

Abstract
The efficacy of pEGFP (plasmid expressing enhanced green fluorescent protein)-encapsulated PEGylated (meaning polyethylene glycol coated) magnesium phosphate nanoparticles (referred to as MgPi-pEGFP nanoparticles) for the induction of immune responses was investigated in a mouse model. MgPi-pEGFP nanoparticles induced enhanced serum antibody and antigen-specific T-lymphocyte responses, as well as increased IFN-? and IL-12 levels compared to naked pEGFP when administered via intravenous, intraperitoneal or intramuscular routes. A significant macrophage response, both in size and activity, was also observed when mice were immunized with the nanoparticle formulation. The response was highly specific for the antigen, as the increase in interaction between macrophages and lymphocytes as well as lymphocyte proliferation took place only when they were re-stimulated with recombinant green fluorescence protein (rGFP). Thus the nanoparticle formulation elicited both humoral as well as cellular responses. Cytokine profiling revealed the induction of Th-1 type responses. The results suggest DNA-encapsulated magnesium phosphate (MgPi) nanoparticles may constitute a safer, more stable and cost-efficient DNA vaccine formulation.
Keywords: Inorganic phosphate nanoparticles, Microemulsion, pEGFP, Vaccine, Antigen-presenting cells
Graphical abstract
1. Introduction
DNA vaccines are promising vehicles for immunization against a variety of human pathogens, including HIV [1], Mycobacterium tuberculosis [2] and malarial parasites [3]. Such immunization with DNA can elicit both cellular and humoral immune responses [4,5], and can be administered repeatedly without inducing any anti-vector immunity. Other benefits of a DNA based vaccine include its ability to polarize T-cells, especially to a Th1 immunological response. DNA vaccine formulations are generally more stable and possess longer shelf-life, which in turn facilitates their cheaper manufacturing, storage, and shipping compared to that of protein-based vaccines. Nonetheless, the immunogenicity of DNA vaccines has been limited by several problems associated with their delivery, such as poor cellular uptake of DNA, degradation of the DNA by DNases and lysosomes, and transient DNA expression. A number of strategies have been used to improve their potency, including, electroporation, infusion, sonication and the gene gun [6,7]. Microparticles and nanoparticles that have been exploited as carriers for such DNAs include polylactidecoglycolide (PLGA) [8,9], alginate microparticles [10], chitosan nanoparticles [11,12], liposomes [13,14], and virosomes [15]. These methods are, however, not acceptable in practice because of a number of crucial limitations, including the requirement for large amounts of DNA, as well as their low expression levels and cytotoxicity. As a result, current non-viral genetic vaccine systems do not efficiently activate antigen-presenting cells (APCs) [16], and so lack the equivalent potency of viral vectors.
It has been suggested that the use of inorganic nanoparticles, such as phosphates of Ca2+, Mg2+, Mn2+, Ba2+, Sr2+, might eliminate these limitations, yet they remain largely unexplored. Bulk-precipitated complexes using these ions have been shown to stimulate varying degrees of DNA transfer efficiency across the cell membrane [17]. Calcium phosphate (CaPi) nanoparticles of average diameters greater than 400 nm have already been reported to serve as non-toxic, biocompatible carriers for DNA delivery [18,19] notwithstanding these particles are too large for efficient intracellular uptake. Our group has previously demonstrated the potential of ultra low size (<100 nm diameter) CaPi nanoparticles as efficient vectors for gene delivery in vitro [20–22]. Moreover, in relation to the induction of immune responses, it has been observed that smaller particles (<300 nm), when complexed with DNA, induced better immune responses than did larger microparticles (~1 µm) [23]; this could be partially attributed to the ability of smaller particles to be taken up more readily by APCs. There is also evidence that particle size plays a critical role in the transfer of nanoparticles in the lymphatic system [24,25]. Our observations of the greater transfection efficiency, in vitro as well as in vivo, of DNA-encapsulated ultra-low size magnesium phosphate nanoparticles [26,27] prompted us to further investigate the potential of these nanoparticles as DNA vaccine carriers.
Here, we report an investigation of the levels of immunogenicity triggered by either a naked pEGFP, or MgPi-pEGFP nanoparticles, via intramuscular (i.m.), intraperitoneal (i.p.) or intravenous administrations (i.v.) in BALB/c mice. The immune response to the expressed antigen was studied through a combination of antibody (IgG) titration, cytokine profile measurement, macrophage (antigen-presenting cell) activation, and lymphocyte proliferation upon in vitro re-stimulation with recombinant green fluorescence protein (rGFP). The immune response so induced was markedly superior to that triggered by either naked pEGFP.
2. Materials and methods
2.1. Materials
All reagents and chemicals were purchased from Sigma unless otherwise stated. Anti-mouse IgG antibody was obtained from Bangalore Genei, India. Interleukin-12 (IL-12) and Interferon-? (IFN-?) were procured from Promega, USA. pEGFP was a gift of Prof. Debi P. Sarcar, Department of Biochemistry, University of Delhi, India. Recombinant green fluorescence protein was a gift of Prof. Anirban Maitra, Department of Pathology, Johns Hopkins Medical Institute, Baltimore, USA.
2.2. Mice
Inbred strains of pathogen-free female BALB/c mice (6–8 weeks old; 20–25 g) were obtained from the Animal House Facility, Department of Zoology, University of Delhi, India. The animals were reared in uniform hygienic conditions under a controlled environment (at 20–25 °C and 12 h dark/light cycle) following the guidelines of the Animal Ethics Committee, University of Delhi, India. The animal experiments were also executed in strict accordance to guidelines approved by the Animal Ethics Committee of the university.
2.3. Preparation of pEGFP-encapsulated MgPi nanoparticles
pEGFP-encapsulated MgPi nanoparticles were prepared using a water-in-oil microemulsion method exactly as reported in our previous work [26,27]. Briefly, 25 ml of an AOT (Aerosol OT or sodium bis(2-ethylhexyl) sulfosuccinate) in hexane solution (0.1 M) was prepared, into which 70 µl of an aqueous solution of magnesium chloride (1.0 M) and 2.94 µg of pEGFP were dissolved by continuous stirring for 12 h to form microemulsion A. In another 25 ml of AOT in hexane solution, 70 µl of aqueous solution of (NH4)2HPO4 (1.0 M) and 2.94 µg of pEGFP, were dissolved by continuous stirring for 12 h to form microemulsion B. Additional buffer (0.1 M Tris HCl buffer, pH 8) was added to both microemulsions before stirring so that the aqueous volume in each microemulsion could reach 450 µl so as to adjust the Wo (the molar ratio of water to AOT) of each microemulsion to 10. Wo governs the size of aqueous core in such microemulsion systems and thus govern the size of the particle formed in these microemulsions. Both the microemulsions were optically clear solutions after 12 h stirring. Microemulsion B was then slowly added to microemulsion A at a rate of 4 ml/h with continuous stirring at 4 °C. The resulting solution was further stirred for another 12 h. The development of translucency indicated magnesium phosphate nanoparticle formation within its aqueous core. Dry ethanol (2 ml) was then added to break the microemulsion. The mixture was centrifuged for 30 min at 13,000 rpm at 4 °C. The pelleted nanoparticles were washed (4×) with 15 ml n-hexane and the particles dispersed in PBS (pH 7.2) by vortexing. The dispersed nanoparticles were dialyzed for 12 h in a 12 kD cut-off dialysis membrane bag to yield a clear dispersion. The dispersed nanoparticles were characterized by particle size determination. The void (placebo) nanoparticles were also prepared using exactly the same protocol without adding pEGFP solution.
2.4. Tagging of methoxy-PEGamine to pEGFP-encapsulated MgPi nanoparticles
In order to render the pEGFP-encapsulated MgPi nanoparticles long circulating inside the body upon their administration via the different routes, their surfaces were modified to acquire polyethylene glycol (PEG) terminals. This process is referred to as “PEGylation” of the surface. To obtain PEGylated nanoparticles, both void as well as pEGFP-encapsulated MgPi nanoparticles were first coated with the highly adhesive polymer, polyacrylic acid (PAA). Acid-coated MgPi nanoparticles were then conjugated with methoxy PEG-amine (Mol Wt 5000) to create the PEGylated nanoparticles. Briefly, a 10 ml dispersion of MgPi nanoparticles in PBS (pH 7.4) obtained from the above process was incubated with 10 µl of acid neutralized (pH 8) PAA (5 kD, 0.5% V/V) for 2–3 h with stirring, followed by a dialysis (12 kD membrane) to remove excess polymer. The carboxylate groups of PAA were conjugated to amine groups of methoxy PEG-amine using EDCI (1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride). Methoxy PEG-amine (50 µl of 40 mg/ml) was added to the nanoparticle suspension with continuous stirring and to this, 50 µl of EDCI (20 mg/ml) was added. Stirring was continued for 8 h, followed by 2–3 h of dialysis to remove all the unconjugated molecules. The particle size of these PEGylated nanoparticles was again measured by DLS to reconfirm whether the PEGylation process had caused any change in the nanoparticles sizes. Lyophilized product was stored at 4 °C until further use. The PEGylated nanoparticle formulation was readily dispersible in an appropriate injectable volume of PBS (pH 7.4). We refer pEGFP-encapsulated PEGylated MgPi nanoparticles to as MgPi-pEGFP nanoparticles in this study.
2.5. Determination of the size of the nanoparticles
The particle sizes of both the void as well as the pEGFP-encapsulated nanoparticles in water-in-oil microemulsions as well as in aqueous solutions were determined by a dynamic light scattering (DLS) technique. Briefly, the measurements were done with a Brookhaven BI8000 instrument fitted with a BI200SM goniometer. An argon-ion air-cooled laser was operated at 488 nm as the light source and the intensity of scattered light were recorded on a scattering angle of 90°. The time-dependent autocorrelation function was derived using a 136-channel digital photon correlator. The particle size was calculated from the auto correlation function using the Stokes–Einstein equation: d = kt/3p?D, where D is the translational diffusion coefficient, d is the particle diameter, ? is the viscosity of the liquid in which particles are suspended, k is Boltzmann’s constant and T is absolute temperature.
2.6. Entrapment efficiency (E%)
The pEGFP-encapsulated nanoparticles in AOT microemulsion were separated after ultracentrifugation (40,000 rpm for 4 h at 4 °C) and the pellet, after washing with hexane, was dissolved in acidic buffer (pH 3). The amount of DNA released from the nanoparticles, [DNA]r, was estimated spectrophotometrically by measuring the optical density at ?260nm. The entrapment efficiency (E) was then calculated from the amount of DNA originally added to the microemulsion ([DNA]0) using the equation E% = [DNA]r /[DNA]0 × 100.
2.7. Agarose gel electrophoresis of free, encapsulated, and adsorbed pEGFP
Agarose gels were used for electrophoresis. In order to demonstrate the encapsulation of pEGFP inside particles and its protection from external DNase, MgPi-pEGFP nanoparticles were run onto agarose gels (1%). Briefly, 10 µl of an aqueous dispersion of MgPi-pEGFP nanoparticles (5 mg/100 µl) solution was incubated with 5 µl of DNase1 (5 mg/ml in Tris buffer) for 15–20 min at 4–8 °C and was finally loaded onto a gel. As a control, the same volume of untreated nanoparticles containing the same amount of pEGFP was also loaded onto the gel. Naked pEGFP (2 µl of 0.5 µg/µl) was either loaded fresh or after incubation with placebo MgPi for adsorption onto particle surfaces overnight at 4–8 °C. In both cases, control experiments involving treatment with DNase1 for 30 min prior to loading were also conducted.
2.8. In vivo gene expression
Two groups of young BALB/c mice (n = 6) were injected with either 1.8 µg of naked pEGFP or 1.8 µg of pEGFP delivered via nanoparticle formulation. Both groups of mice were injected intraperitoneally. Seven days post-injection, mice were sacrificed and their lungs, livers, spleens and lymph nodes were harvested under aseptic conditions. The tissue extracts were prepared in PBS by homogenization and centrifugation (12,000 rpm/4 °C). The tissue homogenates were assayed for total protein using Lowry’s method. Each tissue was normalized for protein and assayed for the expressed green fluorescence protein (GFP) using fluorimeter (excitation filter 365 nm and emission filter 510 nm). The background fluorescence (RFU) obtained from the tissue homogenates of untreated mice was subtracted from the RFU obtained from each of the tissue homogenates of pEGFP treated mice.
2.9. Immunization
BALB/c mice were immunized three times each with the MgPi-pEGFP nanoparticle as well as with control particles and naked pEGFP at weeks 0, 2 and 4. Immunizations were carried out via three routes – intravenous, intraperitoneal or intramuscular. A total of 36 animals were immunized (12 animals per route; 6 with naked pEGFP and 6 with MgPi-pEGFP nanoparticles). The nanoparticle formulations (MgPi-pEGFP) were dispersed in phosphate buffer saline (pH 7.4) and injected (100 µl containing 0.6 µg pEGFP) into each mouse (total dose of 1.8 µg of pEGFP/animal with three injections at weeks 0, 2 and 4). Equivalent amounts of naked pEGFP were also injected in similar ways into animals as positive controls. A group of 6 mice were also injected in a similar way with void PEGylated MgPi nanoparticles to study the effect of nanoparticles themselves on mice.
2.10. Sera and tissue collection
Mice were bled through the retro-orbital plexus and the sera separated by centrifugation for immunoglobulin assessment. The liver, lung, thymus and spleen of these animals were carefully dissected out and then washed in sterile PBS for further studies.
2.11. Antibody assays
Serum anti-GFP antibody (IgG) titers were measured by ELISA using green fluorescence protein (GFP) as the solid phase antigen. Briefly, ELISA plates (96-well U bottom, Tarson, India) were coated with recombinant GFP overnight (200 µl of 5 µg/ml) and non-specific sites blocked with 5% bovine serum albumin in PBS. After washing twice with PBS/0.5% Tween-20, 100 µl of serum samples diluted in PBS were added to the wells. After overnight incubation at 4 °C, the plates were successively washed with Tween-20/PBS and incubated with 1:2000 dilutions of alkaline HRP-conjugated goat anti-mouse IgG antibodies(100 µl) for 2 h at room temperature. Plates were washed again and orthophenylenediamine dihydrochloride (OPD) in 0.05 M citrate buffer (pH 5.0, 100 µl of 1 mg/ml) and 2 µl 30% H2O2 were added. Absorbance at 490 nm was recorded after the addition of oxalic acid (10 µl) as stop solution.
2.12. Macrophage (antigen presenting cells) activation
Splenocytes, consisting of both macrophages and lymphocytes, were prepared from all the experimental mice using the standard protocol. Briefly, the spleens were dissected out and minced in PBS on a stainless steel mesh (~ 4 µm) to make single cell suspensions and then, upon centrifugation, the cells were collected and resuspended in complete RPMI media containing antibiotics (streptomycin and penicillin). The collected cells were then used to analyze the changes in the number of macrophages and their activations after the differing immunization protocols. For determining the change in the number of macrophages in spleens, 1 × 107 splenocytes were plated into 100 mm culture plates in complete RPMI and incubated at 37 °C. After 2 h of incubation, non-adherent cells were washed 3 times with PBS and the adherent cells (about 98% cells were macrophages based both on their morphology and non-specific esterase staining) were detached and counted using hemocytometer. To measure the difference in the activation of macrophages with differing immunization routes, we incubated the 1 × 106 splenocytes with or with rGFP for 24 h at 37 °C in a CO2 incubator. After 24 h, the non-adherent cells were washed and the adherent macrophages were analyzed for change in morphology and phagocytic activity.
2.13. Lymphocyte proliferation assay
Lymphocyte proliferation of the immunized mice was carried out using MTT colorimetric assay as previously described [28]. Splenic lymphocytes were prepared from all experimental mice using the standard protocol. Briefly, the spleens were dissected out and minced in PBS on a stainless steel mesh (~ 4 µm) to make a single cell suspension. The erythrocytes were lysed by 0.54% NH4Cl (pH 7.4). After centrifugation, the cells were re-suspended in complete RPMI media supplemented with antibiotics (streptomycin and penicillin) and 1 × 106 cells were seeded into each well of 96-well culture plates. rGFP (5 µg/ml) was used as a specific stimulating antigen. Wells without stimulating antigen were used as negative control. All the cells were cultured at 5% CO2 and 37 °C for 72 h. Two hour prior to termination, 20 µl MTT (5 mg/ml) was added into each well. After the appearance of purple formazan crystal, the culture plate was centrifuged. The supernatant was removed and the crystals solubilized in 100 µl of dimethyl sulphoxide (DMSO) and the absorbance measured at 570 nm to determine the stimulation index. All experiments were done in triplicate and repeated twice with 3 animals each.
2.14. Assays for IFN-? and IL-12
Splenocytes prepared from mice were seeded into 96-well culture plates (1 × 106 cells/well) in complete RPMI media supplemented with antibiotics. The splenocytes were cultured in the presence of rGFP for 24 h. The culture medium was collected and assayed for the presence of IFN-? and IL-12 by Sandwich ELISA, as per the protocol provided for each cytokine by BD Pharmingen (CA, USA).
2.15. Statistical analysis
The statistical significance of the different data points between naked pEGFP treated and MgPi-pEGFP nanoparticles treated mouse groups was determined using One-way ANOVA with Tukey post-hoc testing. This analysis was performed with SPSS software (version 13.0, SPSS Inc., Chicago, IL). In all cases, values represent mean ± S.D. (n = 6) and differences were considered significant at p < 0.05.
3. Results
3.1. Preparation and characterization of the nanoparticles
Void and pEGFP-encapsulated MgPi nanoparticles were formed in the aqueous core of the AOT/hexane microemulsion. The strategy involved the precipitation of the phosphate salts of magnesium in the absence or presence of pEGFP to obtain void or pEGFP-encapsulated MgPi nanoparticles respectively. The nanoparticle pellet obtained upon centrifugation of the microemulsion was easily dispersible in aqueous solution. The calculated loading/encapsulation efficiency (E%) as defined earlier was found to be nearly 99%. The mean size distributions of the MgPi-pEGFP nanoparticles was in the range of 30–50 nm in water-in-oil microemulsion and 110–130 nm in aqueous dispersion. The increase in sizes of nanoparticles in aqueous solution can be attributed to the slight aggregation of nanoparticles in aqueous media. A representative size distribution profile of MgPi-pEGFP nanoparticles is shown in Fig. 1. No differences between the sizes of the void and pEGFP-encapsulated MgPi nanoparticles were observed, indicating that DNA incorporation does not lead to an increase in particle size. These observations are also corroborated by our previous publication [26], so confirming the reproducibility of our fabrication and characterization methods. The PEGylation process did not contribute to any change in the particle sizes of void and pEGFP-encapsulated MgPi nanoparticles either (data not shown).
Fig. 1.
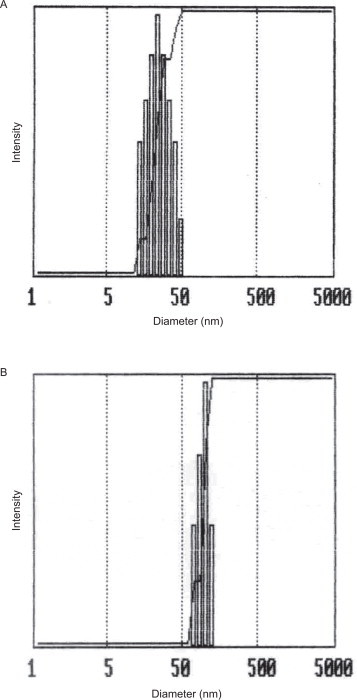
Representative particle size distribution or dynamic light scattering of MgPi-pEGFP nanoparticles in (A) water-in-oil microemulsion and (B) aqueous buffer.
3.2. Intracellular protection of pEGFP by nanoparticle matrices
To test whether the MgPi nanoparticles could protect encapsulated pEGFP from nuclease digestion, MgPi particles with encapsulated pEGFP were subjected to extensive DNase treatment before undergoing gel electrophoresis (Fig. 2). It was found that while naked pEGFP migrated to its usual position (lane 2), pEGFP encapsulated inside the nanoparticles remained at the top, and hardly entered into the gel (lane 4). Although DNase 1 completely digested the naked pEGFP, as demonstrated in lane 3, the pEGFP in MgPi nanoparticles was totally protected, as seen in lane 5. However, when the pEGFP was adsorbed only onto the surface of void nanoparticles, it migrated under the applied current almost like naked pEGFP (lane 6), becoming completely degraded by DNase as seen in lane 7. This was expected, as nanoparticle surfaces clearly do not offer enough protection in and of themselves. These results clearly demonstrate that DNA that is completely encapsulated within the rigid matrix of the magnesium phosphate nanoparticles is offered significant protection. This result confirms our previous results, obtained both with this nanoparticle [26] as well as with CaPi nanoparticles [21,22].
Fig. 2.
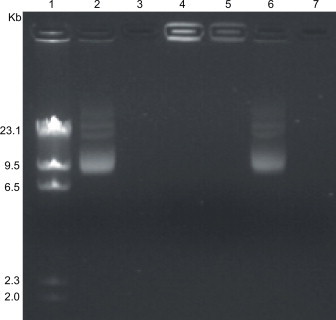
Photomicrograph of agarose (1%) gels revealing free, encapsulated and adsorbed pEGFP. Lane 1: after digestion with HindIII enzyme. Lane 2: Free pEGFP. Lane 3: pEGFP treated with DNase I. Lane 4: MgPi-pEGFP nanoparticles. Lane 5: MgPi-pEGFP nanoparticles treated with DNase I. Lane 6: pEGFP adsorbed on void magnesium phosphate nanoparticles. Lane 7: pEGFP adsorbed on void magnesium phosphate nanoparticles and then treated with DNaseI.
3.3. In vivo green fluorescence protein (GFP) expression
To test the utility of MgPi nanoparticle-mediated gene delivery in vivo, both generally and to specific organs in particular, immature BALB/c mice were injected with MgPi-pEGFP nanoparticles and the expression of green fluorescence protein within different body tissues measured (Fig. 3). GFP expression was observed in all the major tissues of the body, but especially in the immunologically-key spleen and lymph nodes. The level of GFP expression for all tissues examined was greater for nanoparticle-mediated delivery than after naked pEGFP administration, probably due to the protection from DNase degradation. Interestingly, the nanoparticle-mediated GFP expression was significantly higher (p < 0.05) in spleen, lungs, and lymph nodes. The highest GFP expression was observed in liver.
Fig. 3.
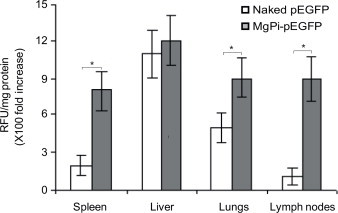
Expression of green fluorescence protein (GFP) in various body tissues. Seven days after intraperitoneal administration of MgPi-pEGFP nanoparticles encapsulating 1.8 µg pEGFP in mice, various body tissues were excised and processed to estimate the amount of GFP expressed. Values represent mean ± S.D. (n = 6). *P < 0.05, significantly different when compared to naked pEGFP.
3.4. Enhanced antibody response to pEGFP delivered via MgPi-pEGFP nanoparticles
Enhanced green fluorescent protein (EGFP) is a marker gene and it has been previously reported to have immunogenic potential [29,30] with an advantage of being traced via multiple techniques. Thus in order to evaluate the efficacy of MgPi as a novel carrier for delivery of DNA vaccine we opted to use pEGFP. The MgPi-pEGFP nanoparticles induced significant antibody responses in BALB/c mice when they were immunized either intravenously, intraperitoneally or intramuscularly (Fig. 4). Mice immunized i.p and i.v. produced higher titers of anti-GFP IgG than those immunized i.m. The MgPi-pEGFP nanoparticles yielded a 1000–5000-fold increase in the antibody titers in the case of intravenous immunization, and only a 100–500-fold in the case of intraperitoneal immunization. But, there was little increase between antibody titers of MgPi-pEGFP nanoparticles and naked pEGFP when injected into muscle.
Fig. 4.
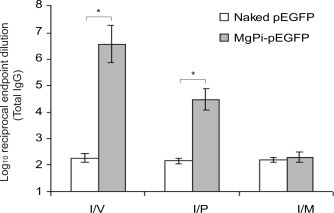
Serum GFP-specific total IgG titer following intravenous, intraperitoneal or intramuscular administration of naked pEGFP and MgPi-pEGFP nanoparticles. Values represent mean ± S.D. (n = 6). *P < 0.05, significantly different when compared to naked pEGFP.
3.5. Macrophage activation
Antigen presenting cells play a pivotal role in induction of immune response. Since uptake of vaccine and presentation of expressed protein is key to the success of immunization. We next examined changes in macrophage activity after immunization with the MgPi-pEGFP nanoparticles, naked pEGFP and void MgPi vectors. There was an increase in the overall number of macrophages (APCs) in spleens of mice immunized with the MgPi-pEGFP vector, compared to those after immunization with naked pEGFP or those in the unimmunized (control with void PEGylated MgPi) mice (Fig. 5A). Immunization via i.v. and i.p. administration was more efficient than via i.m. administration. Upon i.v. administration, the nanoparticles induced significantly more macrophages (p < 0.05) than the naked pEGFP or control treatments. Upon i.p. administration, the nanoparticles induced significantly more macrophages (p < 0.05) than that only of the control group. As shown in Fig. 5B the macrophage obtained from mice immunized with MgPi-pEGFP via i.v. route were much enlarged in its size, a hallmark of their activation. Additionally, we also observed increased phagocytic activity in macrophage from MgPi-pEGFP immunized mice compared to those from immunized with naked pEGFP or control mice.
Fig. 5.
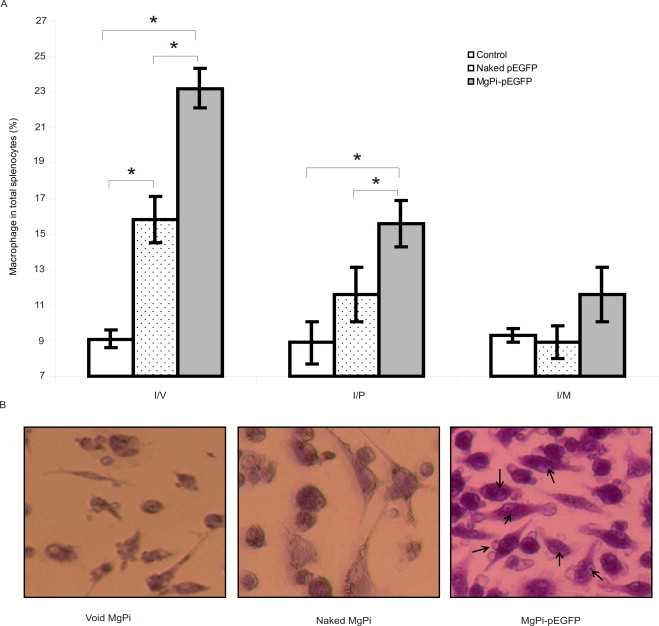
(A) Macrophages (%) in the splenocytes obtained from harvested spleens of control, naked pEGFP or MgPi-pEGFP administered mice. Values represent mean ± S.D. (n = 6). *P < 0.05, significantly different when compared to naked pEGFP as well as control. (B) Photomicrograph for macrophage activation. Macrophages were obtained from the spleen of control and pEGFP (naked or MgPi-encapsulated) i.v. immunized mice and co-cultured with lymphocytes from the same mice in presence of rGFP. Arrows indicating phagocytosis of dead cells (200× magnification, Nikon 100 microscope).
3.6. Lymphocyte proliferation
To check the specificity of the immune response generated in the mice, we re-stimulated the lymphocytes collected from immunized mice with rGFP in vitro and looked for increases in their proliferation. Fig. 6 reveals a significantly (p < 0.05) enhanced proliferation of splenic lymphocytes obtained from mice immunized with MgPi-pEGFP vector upon re-stimulation with rGFP as compared to those obtained from mice immunized with naked pEGFP or from unimmunized control mice. Immunization with MgPi-pEGFP nanoparticles led to greater lymphocyte proliferation via all routes of immunization, albeit not so pronounced as in the case of the i.m. route.
Fig. 6.
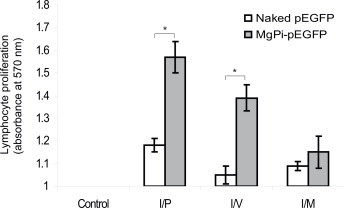
Analysis of cell proliferation upon re-challenge with recombinant GFP of lymphocytes obtained from control, naked pEGFP or MgPi-pEGFP administered mice. Values represent mean ± S.D. (n = 6). *P < 0.05, significantly different when compared to naked pEGFP.
3.7. Cytokine production
The production of the cytokines IFN-? and IL-1 by in vitro splenocytes isolated from immunized mice that had been re-stimulated with recombinant green fluorescence protein (rGFP) antigen are shown in Fig. 7. The nanoparticles delivered i.v. and i.p. triggered significantly (p < 0.05) more IFN-? and IL-12 than immunization with naked pEGFP or the control mice. However, no appreciable production of either of these cytokines was observed when the nanoparticles were delivered i.m. However, when administered i.m., the naked pEGFP resulted in more cytokines than the nanoparticles.
Fig. 7.
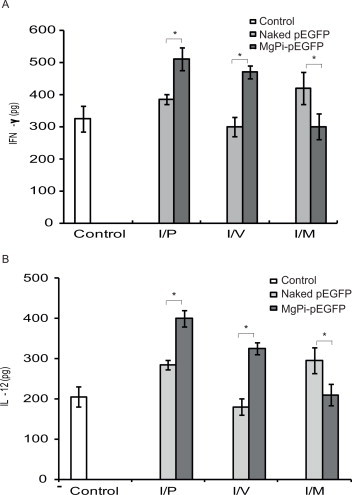
In vitro release of (A) IFN-? and (B) IL-12 by stimulated splenocytes. Splenocytes isolated from experimental animals (control, pEGFP, or MgPi-pEGFP injected) were stimulated with recombinant green fluorescence protein (rGFP) antigen for 24 h. Culture supernatants were collected and assayed for presence of (A) IFN-? and (B) IL-12 using sandwich ELISA kit from BD Pharmingen, USA. Values represent mean ± S.D. (n = 6). *P < 0.05, significantly different when compared to naked pEGFP as well as control.
4. Discussion
This study serves to demonstrate that inorganic phosphate nanoparticles such as magnesium phosphate can serve not only as an efficient DNA delivery system, but also act as potent adjuvants for the induction of effective DNA vaccine immune responses. Although an array of microparticles and nanoparticles have shown potential as pDNA delivery systems for the boosting of immune responses, MgPi nanoparticles appear to offer significant advantages from the point of view of both efficacy and toxicity. In a previous study, we have shown these nanoparticles demonstrate high transfection efficiency [26], and did not show any cytotoxicity in cell culture assays [27]. They triggered no observable adverse effects when injected into mice. As an important constituent of viable bone substitutes, as well as an important and normal normal tissue constituent in vivo [31,32], magnesium hydroxyapatite has long been shown to be biocompatible, and is regarded as very safe for human use. Magnesium phosphate is also in the FDA’s GRAS list [33].
Due to the low transfection rates elicited by other particulate carriers, high doses of DNA have usually been required to trigger sufficient immunization. Effective induction of robust T-cell responses are generally only achieved with a minimum of 50–200 µg doses of DNA [34,35], as seen in the recent study by Meerak et al., wherein they immunized Balb/c mice with 50 µg DNA together with chitosan nanoparticles [35]. However here, in the case of magnesium phosphate nanoparticles, the total effective doses of DNA administered to animals were as small as 1–2 µg, most probably due to the very high transfection efficiency, which was comparable to that of Polyfect® [26].
This study also provides several other improvements and advantages for genetic immunization: the MgPi-pEGFP nanoparticles are smaller in size than particles used in previous studies, which were either larger than 300 nm [35] or in the micron range [23]. The average diameter of the nanoparticles in this study was considerably less than 150 nm, and was thus ideal for engaging the clathrin-coated pit pathway for entry into the cytosol and endosomal compartments [36–44].
These nanoparticles were also able to provide a very high level of protection for DNA from degradation (Fig. 2), which is crucial for efficacious genetic therapy. Naked DNA is highly prone to extracellular and intracellular nuclease attack, a major challenge for efficient DNA transfection both in vitro and in vivo. Lechardeur et al. [45] showed that naked DNA microinjected into the cytoplasm of HeLa and COS-cells is degraded by cytosolic nucleases. Co-injected TRITC-dextran spread throughout the cytosol, but naked plasmid DNA progressively disappeared from the cytoplasm with a half life of 90 min. They concluded that protection of DNA from endonucleases, either by complexing or encapsulating it was necessary. However all subsequent vectors have been able to offer only partial in vivo protection.
EGFP is a commonly used reporter protein used in diverse array of scientific disciplines, for its ease of detection. In addition, previous studies have identified an immunodominant H2-Kd restricted CTL epitope present withing EGFP protein recognized by Balb/c mice, making it a suitable candidate for evaluation of vaccine- induced immune response [29]. Thus in order evaluate the efficacy of Mg-Pi nanoparticles as an optimal carrier for DNA vaccine we preferred pEGFP. In our previous study we have reported that MgPi nanoparticles shows comparable or may be better transfection efficiency in MCF-7, U87, Hela, COS-7 cells than the commonly used PolyFect reagent [26,27]. And it also has the advantage of being highly biocompatible and non-toxic.
The surfaces of the MgPi nanoparticles can be easily modified for prolonged DNA retention and circulation, and thus expression. The GFP expression in the various harvested tissues clearly demonstrated that DNA encapsulated in MgPi nanoparticles could efficiently traverse all paths to reach their respective cellular sites without degradation. The small size of the particles also facilitated their efficient uptake by macrophages, as demonstrated both by the increased expression of GFP in the spleen, as well as the increase in the number of macrophages in the spleens of mice immunized with MgPi-pEGFP (Fig. 3). However, the high GFP expression in liver could be due to the fact that a large proportion of the particles, as well as the pEGFP, was taken up by liver via parenchymal cells rather than macrophages [34].
MgPi nanoparticles are also clearly capable of inducing potent adjuvant effects for antibody induction against encoded protein, so facilitating protection against pathogen challenge. The antibody response triggered by the encapsulated pEGFP is many fold-higher than for naked pEGFP, especially when administered via i.v and i.p routes. The modest cellular and humoral immune response triggered by intramuscularly injected DNA has also been remarked on previously [46]. Cherif et al. [47] studied the immunogenicity of novel nanoparticle-coated MSP-1 C-terminus malaria DNA vaccine using different routes of administration and they also highlighted that the better protection was observed in the following order: i.p. > i.v. > s.c. Various studies, using the same formulation, have demonstrated that route of injection influenced the immune response. However, in the larger number of studies that have evaluated DNA-based immunization, only few have directly compared the immune responses generated by different routes of delivery. Although the mechanism is not clearly understood, we hypothesize that the better response in case of i.p. and i.v. over i.m. immunization with MgPi-pEGFP could be because, for these routes, there is comparatively greater opportunity for the macrophages to ingest the MgPi-pEGFP particles. It might also be because of the poor distribution, inefficient expression or rapid degradation of intramuscularly injected DNA [48].
The MgPi-pEGFP nanoparticles might also be activating macrophages or antigen presenting cells (APCs) upon immunization via the i.v. and i.p. routes. The poor macrophage response in the case of the intramuscular route might be due to the poor uptake of the nanoparticle formulation by the macrophages in this tissue. Further, the enhanced lymphocyte proliferation seen upon re-challenge with rGFP corroborates the idea that the response generated is specific against the antigen expressed by the pEGFP. Increases in lymphocyte proliferation and enhanced APC activity take place only when they are re-stimulated with specific antigen, such as the rGFP here. The enhanced cellular response is also documented in the cytokine profiles, which indicated a better induction of Th-1 type responses.
5. Conclusions
The MgPi-pEGFP vaccine is expressed in all the major tissues of the body, but especially in the immunologically relevant spleen and thymus. It elicit both humoral (as confirmed by increases in antibody titer), as well as cell-mediated responses (as demonstrated by lymphocyte proliferation). The cytokine study suggests a better induction of Th-1 type responses upon nanoparticle-mediated delivery of DNA, and the increased lymphocyte proliferation upon re-challenge with antigen confirmed the specificity of the response. Intravenous and intraperitoneal routes of administration were superior to intramuscular routes, as indicated by immunoglobulin assays, lymphocyte proliferation and APC activation studies. Thus, magnesium phosphate nanoparticles show great promise as efficient carriers for DNA, as well as for effective immunization with encoded protein.
Acknowledgments
The authors acknowledge the financial assistance in the form of a Research Project from the Department of Science and Technology, Government of India and University of Delhi for DU-DST PURSE Grant. One of the authors (ANM) thanks Indian National Science Academy, New Delhi for INSA Senior Scientist Award. We also thank Rajiv Ranjan and Beena Bhandari for their kind assistance with the animal experiments.
Contributor Information
Gajadhar Bhakta, Email: gajadhar.bhakta@imb.a-star.edu.sg.
Anju Shrivastava, Email: ashrivastava@zoology.du.ac.in.
References
- 1.Mascola J.R., Stiegler G., VanCott T.C., Katinger H., Carpenter C.B., Hanson C.E. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nature Medicine. 2000;6:207–210. doi: 10.1038/72318. 10655111 [DOI] [PubMed] [Google Scholar]
- 2.Flynn J., Triebold K., Koller B., Bloom B. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:12013–12017. doi: 10.1073/pnas.89.24.12013. 1465432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bojang K.A., Milligan P.J., Pinder M., Vigneron L., Alloueche A., Kester K.E. Efficacy of RTS, S/AS02 malaria vaccine against Plasmodium falciparum infection in semi-immune adult men in The Gambia:a randomised trial. Lancet. 2001;358:1927–1934. doi: 10.1016/S0140-6736(01)06957-4. 11747915 [DOI] [PubMed] [Google Scholar]
- 4.Tyagi R.K., Garg N.K., Sahu T. Vaccination strategies against malaria: novel carrier(s) more than a tour de force. Journal of Controlled Release: Official Journal of the Controlled Release Society. 2012;162:242–254. doi: 10.1016/j.jconrel.2012.04.037. 22564369 [DOI] [PubMed] [Google Scholar]
- 5.Kathuria N., Kraynyak K.A., Carnathan D., Betts M., Weiner D.B., Kutzler M.A. Generation of antigen-specific immunity following systemic immunization with DNA vaccine encoding CCL25 chemokine immunoadjuvant. Human Vaccines andImmunotherapeutics. 2012;8:1607–1619. doi: 10.4161/hv.22574. 23151454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heller L.C., Ugen K., Heller R. Electroporation for targeted gene transfer. Expert Opinion on Drug Delivery. 2005;2:255–268. doi: 10.1517/17425247.2.2.255. 16296752 [DOI] [PubMed] [Google Scholar]
- 7.Herweijer H., Wolff J.A. Progress and prospects: naked DNA gene transfer and therapy. Gene Therapy. 2003;10:453–458. doi: 10.1038/sj.gt.3301983. 12621449 [DOI] [PubMed] [Google Scholar]
- 8.Kim I.S., Lee S.K., Park Y.M., Lee Y.B., Shin S.C., Lee K.C. Physicochemical characterization of poly(L-lactic acid) and poly(D,L-lactide-co-glycolide) nanoparticles with polyethylenimine as gene delivery carrier. International Journal of Pharmaceutics. 2005;298:255–262. doi: 10.1016/j.ijpharm.2005.04.017. 15941631 [DOI] [PubMed] [Google Scholar]
- 9.Kumar M., Mohapatra S.S., Kong X., Jena P.K., Bakowsky U., Lehr C.M. Cationic poly(lactide-co-glycolide) nanoparticles as efficient in vivo gene transfection agents. Journal of Nanoscience and Nanotechnology. 2004;4:990–994. doi: 10.1166/jnn.2004.130. 15656192 [DOI] [PubMed] [Google Scholar]
- 10.Chuang V.T.G., Kragh-Hansen U., Otagiri M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharmaceutical Research. 2002;19:569–577. doi: 10.1023/a:1015396825274. 12069157 [DOI] [PubMed] [Google Scholar]
- 11.Cambridge C.D., Singh S.R., Waffo A.B., Fairley S.J., Dennis V.A. Formulation, characterization, and expression of a recombinant MOMP Chlamydia trachomatis DNA vaccine encapsulated in chitosan nanoparticles. International Journal of Nanomedicine. 2013;8:1759–1771. doi: 10.2147/IJN.S42723. 23690681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eroglu E., Tiwari P.M., Waffo A.B., Miller M.E., Vig K., Dennis V.A. A nonviral pHEMA+chitosan nanosphere-mediated high-efficiency gene delivery system. International Journal of Nanomedicine. 2013;8:1403–1415. doi: 10.2147/IJN.S43168. 23610520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrillo C., Sanchez-Hernandez N., Garcia-Montoya E., Perez-Lozano P., Sune-Negre J.M., Tico J.R. DNA delivery via cationic solid lipid nanoparticles (SLNs) European Journal of Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical Sciences. 2013;49:157–165. doi: 10.1016/j.ejps.2013.02.011. 23454134 [DOI] [PubMed] [Google Scholar]
- 14.Saljoughian N., Zahedifard F., Doroud D., Doustdari F., Vasei M., Papadopoulou B. Cationic solid–lipid nanoparticles are as efficient as electroporation in DNA vaccination against visceral leishmaniasis in mice. Parasite Immunology. 2013;35:397–408. doi: 10.1111/pim.12042. 23710803 [DOI] [PubMed] [Google Scholar]
- 15.Gluck R., Moser C., Metcalfe I.C. Influenza virosomes as an efficient system for adjuvanted vaccine delivery. Expert Opinion on Biological Therapy. 2004;4:1139–1145. doi: 10.1517/14712598.4.7.1139. 15268680 [DOI] [PubMed] [Google Scholar]
- 16.McKeever U., Barman S., Hao T., Chambers P., Song S., Lunsford L., Hsu Y.Y., Roy K., Hedley M.L. Protective immune responses elicited in mice by immunization with formulations of poly(lactide-co-glycolide) microparticles. Vaccine. 2002;20:1524–1531. doi: 10.1016/s0264-410x(01)00509-6. 11858858 [DOI] [PubMed] [Google Scholar]
- 17.Shack J., Thompsett J.M. The interaction of ions and desoxypentose nucleic acid of calf thymus. Journal of Biological Chemistry. 1953;203:373–387. 13069521 [PubMed] [Google Scholar]
- 18.He Q., Mitchell A., Morcol T., Bell S.J.D. Calcium phosphate nanoparticles induce mucosal immunity and protection against herpes simplex virus type 2. Clinical and Diagnostic Laboratory Immunology. 2002;9:1021–1024. doi: 10.1128/CDLI.9.5.1021-1024.2002. 12204953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Q., Johnson S., Lagner-Bartak C.W., Morcol T., Bell S.J.D. Calcium phosphate nanoparticles as adjuvant. Clinical and Diagnostic Laboratory Immunology. 2000;7:899. doi: 10.1128/cdli.7.6.899-903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maitra A. Calcium phosphate nanoparticles: second-generation nonviral vectors in gene therapy. Expert Review of Molecular Diagnostics. 2005;5:893–905. doi: 10.1586/14737159.5.6.893. 16255631 [DOI] [PubMed] [Google Scholar]
- 21.Bisht S., Bhakta G., Mitra S., Maitra A. pDNA loaded calcium phosphate nanoparticles: highly efficient non-viral vector for gene delivery. International Journal of Pharmaceutics. 2005;288:157–168. doi: 10.1016/j.ijpharm.2004.07.035. 15607268 [DOI] [PubMed] [Google Scholar]
- 22.Roy I., Mitra S., Maitra A., Mozumdar S. Calcium phosphate nanoparticles as novel non-viral vectors for targeted gene delivery. International Journal of Pharmaceutics. 2003;250:25–33. doi: 10.1016/s0378-5173(02)00452-0. 12480270 [DOI] [PubMed] [Google Scholar]
- 23.Singh M., Briones M., Ott G., O’Hagan D. Cationic microparticles:a potent delivery system for DNA vaccines. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:811–816. doi: 10.1073/pnas.97.2.811. 10639162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moghimi S.M., Hawley A.E., Christy N.M., Gray T., Illum L., Davis S.S. Surface engineered nanospheres with enhanced drainage into lymphatics and uptake by macrophages of the regional lymph nodes. FEBS Letters. 1994;344:25–30. doi: 10.1016/0014-5793(94)00351-3. 8181558 [DOI] [PubMed] [Google Scholar]
- 25.Hawley A.E., Illum L., Davis S.S. Lymph node localisation of biodegradable nanospheres surface modified with poloxamer and poloxamine block co-polymers. FEBS Letters. 1997;400:319–323. doi: 10.1016/s0014-5793(96)01408-1. 9009222 [DOI] [PubMed] [Google Scholar]
- 26.Bhakta G., Mitra S., Maitra A. DNA encapsulated magnesium and manganous phosphate nanoparticles: potential non-viral vectors for gene delivery. Biomaterials. 2005;26:2157–2163. doi: 10.1016/j.biomaterials.2004.06.039. 15576191 [DOI] [PubMed] [Google Scholar]
- 27.Bhakta G., Shrivastava A., Maitra A. Magnesium phosphate nanoparticles can be efficiently used in vitro and in vivo as non-viral vectors for targeted gene delivery. Journal of Biomedical Nanotechnology. 2009;5:106–114. doi: 10.1166/jbn.2009.029. 20055113 [DOI] [PubMed] [Google Scholar]
- 28.Zhao K., Shi X., Zhao Y., Wei H., Sun Q., Huang T. Preparation and immunological effectiveness of a swine influenza DNA vaccine encapsulated in chitosan nanoparticles. Vaccine. 2011;29:8549–8556. doi: 10.1016/j.vaccine.2011.09.029. 21945253 [DOI] [PubMed] [Google Scholar]
- 29.Gambotto A., Dworacki G., Cicinnati V., Kenniston T., Steitz J., Tuting T. Immunogenicity of enhanced green fluorescent protein (EGFP) in BALB/c mice: identification of an H2-Kd-restricted CTL epitope. Gene Therapy. 2000;7:2036–2040. doi: 10.1038/sj.gt.3301335. 11175316 [DOI] [PubMed] [Google Scholar]
- 30.Steitz J., Soloff A.C., Barratt-Boyes S.M., Alber S.M., Watkins S.C., Okada H. BALB/c EGFP mice are tolerant against immunization utilizing recombinant adenoviral-based vectors encoding EGFP:a novel model for the study of tolerance mechanisms and vaccine efficacy. Molecular immunology. 2010;47:1149–1153. doi: 10.1016/j.molimm.2009.11.018. 20022379 [DOI] [PubMed] [Google Scholar]
- 31.Landi E., Logroscino G., Proietti L., Tampieri A., Sandri M., Sprio S. Biomimetic MG-substituted hydroxyapatite: from synthesis to in vivo behaviour. Journal of Materials Science: Materials in Medicine. 2008;19:239–247. doi: 10.1007/s10856-006-0032-y. 17597369 [DOI] [PubMed] [Google Scholar]
- 32.Ryu H.S., Hong K.S., Lee J.K., Kim D.J., Lee J.H., Chang B.S. Magnesia-doped HA/beta-TCP ceramics and evaluation of their biocompatibility. Biomaterials. 2004;25:393–401. doi: 10.1016/s0142-9612(03)00538-6. 14585687 [DOI] [PubMed] [Google Scholar]
- 33.http://www.fda.gov/Food/IngredientsPackagingLabeling/GRAS/SCOGS/ucm261491.htm#.
- 34.Baldwin S.L., D’Souza C.D., Orme I.M., Liu M.A., Huygen K., Denis O. Immunogenicity and protective efficacy of DNA vaccines encoding secreted and non-secreted forms of Mycobacterium tuberculosis Ag85A. Tubercle and Lung Disease: the Official Journal of the International Union Against Tuberculosis and Lung Disease. 1999;79:251–259. doi: 10.1054/tuld.1998.0196. 10692994 [DOI] [PubMed] [Google Scholar]
- 35.Meerak J., Wanichwecharungruang S.P., Palaga T. Enhancement of immune response to a DNA vaccine against Mycobacterium tuberculosis Ag85B by incorporation of an autophagy inducing system. Vaccine. 2013;31:784–790. doi: 10.1016/j.vaccine.2012.11.075. 23228812 [DOI] [PubMed] [Google Scholar]
- 36.Hommelgaard A.M., Roepstorff K., Vilhardt F., Torgersen M.L., Sandvig K., van Deurs B. Caveolae: stable membrane domains with a potential for internalization. Traffic. 2005;6:720–724. doi: 10.1111/j.1600-0854.2005.00314.x. 16101676 [DOI] [PubMed] [Google Scholar]
- 37.Heath W.R., Carbone F.R. Cross-presentation, dendritic cells, tolerance and immunity. Annual Review of Immunology. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. 11244030 [DOI] [PubMed] [Google Scholar]
- 38.Houde M., Bertholet S., Gagnon E., Brunet S., Goyette G., Laplante A. Phagosomes are competent organelles for antigen cross-presentation. Nature. 2003;425:402–406. doi: 10.1038/nature01912. 14508490 [DOI] [PubMed] [Google Scholar]
- 39.Johannes L., Lamaze C. Clathrin-dependent or not: is it still the question? Traffic. 2002;3:443–451. doi: 10.1034/j.1600-0854.2002.30701.x. 12047552 [DOI] [PubMed] [Google Scholar]
- 40.Aderem A., Underhill D.M. Mechanisms of phagocytosis in macrophages. Annual Review of Immunology. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. 10358769 [DOI] [PubMed] [Google Scholar]
- 41.Rock K.L., Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunological Reviews. 2005;207:166–183. doi: 10.1111/j.0105-2896.2005.00301.x. 16181335 [DOI] [PubMed] [Google Scholar]
- 42.Gamvrellis A., Leong D., Hanley J.C., Xiang S.D., Mottram P., Plebanski M. Vaccines that facilitate antigen entry into dendritic cells. Immunology and Cell Biology. 2004;82:506–516. doi: 10.1111/j.0818-9641.2004.01271.x. 15479436 [DOI] [PubMed] [Google Scholar]
- 43.Foti M., Granucci F., Ricciardi-Castagnoli P. A central role for tissue-resident dendritic cells in innate responses. Trends in Immunology. 2004;25:650–654. doi: 10.1016/j.it.2004.10.007. 15530834 [DOI] [PubMed] [Google Scholar]
- 44.Xiang S.D., Scholzen A., Minigo G., David C., Apostolopoulos V., Mottram P.L. Pathogen recognition and development of particulate vaccines: does size matter? Methods. 2006;40:1–9. doi: 10.1016/j.ymeth.2006.05.016. 16997708 [DOI] [PubMed] [Google Scholar]
- 45.Lechardeur D., Lukacs G.L. Intracellular barriers to non-viral gene transfer. Current Gene Therapy. 2002;2:183–194. doi: 10.2174/1566523024605609. 12109215 [DOI] [PubMed] [Google Scholar]
- 46.Otten G.R., Doe B., Schaefer M., Chen M., Selby M.J., Goldbeck C. Relative potency of cellular and humoral immune responses induced by DNA vaccination. Intervirology. 2000;43:227–232. doi: 10.1159/000053990. 11251378 [DOI] [PubMed] [Google Scholar]
- 47.Cherif M.S., Shuaibu M.N., Kurosaki T., Helegbe G.K., Kikuchi M., Yanagi T. Immunogenicity of novel nanoparticle-coated MSP-1 C-terminus malaria DNA vaccine using different routes of administration. Vaccine. 2011;29:9038–9050. doi: 10.1016/j.vaccine.2011.09.031. 21939717 [DOI] [PubMed] [Google Scholar]
- 48.Dubensky T.W., Jr, Ulmer J.B. Delivery systems for gene-based vaccines. Molecular Medicine. 2000;6:723–732. 11071268 [PMC free article] [PubMed] [Google Scholar]