurrent chromosome aberration, namely the Robertsonian translocation of chromosome 6. It would be interesting to investigate if this translocation is somehow positively selected, maybe because it engenders the dosage increase of genes conferring a growing advantage (Nanog, Kras, or Raf1, for instance).
Given the broad range of SIRT1 target proteins, many putative players could be possibly involved, directly or indirectly, in the abnormalities we observe in Sirt1−/− iPSCs. For example, SIRT1 deacetylates p53 (Han et al., 2008), preventing its transactivation and translocation into the nucleus, and in specific conditions positively regulates SIRT6, another member of mammalian sirtuin family, whose deficiency correlates with telomeric dysfunctions in human fibroblasts (Michishita et al., 2008). Deregulation of p53 and/or SIRT6 could result in genomic instability, telomere fragility, and abnormal chromatin structure. However, in Sirt1−/− iPSCs, we could not observe hyperexpression of p53 or downregulation of SIRT6.
In conclusion, this report demonstrates a role of SIRT1 deacetylase in the maintenance of “good-quality” iPSCs, with proper telomere elongation, TERRA transcription, telomeric chromatin remodeling, and genome integrity.
Experimental Procedures
Generation of Mouse iPSCs
Reprogramming of primary MEFs (passage 2) was performed as previously described (Marion et al., 2009), following modifications of a previous protocol (Blelloch et al., 2007). The efficiency of the reprogramming process was assessed 2 weeks postinfection by scoring, by means of an ImageJ software routine, the number of iPSC colonies obtained at day 14 postinfection, either alkaline phosphatase stained (Alkaline Phosphatase Detection kit, Millipore) or NANOG stained (antibody #8822 1:250, Cell Signaling). This count was normalized to the total number of cells infected, obtained by flow cytometry analysis of an identical sample infected with the three factors plus GFP. Colonies were picked after 2 weeks and expanded on feeder fibroblasts using standard procedures. All procedures performed on mice were revised and approved by the institutional ethical committee.
Differentiation Assay through EB Aggregation
Four clones of each Sirt1+/+ and Sirt1−/− iPSCs on feeder layers were trypsinized and resuspended at a density of 2.5 × 106/ml in LIF-free medium. A total of 100 hanging drops of 5,000 cells (20 μl) per clone were suspended for 48 hr and transferred to a nonadherent Petri dish. To perform immunohistochemistry, EBs were embedded in 5% gelatin and formalin fixed.
Histopathology and Immunohistochemistry
After excision, teratomas were fixed in formalin, embedded in paraffin and dissected at a thickness of 5 μm. For pathological examination sections were stained with hematoxylin and eosin, according to standard procedures. For immunohistochemical analysis, sections were processed with antibodies against mouse Phospho(Ser10)-histone 3 (Millipore, 06-570), OCT3/4 (Santa Cruz sc-9081), and NESTIN (Millipore MAB 353).
Quantification of Immunohistochemistry Staining
To quantify P-H3-positive cells in teratoma sections, ten pictures of random fields were taken from one section per clone (n = 3 per genotype) and the number of positive cells manually counted. To quantify the percentage of positive area in sections stained for OCT4 or NESTIN, one section of each teratoma (n = 3 per genotype) was divided into a sufficient number of pictures in order to cover it all (using the Pannoramic Viewer software). In every picture, the percentage of positive area was scored through an ImageJ software routine. The percentage of positive area was then calculated for the whole section.
To quantify the percentage of positive area in EB sections stained for OCT4 or NESTIN, three pictures per clone (n = 12 per genotype) were taken using the Pannoramic Viewer software. Positive area and total EB area were scored in every picture through an ImageJ software routine. Percentage of positive area was calculated per every picture and then grouped per genotype.
Coimmunoprecipitation c-MYC-FLAG-SIRT1
293T cells were transfected with 4 μg of murine Sirt1 or a FLAG-Tagged c-Myc-expressing vectors. After 72 hr, the cells were lysed, sonicated, and precleared. A total of 1 mg of every sample was immunoprecipitated with a SIRT1 antibody (Abcam ab12193), followed by the capture of the complex by a G-Ultralink Resin beads (Thermo Scientific, #53125) or an anti-FLAG M2 affinity gel (Sigma, #A2220).
Acknowledgments
We thank Rosa Serrano and Esther Collado for mouse care, Marta Cañamero for teratoma section analysis, Diego Megías for help with confocal microscopy, and Manuel Serrano for helpful discussion. M.L.D.B. holds a contract linked to Reprogramming in Cáncer and Regeneration project (ReCaRe, S2011/BMD-2303). Research in the Blasco lab was funded by European Research Council (ERC) project TEL STEM CELL (GA#232854), European Union FP7 projects 2007-A-20088 (MARK-AGE) and 2010-259749 (EuroBATS), Spanish Ministry of Economy and Competitiveness projects SAF2008-05384 and CSD2007-00017, Regional of Government of Madrid project S2010/BMD-2303 (ReCaRe), the AXA Research Fund (Life Risks Project), a Lilly 2010 Preclinical Biomedicine Research Award (Fundación Lilly, Spain), and Fundación Botín (Spain).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Azzalin C.M., Reichenbach P., Khoriauli L., Giulotto E., Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007;318:798–801. doi: 10.1126/science.1147182. [DOI] [PubMed] [Google Scholar]
- Benetti R., García-Cao M., Blasco M.A. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat. Genet. 2007;39:243–250. doi: 10.1038/ng1952. [DOI] [PubMed] [Google Scholar]
- Blander G., Bhimavarapu A., Mammone T., Maes D., Elliston K., Reich C., Matsui M.S., Guarente L., Loureiro J.J. SIRT1 promotes differentiation of normal human keratinocytes. J. Invest. Dermatol. 2009;129:41–49. doi: 10.1038/jid.2008.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blelloch R., Venere M., Yen J., Ramalho-Santos M. Generation of induced pluripotent stem cells in the absence of drug selection. Cell Stem Cell. 2007;1:245–247. doi: 10.1016/j.stem.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvanese V., Lara E., Suárez-Alvarez B., Abu Dawud R., Vázquez-Chantada M., Martínez-Chantar M.L., Embade N., López-Nieva P., Horrillo A., Hmadcha A. Sirtuin 1 regulation of developmental genes during differentiation of stem cells. Proc. Natl. Acad. Sci. USA. 2010;107:13736–13741. doi: 10.1073/pnas.1001399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S.R., Blackburn E.H. Telomeres and telomerase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004;359:109–121. doi: 10.1098/rstb.2003.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Shi X., Rudolph C., Yu Y., Zhang D., Zhao X., Mai S., Wang G., Schlegelberger B., Shi Q. Recurrent trisomy and Robertsonian translocation of chromosome 14 in murine iPS cell lines. Chromosome Res. 2011;19:857–868. doi: 10.1007/s10577-011-9239-y. [DOI] [PubMed] [Google Scholar]
- Cheng H.L., Mostoslavsky R., Saito S., Manis J.P., Gu Y., Patel P., Bronson R., Appella E., Alt F.W., Chua K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K.F., Mostoslavsky R., Lombard D.B., Pang W.W., Saito S., Franco S., Kaushal D., Cheng H.L., Fischer M.R., Stokes N. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- Deng Z., Norseen J., Wiedmer A., Riethman H., Lieberman P.M. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell. 2009;35:403–413. doi: 10.1016/j.molcel.2009.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores I., Benetti R., Blasco M.A. Telomerase regulation and stem cell behaviour. Curr. Opin. Cell Biol. 2006;18:254–260. doi: 10.1016/j.ceb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Flores I., Evan G., Blasco M.A. Genetic analysis of myc and telomerase interactions in vivo. Mol. Cell. Biol. 2006;26:6130–6138. doi: 10.1128/MCB.00543-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D., Valenti L., Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- Greider C.W., Blackburn E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Haigis M.C., Sinclair D.A. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M.K., Song E.K., Guo Y., Ou X., Mantel C., Broxmeyer H.E. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D., Maraver A., Cañamero M., Gómez-López G., Inglada-Pérez L., Robledo M., Castelblanco E., Matias-Guiu X., Serrano M. SIRT1 promotes thyroid carcinogenesis driven by PTEN deficiency. Oncogene. 2013;32:4052–4056. doi: 10.1038/onc.2012.407. [DOI] [PubMed] [Google Scholar]
- Hisahara S., Chiba S., Matsumoto H., Tanno M., Yagi H., Shimohama S., Sato M., Horio Y. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc. Natl. Acad. Sci. USA. 2008;105:15599–15604. doi: 10.1073/pnas.0800612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabra N., Li Z., Chen L., Li B., Zhang X., Wang C., Yeatman T., Coppola D., Chen J. SirT1 is an inhibitor of proliferation and tumor formation in colon cancer. J. Biol. Chem. 2009;284:18210–18217. doi: 10.1074/jbc.M109.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.S., Xiao C., Wang R.H., Lahusen T., Xu X., Vassilopoulos A., Vazquez-Ortiz G., Jeong W.I., Park O., Ki S.H. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.L., Peng Q., Fong S.W., Chen A.C., Lee K.F., Ng E.H., Nagy A., Yeung W.S. Sirtuin 1 facilitates generation of induced pluripotent stem cells from mouse embryonic fibroblasts through the miR-34a and p53 pathways. PLoS ONE. 2012;7:e45633. doi: 10.1371/journal.pone.0045633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Wu H., Loring J., Hormuzdi S., Disteche C.M., Bornstein P., Jaenisch R. Trisomy eight in ES cells is a common potential problem in gene targeting and interferes with germ line transmission. Dev. Dyn. 1997;209:85–91. doi: 10.1002/(SICI)1097-0177(199705)209:1<85::AID-AJA8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Liu L., Bailey S.M., Okuka M., Muñoz P., Li C., Zhou L., Wu C., Czerwiec E., Sandler L., Seyfang A. Telomere lengthening early in development. Nat. Cell Biol. 2007;9:1436–1441. doi: 10.1038/ncb1664. [DOI] [PubMed] [Google Scholar]
- Maicher A., Kastner L., Luke B. Telomeres and disease: enter TERRA. RNA Biol. 2012;9:843–849. doi: 10.4161/rna.20330. [DOI] [PubMed] [Google Scholar]
- Marion R.M., Strati K., Li H., Tejera A., Schoeftner S., Ortega S., Serrano M., Blasco M.A. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009;4:141–154. doi: 10.1016/j.stem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Martínez P., Thanasoula M., Muñoz P., Liao C., Tejera A., McNees C., Flores J.M., Fernández-Capetillo O., Tarsounas M., Blasco M.A. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E., Yellajoshula D., George E., Scambler P.J., Brown D.T., Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E., McCord R.A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T.L., Barrett J.C. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narala S.R., Allsopp R.C., Wells T.B., Zhang G., Prasad P., Coussens M.J., Rossi D.J., Weissman I.L., Vaziri H. SIRT1 acts as a nutrient-sensitive growth suppressor and its loss is associated with increased AMPK and telomerase activity. Mol. Biol. Cell. 2008;19:1210–1219. doi: 10.1091/mbc.E07-09-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer P., Michan S., McVay M., Mostoslavsky R., Vann J., Park S.K., Hartlerode A., Stegmuller J., Hafner A., Loerch P. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios J.A., Herranz D., De Bonis M.L., Velasco S., Serrano M., Blasco M.A. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 2010;191:1299–1313. doi: 10.1083/jcb.201005160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfluger P.T., Herranz D., Velasco-Miguel S., Serrano M., Tschöp M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R.P., Garrobo I., Foronda M., Palacios J.A., Marión R.M., Flores I., Ortega S., Blasco M.A. TRF1 is a stem cell marker and is essential for the generation of induced pluripotent stem cells. Nat. Commun. 2013;4:1946. doi: 10.1038/ncomms2946. [DOI] [PubMed] [Google Scholar]
- Schoeftner S., Blasco M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008;10:228–236. doi: 10.1038/ncb1685. [DOI] [PubMed] [Google Scholar]
- Sfeir A., Kosiyatrakul S.T., Hockemeyer D., MacRae S.L., Karlseder J., Schildkraut C.L., de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair D.A., Guarente L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Wang J., Xie L.Y., Allan S., Beach D., Hannon G.J. Myc activates telomerase. Genes Dev. 1998;12:1769–1774. doi: 10.1101/gad.12.12.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.H., Sengupta K., Li C., Kim H.S., Cao L., Xiao C., Kim S., Xu X., Zheng Y., Chilton B. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F., Zhang Q., Zhang K., Xie W., Grunstein M. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol. Cell. 2007;27:890–900. doi: 10.1016/j.molcel.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung F., Hoberg J.E., Ramsey C.S., Keller M.D., Jones D.R., Frye R.A., Mayo M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.