

Delineate a new mechanism where systemic poly I:C administration boosts local T cell immunity, and how IL-7 bridges TLR3 signal to adaptive immunity.
Keywords: poly I:C, IFN-γ, M1 macrophage, chemokine, effector T cells, NK cells
Abstract
In this study, we tested the hypothesis that systemic administration of TLR3 agonist poly I:C can enhance T cell infiltration of lung through up-regulating IL-7 expression. poly I:C, a synthetic analog of viral dsRNA and a TLR3 agonist, is studied extensively as vaccine adjuvant as a result of its pleotropic immune-stimulatory effects. Here, we show that systemic poly I:C administration induces substantial IL-7 production in the lung in a type 1 IFN- and IFN-γ-dependent fashion. Blockade of the IL-7Rα signal with a neutralizing antibody abrogated poly I:C-induced MCP-1 up-regulation, macrophage recruitment, and CXCR3 ligand expression in the lung. Conversely, administration of IL-7 enhances these events, and it does so by enhancing T cell IFN-γ production. We also show that the initial up-regulation of CXCR3 ligands and infiltration of T cells in the lung are mediated by poly I:C-induced IFN-γ from NK cells; however, the sustained and optimal CXCR3 ligand expression and T cell infiltration require poly I:C-induced IL-7 and T cell-derived IFN-γ. In a model of multiorgan inflammation elicited by adoptive transfer of immune cells into RAG1−/− mice, we show that poly I:C enhances IL-7 production in the lung and promotes expression of CXCR3 ligands and recruitment of IFN-γ+ T cells in an IL-7-dependent fashion. Collectively, these results strongly support our hypothesis and delineate a new mechanism by which poly I:C boosts the T cell immune response in the lung by inducing local IL-7 production, which in turn, enhances T cell-derived IFN-γ to promote macrophage recruitment, CXCR3 ligand expression, and T cell infiltration.
Introduction
The innate immune system provides the first line of defense against microbial infections and also plays a pivotal role in activating and shaping the adaptive immune response. Innate immune cells and tissue cells receive signals from pathogens through PRRs. Signals transduced through PRRs induce the maturation of APCs and also activate the production of a variety of proinflammatory cytokines. Through these mechanisms, PRR signals play a crucial role in activating innate and adaptive immune responses to combat pathogens and cancer [1, 2]. TLRs are a major group of PRRs and recognize conserved PAMPs expressed by viruses and bacteria [1–4]. Among the TLRs, TLR3 is triggered by viral dsRNA [5, 6] and is expressed in many cell types, including epithelial cells [7], fibroblasts [8], endothelial cells [9], and macrophages [10]. Activation of TLR3 by poly I:C, a synthetic analog of dsRNA, potently induces production of type 1 IFN in various innate immune cells and nonimmune tissue cells, which in turn, induces the maturation of DCs and activates NK cells [11, 12]. Moreover, poly I:C can induce a panel of pro- and anti-inflammatory cytokines, such as IFN-γ, TNF-α, IL-12p35, IL-23, IL-6, and IL-10 [6, 13, 14], which further facilitate adaptive immune responses [2, 6]. Because of these immune-stimulatory effects, TLR3 agonist poly I:C was studied extensively as a vaccine adjuvant for T cell-mediated antiviral and antitumor immune response [13–18]. However, TLR3 agonist-induced inflammatory responses can also elicit or facilitate autoimmune diseases or other chronic inflammatory diseases [3, 4]. poly I:C administration was reported to enhance tissue inflammation and damage and exacerbate T cell-mediated liver damage [19], lung inflammation [20], and lupus nephritis [21].
The cellular and molecular mechanisms underlying the effect of the TLR3 agonist on T cell immunity in different organs remain to be fully investigated. In a model of autoimmune liver disease, poly I:C potently induces CXCL9 expression by liver cells, resulting in enhanced infiltration of autoreactive CXCR3+ CD8 T cells in the liver and exacerbated tissue damage [19]. Hence, tissue expression of CXCR3 ligands may be an important mechanism by which the TLR3 agonist enhances local T cell immunity. CXCR3 is a chemokine receptor that is expressed on IFN-γ-producing effector CD4 and CD8 T, NK, and NKT cells [22–24]. The ligands for CXCR3 include CXCL9 (MIG), CXCL10 (IFN-induced protein-10), and CXCL11 (IFN-inducible T cell α-chemoattractant). CXCR3 ligands are highly expressed at the target tissues in many cancer, infections, and autoimmune diseases [22–24] and play a crucial role in the migration of IFN-γ-producing T and NK cells to these sites. Expression of CXCR3 ligands can be induced by external stimuli in DCs, macrophages, and various nonimmune cells [22–24]. IFN-γ is the predominant signal for the induction of CXCL9, whereas IFN-γ, type 1 IFNs, and TNF-α can all induce CXCL10 and CXCL11 expression [23, 24].
Poly I:C treatment has been reported to induce IL-7 expression in hepatocytes in a type 1 IFN-dependent fashion [25]. IL-7 is an essential factor for the development and homeostasis of T cells [26–28]. In recent years, growing evidence has demonstrated expanding, new roles of IL-7 in enhancing the magnitude and intensity of the T cell immune response, especially those mediated by Th1, Th17, and activated CD8 T cells [29–32]. Administration of IL-7 in mice leads to expansion of IFN-γ- and IL-17-producing T cells and subsequent enhancement of antiviral and antitumor immune responses [30, 33, 34]. Not surprisingly, IL-7 was reported to play a pathogenic role in a number of autoimmune diseases, including EAE [29], rheumatoid arthritis [32, 34, 35], and inflammatory bowel disease [36]. The regulation of IL-7 gene expression and production by external signals is poorly understood. The main cellular sources of IL-7 are stromal cells [37–40], keratinocytes [41], intestinal epithelial cells [42], and fibroblasts [35, 40]. It is believed that in central lymphoid organs, IL-7 is constitutively expressed by stromal cells at a constant rate [38, 43, 44]. However, recent research has shown that production of IL-7 in some tissues and organs can be regulated by external signals. TLR signals induce IL-7 production in hepatocytes, which in turn, modulates T cell homeostasis and facilitates the development of EAE [25]. Moreover, IFN-γ and IL-6 promote IL-7 production in intestinal epithelial cells [45, 46] and fibroblasts [47], respectively. These findings suggest an important new link between the innate and the adaptive immune response through the induction and action of IL-7.
Lung is a common target site of microbial infection and cancer. Lung alveolar and bronchial epithelial cells constitutively express TLR3, which can be up-regulated further by viruses, such as influenza A, and by poly I:C [7, 48]. In the present study, we investigated the effect of systemic administration of poly I:C on IL-7 production in the lung and the subsequent events induced by IL-7. We show that systemic administration of poly I:C can induce IL-7 production in the lung, and IL-7, in turn, increases T cell-derived IFN-γ to promote recruitment of macrophages and production of CXCR3 ligands. Moreover, by using an in vivo model of multiorgan inflammation, we showed that systemic administration of poly I:C enhances IL-7 production, and IL-7 is required for the optimal macrophage recruitment, CXCL9 expression, and T cell infiltration in the lung. We also demonstrate that the early, initial up-regulation of CXCR3 ligands and T cell infiltration in the lung is mediated by poly I:C-induced IFN-γ from NK cells. Our results elucidate a new mechanism by which TLR3 agonist poly I:C enhances local T cell immunity in the lung, which is through induction of IL-7 and IL-7-dependent amplification of T cell-derived IFN-γ and reinforcement of CXCR3 ligand expression. These findings delineate new pathways linking TLR signal-elicited innate and adaptive immune responses and could also facilitate the design of better strategies to use TLR agonists as vaccine adjuvants for cancer and infections.
MATERIALS AND METHODS
Mice
C57BL/6, RAG1−/−, and IFN-γ−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and were kept under pathogen-free conditions. At the time of experiments, mice were 6–8 weeks of age. All experiments were carried out under the guidelines of the Institutional Animal Care and Use Committee at The Forsyth Institute.
Antibodies and flow cytometry
Cells were stained and analyzed on a FACSAria III cell sorter (Becton Dickinson, Franklin Lakes, NJ, USA). Dead cells were excluded by forward light-scatter. Fluorescence-conjugated antibodies with the following specificities were used for staining: CD4 (GK1.5), CD8α (536-7), CD25 (PC61), CD122 (5H4), IL-10 (JES5-16E3), F4/80 (BM8), IL-7Rα (A7R34), CD3 (H57-597), and NK1.1 (PK136; from BioLegend, San Diego, CA, USA); IFN-γ (XMG1.2) and CXCL9 (MIG-2F5.5; from eBioscience, San Diego, CA, USA); purified monoclonal rat anti-mouse IL-7Rα (A 55 R34) and its isotype control rat IgG2a (2A3; from Bio X Cell, West Lebanon, NH, USA); and anti-IFNAR-1 (MAR1-5A3) and anti-IFN-γ (XMG1.2; from BioLegend).
In vivo administration of poly I:C, antibodies, and IL-7 C57BL/6 mice were i.p.-injected with 100 μg poly I:C (Sigma-Aldrich, St. Louis, MO, USA), and 6 h later, mice were killed, and lung and liver tissues were collected for further analysis. In some experiments, C57BL/6 mice were i.p.-injected with 100 μg control IgG, anti-IL-7Rα, anti-IFNAR1, or anti-IFN-γ, 2 h prior to poly I:C injection. Mice were killed 6 h or 24 h after poly I:C administration to harvest organs. For in vivo IL-7 administration, mice were i.p.-injected with 20 μg rhIL-7 (kindly provided by Biological Resources Branch, National Cancer Institute, Frederick, MD, USA) and killed 24 h later for organ collection.
Histology and immunofluorescence staining
Tissue samples were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned to 5 μm thickness from different areas across the tissue. Sections were then stained with H&E and examined for leukocytes infiltration under a microscope. Some paraffin sections were subjected to immunofluorescence staining after deparaffinization, rehydration, and antigen retrieval. The sections were incubated with biotin-conjugated anti-F4/80 (BM8), PE-conjugated anti-CXCL9 (MIG-2F5.5), or goat anti-mouse IL-7 at 4°C overnight, followed by Alexa647-conjugated streptavidin or anti-goat IgG. The stained samples were examined with a Leica laser-scanning confocal microscope (Leica Microsystems, Wetzlar, Germany). Images were average projections of three optical sections and processed with the Leica confocal software.
Staining of intracellular cytokines
For intracellular cytokine staining, cells were stained for surface molecules first, then fixed and permeabilized with Cytofix/Cytoperm buffer (eBioscience), and subsequently, incubated with indicated anticytokine antibodies in Perm/Wash buffer (eBioscience) for 30 min. Control staining with isotype control IgGs was performed in all of the experiments.
Induction of multiorgan inflammation
Spleen and LNs were harvested from donor C57B/6 mice and gently meshed into single-cell suspensions with frosted glass slides. Erythrocytes in spleen were removed by ACK lysing buffer (Quality Biological, Gaithersburg, MD, USA). The mixture of splenocytes and LN cells was incubated with biotinylated anti-CD25 (PC61.5) and anti-CD122 (TM-β1), followed by antibiotin-conjugated magnetic beads. CD25− CD122− cells were harvested after negative selection with MACS cell purification system (Miltenyi Biotec, Auburn, CA, USA). CD25−CD122− cells (6×106) were transferred into RAG1−/− mice by i.v. injection. After 2 weeks, poly I:C was administered i.p. into mice, together with control IgG or anti-IL-7Rα, three times/week for 2 more weeks. Mice were then killed and organs harvested for analysis.
Preparation of single-cell suspension
Lung and spleen were cut into small fragments, placed in a grinder, and processed with a tissue homogenizer. Tissue homogenates were filtered through a 100-μm nylon mesh, washed twice in PBS, and removed of erythrocytes with ACK lysing buffer. The single cells were resuspended in culture medium.
Ex vivo T cell stimulation
Mice were killed, and tissue cells were isolated and stimulated in vitro for 4 h with PMA (50 ng/ml) and ionomycin (1 μM; both from Calbiochem, EMD Millipore, Billierica, MA, USA), with the addition of monensin (eBioscience) during the final 2 h. Cells were then stained for surface markers and intracellular cytokines with the intracellular cytokine staining kit (eBioscience and BioLegend).
ELISA
IL-7 and IFN-γ concentration in mouse serum was determined with ELISA MAX kits (BioLegend), according to the manufacturer's protocols.
Real-time RT-PCR
Total RNA was reverse-transcribed into cDNA using Oligo (dT) and MMLV RT (Promega, Madison, WI, USA). The cDNA was subjected to real-time PCR amplification (Qiagen, Valencia, CA, USA) for 40 cycles with annealing and extension temperature at 60°C, on a LightCycler 480 Real-Time PCR System (Roche Diagnostics, Minneapolis, MN, USA). Primers were synthesized at Sigma-Aldrich, and their sequences are: IL-7 F, 5′-GGAACTGATAGTAATTGCCCG-3′, R, 5′-TTCAACTTGCGAGCAGCACG-3′; IFN-α1 F, 5′-ACCTCAGGAACAAGAGAGCC-3′, R, 5′-CTGCGGGAATCCAAAGTCCT-3′; IFN-β F, 5′-TAAGCAGCTCCAGCTCCAAG-3′, R, 5′-CCCTGTAGGTGAGGTTGATC-3′; IFN-γ F, 5′-GGATGCATTCATGAGTATTGC-3′, R, 5′-CTTTTCCGCTTCCTGAGG-3′; CXCL9 F, 5′-CCCTCAAAGACCTCAAACAGT-3′, R, 5′-AGCCGGATCTAGGCAGGTT-3′; CXCL10 F, 5′-CCAGTGAGAATGAGGGCCAT-3′, R, 5′-CCGGATTCAGACATCTCTGC-3′; CXCL11 F, 5′-GCAGAGATCGAGAAAGCTTCT-3′, R, 5′-GTCCAGGCACCTTTGTCGTT-3′; CXCR3 F, 5′-GAGAGCGACTTCTCTGACTC-3′, R, 5′-ACTCAGTAGCACAGCAGCCA-3′; IL-12p40 F, 5′-CACATCTGCTGCTCCACAAG-3′, R, 5′-CCGTCCGGACTAATTTGGTG-3′; IL-23p19 F, 5′-CTCTCGGAATCTCTGCATGC-3′, R, 5′-ACCTCTTCACACTGGATACG-3′; MIF F, 5′-TTTCTGTCGGAGCTCACCCA-3′, R, 5′-CGCTAAAGTCATGAGCTGGT-3′; F4/80 F, 5′-GAGGCTTCCTGTCCAGCAAT-3′, R, 5′-GGACCACAAGGTGAGTCACT-3′.
Statistical analysis
All statistical significance was determined by Student's t-test (two-tailed, two-sample equal variance). P values <0.05 were considered statistically significant.
Online Supplemental material
Supplemental Fig. 1 shows the induction of IL-7 by poly I:C in the liver. Supplemental Fig. 2 depicts the experimental design of the in vivo inflammation model used in this study.
RESULTS
Poly I:C induces IL-7 expression in the lung
A previous report has shown that systemic challenge with poly I:C induces liver inflammation and IL-7 production by hepatocytes. We therefore examined whether systemic administration of poly I:C can induce similar events in the lung, an important target site of infection and cancer. We administered 100 μg poly I:C i.p. into C57BL/6 mice and measured mRNA levels of IL-7 in the lung and liver 6 h later. Consistent with previous reports, poly I:C induced expression of IL-7 mRNA and protein in the liver, as assessed by real-time RT-PCR and immunofluorescence staining, respectively (Supplemental Fig. 1). Importantly, we found that poly I:C administration also led to a marked increase in IL-7 mRNA level in the lung (Fig. 1A), as well as IFN-γ and type 1 IFNs, known targets of poly I:C [49]. Immunofluorescence staining showed a dramatic increase in IL-7 protein in the lung (Fig. 1B). IL-7 appears to be produced mainly by the epithelial cells lining the pulmonary airways and alveoli (Fig. 1B). Hence, we show that poly I:C rapidly induces IL-7 gene expression and protein production in the lung, similarly to the liver. Serum concentration of IFN-γ showed a marked increase at 6 h and 24 h after poly I:C treatment (Fig. 1C). Although serum level of IL-7 did not show an increase at 6 h after poly I:C administration, it showed a considerable elevation at 24 h post-treatment (Fig. 1C).
Figure 1. poly I:C enhances expression of IL-7 in the lung.
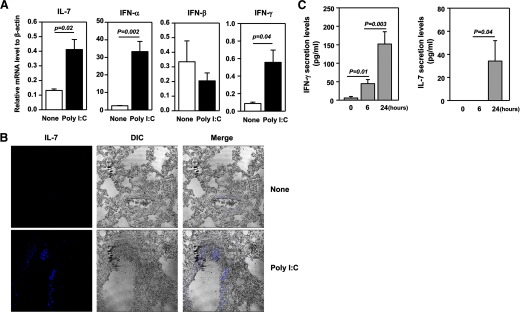
(A) Real-time PCR analysis of gene expression in the lung from C57BL/6 mice that have received i.p. injection of poly I:C, 6 h earlier, presented relative to that of β-actin. (B) Immunofluorescence staining of IL-7 in lung sections from C57BL/6 mice that have received i.p. injection of poly I:C, 24 h earlier. Differential interference contrast (DIC) image of the same sample is also shown. Data are representative of analyses of six individual mice in each group (three mice/experiment; total of two independent experiments). (C) IFN-γ and IL-7 serum concentration at indicated time-points after poly I:C treatment was measured by ELISA (two to three mice/experiment; total of two independent experiments).
Induction of lung IL-7 expression by poly I:C requires type 1 IFNs and IFN-γ
It has been reported that type 1 IFN signals are required for TLR-induced IL-7 production by hepatocytes and that IFN-γ can promote IL-7 production in intestinal epithelial cells [25, 45]. We therefore assessed whether poly I:C induction of IL-7 in the lung is dependent on these cytokines. We pretreated C57BL/6 mice with a neutralizing anti-IFNAR1 or anti-IFN-γ mAb before poly I:C administration and found that the induction of IL-7 gene expression in the lung by poly I:C was abrogated almost completely by anti-IFNAR1 and was reduced markedly by anti-IFN-γ (Fig. 2A). Hence, the induction of IL-7 expression in the lung absolutely depends on poly I:C induction of type 1 IFNs and also, to a large degree, depends on IFN-γ. We next examined whether IL-7 can affect poly I:C-induced expression of type 1 IFNs and IFN-γ. We pretreated mice with a neutralizing anti-IL-7Rα antibody (clone A7R34), which inhibits binding of IL-7 to cell surface IL-7Rα and does not deplete IL-7Rα-expressing cells by active cytotoxic mechanisms [50–52], before administration of poly I:C. We then assessed the expression of the IFNs 24 h after poly I:C injection. Expression of IFN-α, IFN-β, and IFN-γ genes was up-regulated markedly in the lung by poly I:C after 24 h of treatment (Fig. 2B), although IFN-β was not affected by poly I:C at an earlier time (Fig. 1A). Moreover, the induction of IFN-α and IFN-β was not affected by IL-7Rα blockade, whereas the induction of IFN-γ gene expression was reduced considerably (Fig. 2B). Hence, type 1 IFNs and IFN-γ are required for poly I:C-induced IL-7 expression in the lung. Furthermore, IL-7 and IFN-γ form a mutually potentiating loop by enhancing the expression of one another.
Figure 2. poly I:C-induced type 1 IFNs and IFN-γ are required for IL-7 expression.

(A) C57BL/6 mice were pretreated with anti-IFNAR1 or anti-IFN-γ, 2 h prior to poly I:C injection. After 6 h, the relative IL-7 mRNA level in lung tissue was measured by real-time RT-PCR, presented relative to that of β-actin. (B) Mice were pretreated with anti-IL-7Rα before poly I:C injection. After 24 h, the lung was analyzed for expression of indicated genes. Data are from analyses of six individual mice in each group (two mice/experiment; total of three independent experiments).
Optimal induction of lung CXCR3 ligand expression by poly I:C requires IL-7
Previous reports showed that poly I:C can induce expression of a number of chemokines in lung and liver, including chemokines that attract CXCR3-expressing T and NK cells. We thus investigated whether poly I:C-induced IL-7 contributes to the up-regulation of CXCR3 ligands in the lung. We treated C57BL/6 mice with poly I:C, together with control IgG or anti-IL-7Rα, and 24 h later, examined the mRNA levels of CXCR3 ligands. poly I:C treatment induced a substantial increase in CXCL9, CXCL10, and CXCL11 mRNA in the lung (Fig. 3A). Blockade of IL-7Rα significantly reduced the induction of CXCL9 (P=0.04) and CXCL11 (P=0.01) by poly I:C, although it did not affect that of CXCL10 (Fig. 3A). Hence, IL-7 is required for poly I:C induction of CXCR3 ligands in the lung. Accordingly, mRNA levels for CXCR3, which is expressed mainly by IFN-γ-producing effector T cells and NK cells, were also increased in the lung by poly I:C treatment, and this increase was abolished almost completely by anti-IL-7Rα (Fig. 3A).
Figure 3. poly I:C-induced IL-7 promotes expression of CXCR3 ligands in the lung.

C57BL/6 mice were pretreated with anti-IL-7Rα, 2 h prior to poly I:C injection. (A) Levels of CXCL9, -10, and -11 and CXCR3 mRNA, presented relative to that of β-actin, were measured from lung, 24 h after poly I:C injection. (B) Image of lung sections costained for CXCL9 and F4/80. Data are representative of six individual mice in each group (two mice/experiment; total of three independent experiments). (C) Real-time PCR analysis of M1 signature genes in the lung. Data are from analyses of six individual mice in each group (two mice/experiment; total of three independent experiments).
Next, by immunofluorescence staining, we confirmed the up-regulation of CXCL9 levels in the lung by poly I:C in an IL-7-dependent manner (Fig. 3B). To determine whether APCs are the cells that respond to poly I:C–IL-7 to produce CXCL9, we costained the tissue sections with macrophage marker F4/80. poly I:C induced a marked increase in F4/80+ macrophages in the lung, which was abrogated by the IL-7Rα blockade (Fig. 3B). Furthermore, the vast majority of CXCL9 signals after poly I:C treatment colocalized with F4/80 staining, indicating that macrophages in the lung were the main sources of CXCL9 in response to poly I:C–IL-7 (Fig. 3B). It has been shown that the classically activated, M1-type macrophages, but not the alternatively activated, M2 macrophages, are the major producers of CXCR3 ligands. Consistent with this, expression of several functional markers for M1 macrophages, including IL-12p40, IL-23p19, and MIF, was up-regulated in the lung by poly I:C, and the up-regulation was blocked by anti-IL-7Rα (Fig. 3C). This suggests that poly I:C treatment increases the numbers and activities of M1 macrophages in the lung in an IL-7Rα-dependent fashion. IFN-γ is a well-documented activator of M1 macrophages. As poly I:C increases the IFN-γ level in the lung through IL-7 (Fig. 2A), it is therefore conceivable that by potentiating IFN-γ production, IL-7 promotes the activation of M1 macrophages and their production of CXCR3 ligands.
IL-7 administration leads to up-regulation of CXCR3 ligands in the lung by increasing T cell-derived IFN-γ
We next investigate whether administration of IL-7 is sufficient to up-regulate CXCR3 ligands in the lung. We administered rh IL-7 to C57BL/6 mice and analyzed mRNA levels of CXCR3 ligands 1 day later. IL-7 treatment resulted in a marked increase in all three CXCR3 ligands in the lung (Fig. 4A). Consistent with the observations from the IL-7Rα blockade described earlier, IL-7 administration increased the expression of IFN-γ and signature genes of M1 macrophages in the lung tissues (Fig. 4A). We next assessed whether the effect of IL-7 is dependent on IFN-γ by injecting IL-7 into IFN-γ−/− mice. The expression levels of all three CXCR3 ligands in the lung were not detectable by real-time RT-PCR in IFN-γ−/− mice, treated or not treated with rh IL-7 (data not shown). These results indicate that the expression of CXCR3 ligands in the lung is stringently dependent on the presence of IFN-γ and that IL-7 cannot induce these chemokines in the absence of IFN-γ. Activated T cells and NK cells are the major cellular sources of IFN-γ. To assess whether the effect of IL-7 to induce IFN-γ requires the presence of lymphocytes, we administered rh IL-7 into RAG1−/− mice. IL-7 treatment did not up-regulate IFN-γ expression in these mice (Fig. 4B), indicating that the cell sources responding to IL-7 to produce IFN-γ are lymphocytes and particularly, T cells. As a result of abrogated IFN-γ up-regulation, IL-7 administration could not up-regulate expression of any CXCR3 ligand or MCP-1 in the lung of RAG1−/− mice (Fig. 4B). Hence, administration of IL-7 up-regulates CXCR3 ligand production in the lung through increasing T cell-derived IFN-γ.
Figure 4. Exogenous IL-7 induces expression of CXCR3 ligands in the lung.

(A) C57BL/6 mice were treated with 20 μg rh IL-7 for 24 h. Expression level of CXCL9, -10, and -11, IFN-γ, and M1 signature gene mRNA was measured from the lung. Data are from analyses of five individual mice in each group (one to two mice/experiment; total of three independent experiments). (B) RAG1−/− mice were treated with 20 μg rh IL-7 for 24 h. Gene expression in lung tissue was measured. Data are from analyses of five individual mice in each group (two to three mice/experiment; total of two independent experiments).
IL-7 up-regulates MCP-1 expression in the lung in an IFN-γ-dependent fashion
The number of F4/80+ macrophage cells was increased with poly I:C treatment, and this increase was abolished by anti-IL-7Rα treatment (Fig. 3). These results suggest that the recruitment of macrophages into the lung might be enhanced by poly I:C-induced IL-7. We thus measured mRNA levels of MCP-1, an important chemokine for monocytes/macrophage tissue migration [53, 54]. The MCP-1 mRNA level was increased substantially in the lung of C57BL/6 mice, 24 h after poly I:C treatment, and this increase was abrogated completely by the IL-7Rα blockade (Fig. 5A). In comparison, the MCP-2 mRNA level was extremely low and not altered by these treatments (data not shown). Administration of rhIL-7 induced marked elevation in MCP-1 mRNA in the lung of C57BL/6 mice, 24 h post-treatment (Fig. 5B). In contrast, IL-7 did not up-regulate MCP-1 in IFN-γ−/− mice (Fig. 5B). Correlating with a lack of an IFN-γ increase in RAG1−/− mice, IL-7 could not up-regulate MCP-1 expression in these mice (Fig. 5C). Thus, IL-7 enhances MCP-1 expression in the lung by increasing T cell-derived IFN-γ.
Figure 5. poly I:C and IL-7 induce MCP-1 expression in the lung.

(A) C57BL/6 mice were pretreated with anti-IL-7Rα, 2 h prior to poly I:C injection, and mRNA level of MCP-1 in the lung was measured 24 h after poly I:C injection. (B) C57BL/6 or IFN-γ−/− mice were treated with 20 μg rh IL-7 for 24 h, and mRNA level of MCP-1 in the lung was measured. (C) RAG1−/− mice were treated with 20 μg rhIL-7 for 24 h. MCP-1 mRNA in lung tissue was measured. Data are from analyses of five individual mice in each group (two to three mice/experiment; total of two independent experiments).
Initial up-regulation of CXCR3 ligands and T cell infiltration are mediated by poly I:C-induced IFN-γ from NK cells
Having shown that IL-7-induced up-regulation of IFN-γ and chemokines in the lung is dependent on T cells, we next examined how poly I:C induces the initial influx of T cells into the lung before the IL-7–T cell–IFN-γ pathway is established. We first analyzed the kinetics of T cell infiltration of the lung after poly I:C treatment. Flow cytometric analysis showed that the frequency of total TCR-β+ T cells and CD4 and CD8 T cells in the lung increased with time, starting as early as 6 h after poly I:C treatment (Fig. 6A). In comparison, the proportion of γδ T and NK cells did not increase within 24 h post-treatment (Fig. 6A). We next aimed to delineate the early events that lead to the initial T cell recruitment by 6 h after poly I:C treatment. Lung resident NK cells play an important role in early host defense against various microbial agents, in a large part, by producing IFN-γ [6, 55]. Moreover, poly I:C has been shown to induce IFN-γ production in NK cells [6, 56–59]. We therefore postulate that the initial CXCR3 ligand up-regulation and T cell recruitment to the lung in response to poly I:C are driven by NK cell-derived IFN-γ. Indeed, injection of poly I:C to RAG1−/− mice led to a marked up-regulation of IFN-γ, CXCL9, and CXCL10 in the lung at 6 h post-treatment (Fig. 6B), indicating that poly I:C can rapidly induce expression of these molecules, independently of T cells and T cell-derived IFN-γ. Moreover, in RAG1−/− mice that were depleted of NK cells with injection of anti-NK1.1 antibody, poly I:C could not induce IFN-γ and CXCR3 ligands (Fig. 6B). Hence, early induction of IFN-γ from NK cells by poly I:C led to subsequent up-regulation of CXCR3 ligands in the lung, resulting in initial recruitment of T cells.
Figure 6. Initial up-regulation of IFN-γ and CXCR3 ligands induced by poly I:C depends on NK cells.

(A) Flow cytometry of lung-infiltrating leukocyte populations in C57BL/6 mice at indicated time-points after poly I:C administration, with the gating indicated above the plots. Upper-right panel, percentage of NK1.1+, TCR-β+, or TCR-γδ+ cells within total live cells from the lung; lower-right panel, percentage of CD4+ or CD8+ cells within total live cells from the lung. SSC, Side-scatter. (B) C57BL/6 mice were pretreated with anti-NK1.1, 2 days prior to poly I:C injection. IFN-γ and chemokine mRNA levels in the lung were measured 6 h after poly I:C injection. All data are from analyses of five individual mice in each group (two mice/experiment; total of two independent experiments).
Poly I:C enhances CXCR3 ligand expression and inflammation in the lung in an IL-7-dependent manner in a multiorgan inflammation model
We next assessed whether the poly I:C–IL-7 pathway affects CXCR3 ligand expression in the lung and exacerbate T cell infiltration in these organs in an in vivo multiorgan inflammation model. We transferred a mixture of splenocytes and LN cells that were depleted of Tregs (CD25−CD122−) into RAG1−/− mice to induce multiorgan autoimmune inflammation, similarly to that observed in mice deficient in Tregs (Supplemental Fig. 2). Two weeks after the adoptive transfer, we administered poly I:C to the mice, together with a neutralizing anti-IL-7Rα antibody or its isotype control IgG, three times/week for 2 weeks (Supplemental Fig. 2). H&E staining of tissue sections showed that poly I:C-treated mice had a dramatically higher amount of immune cells in the lung than control mice (Fig. 7A). Importantly, the effect of poly I:C to aggravate lung inflammation was abolished by the IL-7Rα blockade (Fig. 7A). Consistent with these observations, the survival rate of mice was decreased considerably by poly I:C treatment and was restored to 100% by the IL-7Rα blockade (Fig. 7B). Hence, poly I:C significantly exacerbates lung inflammation in an IL-7-dependent fashion. Immunofluorescence analysis showed that poly I:C treatment led to a dramatically elevated IL-7 expression in the lung, which was not affected by anti-IL-7Rα (Fig. 7C). Moreover, poly I:C treatment increased the numbers of CXCL9-producing macrophages in the lung, and this increase was reduced greatly by the IL-7Rα blockade (Fig. 7D).
Figure 7. poly I:C up-regulates IL-7 and CXCL9 and exacerbates inflammation of the lung.

CD25−CD122− cells were transferred to RAG1−/− mice. Two weeks later, the recipient mice received regular injections with poly I:C plus IgG or anti-IL-7Rα for another 2 weeks. (A) H&E staining of lung sections 2 weeks after poly I:C and antibody treatment. Upper panels, low-magnitude image of lung section (original scale bars=500 μm); lower panels, high-magnitude image of lung section. (B) Survival rate of mice during the last 2 weeks of the experiment. (C) Immunofluorescence staining of IL-7 in the same lung sections from A. (D) Immunofluorescence staining of CXCL9 and F4/80 in the same lung sections from A. Data are representative of analyses of eight mice in each group (two mice/experiment; total of four independent experiments).
Taken together, these data demonstrate that in a model of multiorgan inflammation, systemic poly I:C administration can up-regulate CXCL9 expression and lung inflammation by inducing IL-7 production.
Poly I:C enhances infiltration of lung by IFN-γ-producing T cells in an IL-7-dependent manner in the in vivo inflammation model
We next investigated whether the poly I:C-induced, IL-7-depedent up-regulation of CXCL9 expression resulted in enhanced infiltration of the lung by IFN-γ-producing T cells, the major T cell populations that express CXCR3. Immunofluorescence staining demonstrated that poly I:C increased the number of CD3+ T cells in the lung, and this increase was abolished by the IL-7Rα blockade (data not shown). Further analysis of cell subsets showed that poly I:C induced a significant increase in the percentages of CD3+ T cells, macrophages, CD4 and CD8 T cells, as well as IFN-γ+ CD4 and CD8 T cells in the lung in an IL-7Rα-dependent fashion (Fig. 8A and B). Collectively, these data indicate that the poly I:C-induced increase in CXCL9 led to enhanced recruitment of IFN-γ+ T cells to the lung.
Figure 8. poly I:C enhances lung infiltration by IFN-γ[supb]+.

T cells in an IL-7-dependent fashion. Lung was harvested from RAG1−/− mice that have received CD25−CD122− cells and treated with poly I:C plus IgG or anti-IL-7Rα, as described in Fig. 6. (A) Surface staining of CD3, F4/80, CD4, CD8α, and IL-7Rα and intracellular cytokine staining on cells from the lung with the gating indicated above the plots. FSC, Forward-scatter. (B) Left panels, percentage of F4/80+ or CD3+ cells within total cells analyzed; middle panels, percentage of CD4 or CD8 T cells in mononuclear cells; right panels, percentage of IFN-γ+ cells within CD4 or CD8 T cells. All data are from analyses of at least eight individual mice in each group (two mice/experiment; total of four independent experiments).
One potential consequence of in vivo anti-IL-7Rα treatment is the overall reduction in the number of transferred T cells in the host RAG1−/− mice, as IL-7, at its physiological level, is essential for the survival and homeostatic proliferation of T cells. To test whether our anti-IL-7Rα treatment regime, a relatively low dose at 50 μg, affects overall T cell numbers, we performed the control experiments, in which IgG or anti-IL-7Rα was injected into host RAG1−/− mice that were transferred with immune cells, in the absence of poly I:C treatment. We found that the dosage of anti-IL-7Rα that we used did not lead to a reduction in the frequency and absolute numbers of transferred T cells, including CD4, CD8, and γδ T cells, and NK cells in the spleen (Supplemental Fig. 3A). Moreover, this dosage of anti-IL-7Rα did not affect the numbers of these T cell populations and NK cells in the lung (Supplemental Fig. 3B). These results indicate that our anti-IL-7Rα injection regime does not affect the normal homeostatic proliferation and survival of the transferred T cells in the absence of poly I:C treatment. However, it is able to reverse poly I:C-induced, excessive IL-7 activity. Therefore, we concluded that the effect of the IL-7Rα blockade on lung inflammation is not a result of an overall loss of T cells but is a result of the blockade of poly I:C-induced, excessive IL-7 activity. Finally, we assessed whether this dosage of anti-IL-7Rα affects intestinal inflammation in the presence or absence of poly I:C treatment. H&E staining showed that anti-IL-7Rα treatment did not affect the degree of colon inflammation in the absence poly I:C treatment (Supplemental Fig. 4). More importantly, anti-IL-7Rα treatment did not affect colon inflammation in the presence of poly I:C treatment (Supplemental Fig. 4), whereas it significantly reduced lung inflammation (Fig. 7A). Hence, the effect of IL-7 on lung inflammation is not likely to be a consequence of the altered inflammatory colitis milieu.
In summary, we define a new mechanism by which systemic administration of the TLR3 agonist enhances T cell-mediated inflammation and immunity in the lung through inducing local IL-7 production (Fig. 9). The initial poly I:C signal induces IFN-γ production from NK cells, likely resident NK cells in the lung, through induction of type 1 IFN. poly I:C also induces IL-7 production from lung epithelial cells in a type 1 IFN- and IFN-γ-dependent fashion. NK cell-derived IFN-γ leads to up-regulation of CXCR3 ligands and the initial influx of T cells in the lung. After the migration of first T cells into the lung, poly I:C-induced IL-7 promotes IFN-γ production by these T cells to sustain the up-regulation of MCP-1, recruitment of macrophages, and production of CXCR3 ligands. In this fashion, poly I:C-induced IL-7 promotes sustained and optimal CXCR3 ligand expression and T cell infiltration in the lung to enhance local T cell immunity (Fig. 9).
Figure 9. Model: poly I:C-induced IL-7 promotes macrophage recruitment, CXCR3 ligand expression, and T cell infiltration in the lung by enhancing IFN-γ production.

Poly I:C treatment rapidly induces IFN-γ production from lung resident NK cells and also induces IL-7 production from lung epithelial cells. IFN-γ derived from NK cells, in turn, induces expression of CXCR3 ligands, leading to initial influx of T cells in the lung. At this point, poly I:C-induced IL-7 promotes IFN-γ production from these early lung-infiltrating T cells to sustain and reinforce the up-regulation of MCP-1, recruitment of macrophages, production of CXCR3 ligands, and infiltration of IFN-γ-producing T cells. Through these mechanisms, poly I:C-induced IL-7 facilitates the infiltration of IFN-γ-producing effector T cells into lung, thereby exacerbating local T cell immune response.
DISCUSSION
The present study defines a new mechanism by which systemic administration of poly I:C enhances local T cell immune response in the lung, in which poly I:C-induced IL-7 promotes T cell-derived IFN-γ to enhance macrophage recruitment and CXCR3 ligand expression. We show that systemic challenge with the TLR3 agonist poly I:C induces IL-7 production in the lung in a type 1 IFN- and IFN-γ-dependent fashion. We further show that poly I:C-induced IL-7, through enhancing T cell IFN-γ production, promotes expression of MCP-1, recruitment of macrophages, and production of CXCR3 ligands by these macrophages. Furthermore, using an in vivo model of multiorgan inflammation, we demonstrate that poly I:C enhances IL-7 production in the lung and promotes CXCR3 ligand expression and T cell infiltration in an IL-7-dependent fashion. Hence, although the initial up-regulation of CXCR3 ligands and infiltration of T cells induced by poly I:C depend on NK cell-derived IFN-γ, the optimal and sustained CXCR3 ligand expression and T cell infiltration require poly I:C-induced IL-7 and T cell-derived IFN-γ.
In addition to the lung, systemic poly I:C administration induces IL-7 and CXCR3 ligands in other organs, such as liver [25] (Supplemental Fig. 1), salivary glands, and lacrimal glands (unpublished results). Interestingly, IL-7 levels in LNs and spleen are reduced by poly I:C (data not shown) and by agonists of other TLRs [25]. Thus, by limiting IL-7 levels in lymphoid tissues but increasing them in nonlymphoid organs, poly I:C treatment may promote redistribution of lymphocytes from lymphoid tissues to nonlymphoid organs, enhancing inflammation and immunity at these sites. In future studies, we will investigate the cellular and molecular mechanisms underlying the differential regulation of IL-7 expression in lymphoid and nonlymphoid tissues and identify other tissues that can be affected by poly I:C.
The predominant cellular sources of IFN-γ are NK, Th1, and activated CD8 T cells. We found that NK cell-derived IFN-γ and T cell-derived IFN-γ are critical for the up-regulation of CXCR3 ligands and T cell infiltration at different stages after poly I:C treatment. At early phase, poly I:C induces IFN-γ and CXCR3 ligands in the lung in a NK cell-dependent but T cell-independent manner. Conceivably, this early induction of CXCR3 ligands leads to initial influx of T cells in the lung, which we observed at 6 h after poly I:C treatment. We speculate that the IFN-γ-producing NK cells responding to poly I:C are lung resident NK cells, as there was no increase in NK cells in the lung at 6 h or even 24 h after poly I:C treatment. Our findings are in accordance with the facts that lung resident NK cells are crucial, early players against microbial infections, largely through their ability to rapidly produce IFN-γ [6, 55]. Our results are also supported by previous studies showing that poly I:C can rapidly induce IFN-γ production in NK cells through multiple mechanisms [6, 56–59] and that i.p. administration of poly I:C can induce activation of NK cells in the lung [15]. In mice, poly I:C-induced activation of NK cells is not mediated by a NK cell-intrinsic mechanism but is dependent on type 1 IFNs produced by multiple other cell types [12]. We speculate that the same mechanism is operating in the lung.
At a later stage after poly I:C treatment, after the initial burst of NK-derived IFN-γ and migration of the first T cells into the lung, poly I:C-induced IL-7 acts on these T cells to increase IFN-γ levels. In this fashion, IL-7 sustains and reinforces the up-regulation of MCP-1 and CXCR3 ligands to promote continuous T cell infiltration. By directly administering IL-7 to RAG1−/− mice, we show that IL-7 requires the presence of T cells to up-regulate IFN-γ in the lung, suggesting that T cells are the major cell sources responding to IL-7 to produce IFN-γ. This is consistent with the fact that IL-7 was not reported to enhance IFN-γ production by NK cells but was widely shown to induce IFN-γ production from T cells and expand IFN-γ-producing T cells [30–34]. IL-7-induced IFN-γ, in turn, enhances the recruitment of more IFN-γ-producing cells to the lung by increasing CXCR3 ligand levels. In summary, we propose that the early, initial up-regulation of CXCR3 ligands and T cell infiltration in the lung is mediated by poly I:C-induced IFN-γ from resident NK cells; however the sustained and optimal CXCR3 ligand expression and T cell infiltration require poly I:C-induced IL-7 and T cell-derived IFN-γ. We will use approaches that cause tissue-specific ablation of NK cells or T cells in the future to definitively characterize the type and location of cytokine-producing cells in response to poly I:C treatment.
The epithelial cells of the lung airways and alveoli appear to be the sources of IL-7 in response to poly I:C treatment, although further studies are required to precisely define the producers of IL-7. The signaling pathways that mediate poly I:C induction of IL-7 expression also needs further determination. Apart from TLR3, poly I:C can also bind to MDA5 to induce type 1 IFN production in a variety of cells, including lung epithelial cells [60–63]. We will investigate the contribution of TLR3 and MDA5 signaling pathways to the induction of IL-7 with mice that are deficient in each molecule in the future. Interestingly, poly I:C induction of IL-7 in the lung not only requires type 1 IFNs, similarly to the liver [25], but also requires IFN-γ. How type 1 IFNs and IFN-γ induce IL-7 expression in the lung epithelial cells awaits further investigation. IFN-γ has been shown to promote IL-7 expression in intestinal epithelial cells by activating transcription factors IRF-1 and IRF-2 [46]. Similar mechanisms may be operating in the lung as well.
Several studies have shown that administration of IL-7 to mice bearing lung cancer leads to up-regulation of CXCR3 ligands and enhancement of anti-tumor immunity in a CXCR3 ligand-dependent fashion [64, 65]. Here, we demonstrate that the TLR3 agonist poly I:C, a widely studied adjuvant for antitumor and antipathogen immune responses, can up-regulate CXCR3 ligand production by inducing IL-7 in the lung. Classically activated M1 type macrophages, but not the alternatively activated M2 macrophages, have been shown to be important sources of CXCR3 ligands [66–69]. We show here that the major cell types in the lung responding to poly I:C–IL-7 to produce CXCL9 are M1 macrophages, as characterized by the expression of IL-12p40, IL-23p19, and MIF. IFN-γ is one of the major stimuli that activates macrophages and confers them the M1 characteristics. Moreover, IFN-γ can also facilitate the recruitment of macrophages to tissue sites by inducing tissue production of MCP-1 [70, 71]. Consistent with these known effects of IFN-γ, we show that IL-7 can up-regulate MCP-1 and the number of macrophages in the lung, in an IFN-γ-dependent fashion. In fact, the recruitment of macrophages into the lung may be the primary event induced by the IL-7–IFN-γ pathway, which leads to subsequent elevation of local CXCR3 ligands. Future studies will investigate this possibility.
Finally, we used a relatively low dose of anti-IL-7Rα (50 μg) in this study. IL-7 is a crucial factor for the survival and homeostatic proliferation of T cells. We found that the dosage of anti-IL-7Rα that we used does not affect the normal homeostatic proliferation and survival of the transferred T cells in the absence of poly I:C treatment. It is, however, able to inhibit poly I:C-induced events that are dependent on the elevated levels and activity of IL-7. Furthermore, we showed that anti-IL-7Rα treatment markedly diminished lung inflammation in the presence of poly I:C treatment without affecting the degree of colon inflammation (Supplemental Fig. 4). Hence, the effect of IL-7 on lung inflammation is not a consequence of an altered inflammatory colitis milieu. Consequently, we are able to conclude that the effects of anti-IL-7Rα treatment that we have observed do not result from an overall loss of T cells or an altered inflammatory colitis environment, but result from a blockade of poly I:C-induced, excessive IL-7 activity in the lung.
In summary, the present study defines a crucial role of IL-7 in promoting a T cell immune response in the lung induced by systemic administration of the TLR3 agonist, in which poly I:C-induced IL-7 up-regulates T cell-derived IFN-γ to promote recruitment of macrophages and their production of CXCR3 ligands. These findings delineate a new, IL-7-dependent mechanism bridging innate and adaptive immune response and will also provide insights into developing better strategies to combat cancer and infectious diseases by using TLR agonists.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the U.S. National Institutes of Health (P30DE020751).
We thank Dr. D. J. Smith for support and advice in this project. We thank Drs. A. Bhandoola and K. Shortman for critical reading of the manuscript, the Forsyth Institute Animal Facility for maintaining animals, and Biological Resources Branch, Division of Cancer Treatment and Diagnosis, National Cancer Institute, for providing rhIL-7.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- −/−
- deficient
- ACK
- ammonium-chloride-potassium
- EAE
- experimental autoimmune encephalomyelitis
- F
- forward
- hIL-7
- human IL-7
- IFNAR-1
- IFN-α/βR 1
- IRF
- IFN regulatory factor
- MDA5
- melanoma differentiation-associated gene 5
- MIF
- macrophage migration inhibitory factor
- MIG
- monokine induced by IFN-γ
- poly I:C
- polyinosinic:polycytidylic acid
- R
- reverse
- Treg
- regulatory T cell
AUTHORSHIP
J-O.J. performed all experiments, collected and analyzed data, and contributed to the writing of the manuscript. Q.Y. conceived of the concept of this project, designed experiments, supervised the study, and wrote the manuscript.
DISCLOSURES
The authors have no competing financial interests.
REFERENCES
- 1. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 2. Arpaia N., Barton G. M. (2011) Toll-like receptors: key players in antiviral immunity. Curr. Opin. Virol. 1, 447–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mills K. H. (2011) TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 11, 807–822 [DOI] [PubMed] [Google Scholar]
- 4. Marshak-Rothstein A. (2006) Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 6, 823–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alexopoulou L., Holt A. C., Medzhitov R., Flavell R. A. (2001) Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 [DOI] [PubMed] [Google Scholar]
- 6. Matsumoto M., Oshiumi H., Seya T. (2011) Antiviral responses induced by the TLR3 pathway. Rev. Med. Virol. doi: 10.1002/rmv.680 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7. Guillot L., Le Goffic R., Bloch S., Escriou N., Akira S., Chignard M., Si-Tahar M. (2005) Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 280, 5571–5580 [DOI] [PubMed] [Google Scholar]
- 8. Brentano F., Schorr O., Gay R. E., Gay S., Kyburz D. (2005) RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum. 52, 2656–2665 [DOI] [PubMed] [Google Scholar]
- 9. Tissari J., Siren J., Meri S., Julkunen I., Matikainen S. (2005) IFN-α enhances TLR3-mediated antiviral cytokine expression in human endothelial and epithelial cells by up-regulating TLR3 expression. J. Immunol. 174, 4289–4294 [DOI] [PubMed] [Google Scholar]
- 10. Malmgaard L., Melchjorsen J., Bowie A. G., Mogensen S. C., Paludan S. R. (2004) Viral activation of macrophages through TLR-dependent and -independent pathways. J. Immunol. 173, 6890–6898 [DOI] [PubMed] [Google Scholar]
- 11. Kumar H., Koyama S., Ishii K. J., Kawai T., Akira S. (2008) Cutting edge: cooperation of IPS-1- and TRIF-dependent pathways in poly IC-enhanced antibody production and cytotoxic T cell responses. J. Immunol. 180, 683–687 [DOI] [PubMed] [Google Scholar]
- 12. Longhi M. P., Trumpfheller C., Idoyaga J., Caskey M., Matos I., Kluger C., Salazar A. M., Colonna M., Steinman R. M. (2009) Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 206, 1589–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seya T., Matsumoto M. (2009) The extrinsic RNA-sensing pathway for adjuvant immunotherapy of cancer. Cancer Immunol. Immunother. 58, 1175–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsumoto M., Seya T. (2008) TLR3: interferon induction by double-stranded RNA including poly(I:C). Adv. Drug Deliv. Rev. 60, 805–812 [DOI] [PubMed] [Google Scholar]
- 15. Ngoi S. M., Tovey M. G., Vella A. T. (2008) Targeting poly(I: C) to the TLR3-independent pathway boosts effector CD8 T cell differentiation through IFN-α/β. J. Immunol. 181, 7670–7680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salem M. L., Diaz-Montero C. M., El-Naggar S. A., Chen Y., Moussa O., Cole D. J. (2009) The TLR3 agonist poly(I: C) targets CD8+ T cells and augments their antigen-specific responses upon their adoptive transfer into naive recipient mice. Vaccine 27, 549–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ainai A., Tashiro M., Hasegawa H. (2011) Cross-protective immunity against influenza virus infections induced by intranasal vaccination together with a TLR3-mucosal adjuvant. Hum. Vaccin. 7 (Suppl.), 174–182 [DOI] [PubMed] [Google Scholar]
- 18. Wick D. A., Martin S. D., Nelson B. H., Webb J. R. (2011) Profound CD8+ T cell immunity elicited by sequential daily immunization with exogenous antigen plus the TLR3 agonist poly(I: C). Vaccine 29, 984–993 [DOI] [PubMed] [Google Scholar]
- 19. Lang K. S., Georgiev P., Recher M., Navarini A. A., Bergthaler A., Heikenwalder M., Harris N. L., Junt T., Odermatt B., Clavien P. A., Pircher H., Akira S., Hengartner H., Zinkernagel R. M. (2006) Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J. Clin. Invest. 116, 2456–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stowell N. C., Seideman J., Raymond H. A., Smalley K. A., Lamb R. J., Egenolf D. D., Bugelski P. J., Murray L. A., Marsters P. A., Bunting R. A., Flavell R. A., Alexopoulou L., San Mateo L. R., Griswold D. E., Sarisky R. T., Mbow M. L., Das A. M. (2009) Long-term activation of TLR3 by poly(I: C) induces inflammation and impairs lung function in mice. Respir. Res. 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patole P. S., Grone H. J., Segerer S., Ciubar R., Belemezova E., Henger A., Kretzler M., Schlondorff D., Anders H. J. (2005) Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J. Am. Soc. Nephrol. 16, 1326–1338 [DOI] [PubMed] [Google Scholar]
- 22. Lacotte S., Brun S., Muller S., Dumortier H. (2009) CXCR3, inflammation, and autoimmune diseases. Ann. N. Y. Acad. Sci. 1173, 310–317 [DOI] [PubMed] [Google Scholar]
- 23. Groom J. R., Luster A. D. (2011) CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89, 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Groom J. R., Luster A. D. (2011) CXCR3 in T cell function. Exp. Cell. Res. 317, 620–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sawa Y., Arima Y., Ogura H., Kitabayashi C., Jiang J. J., Fukushima T., Kamimura D., Hirano T., Murakami M. (2009) Hepatic interleukin-7 expression regulates T cell responses. Immunity 30, 447–457 [DOI] [PubMed] [Google Scholar]
- 26. Park J. H., Adoro S., Guinter T., Erman B., Alag A. S., Catalfamo M., Kimura M. Y., Cui Y., Lucas P. J., Gress R. E., Kubo M., Hennighausen L., Feigenbaum L., Singer A. (2010) Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat. Immunol. 11, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mackall C. L., Fry T. J., Gress R. E. (2011) Harnessing the biology of IL-7 for therapeutic application. Nat. Rev. Immunol. 11, 330–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sprent J., Surh C. D. (2011) Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat. Immunol. 12, 478–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee L. F., Axtell R., Tu G. H., Logronio K., Dilley J., Yu J., Rickert M., Han B., Evering W., Walker M. G., Shi J., de Jong B. A., Killestein J., Polman C. H., Steinman L., Lin J. C. (2011) IL-7 promotes T(H)1 development and serum IL-7 predicts clinical response to interferon-β in multiple sclerosis. Sci. Transl. Med. 3, 93ra68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pellegrini M., Calzascia T., Toe J. G., Preston S. P., Lin A. E., Elford A. R., Shahinian A., Lang P. A., Lang K. S., Morre M., Assouline B., Lahl K., Sparwasser T., Tedder T. F., Paik J. H., DePinho R. A., Basta S., Ohashi P. S., Mak T. W. (2011) IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell 144, 601–613 [DOI] [PubMed] [Google Scholar]
- 31. Hartgring S. A., Bijlsma J. W., Lafeber F. P., van Roon J. A. (2006) Interleukin-7 induced immunopathology in arthritis. Ann. Rheum. Dis. 65 (Suppl 3), iii69–iii74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hartgring S. A., Willis C. R., Alcorn D., Nelson L. J., Bijlsma J. W., Lafeber F. P., van Roon J. A. (2010) Blockade of the interleukin-7 receptor inhibits collagen-induced arthritis and is associated with reduction of T cell activity and proinflammatory mediators. Arthritis Rheum. 62, 2716–2725 [DOI] [PubMed] [Google Scholar]
- 33. Pellegrini M., Calzascia T., Elford A. R., Shahinian A., Lin A. E., Dissanayake D., Dhanji S., Nguyen L. T., Gronski M. A., Morre M., Assouline B., Lahl K., Sparwasser T., Ohashi P. S., Mak T. W. (2009) Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat. Med. 15, 528–536 [DOI] [PubMed] [Google Scholar]
- 34. Kim H. R., Hwang K. A., Park S. H., Kang I. (2008) IL-7 and IL-15: biology and roles in T-cell immunity in health and disease. Crit. Rev. Immunol. 28, 325–339 [DOI] [PubMed] [Google Scholar]
- 35. Churchman S. M., Ponchel F. (2008) Interleukin-7 in rheumatoid arthritis. Rheumatology (Oxford) 47, 753–759 [DOI] [PubMed] [Google Scholar]
- 36. Totsuka T., Kanai T., Nemoto Y., Makita S., Okamoto R., Tsuchiya K., Watanabe M. (2007) IL-7 Is essential for the development and the persistence of chronic colitis. J. Immunol. 178, 4737–4748 [DOI] [PubMed] [Google Scholar]
- 37. Witte P. L., Frantsve L. M., Hergott M., Rahbe S. M. (1993) Cytokine production and heterogeneity of primary stromal cells that support B lymphopoiesis. Eur. J. Immunol. 23, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 38. Alves N. L., Richard-Le Goff O., Huntington N. D., Sousa A. P., Ribeiro V. S., Bordack A., Vives F. L., Peduto L., Chidgey A., Cumano A., Boyd R., Eberl G., Di Santo J. P. (2009) Characterization of the thymic IL-7 niche in vivo. Proc. Natl. Acad. Sci. USA 106, 1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marrack P., Bender J., Hildeman D., Jordan M., Mitchell T., Murakami M., Sakamoto A., Schaefer B. C., Swanson B., Kappler J. (2000) Homeostasis of α β TCR+ T cells. Nat. Immunol. 1, 107–111 [DOI] [PubMed] [Google Scholar]
- 40. Mazzucchelli R. I., Warming S., Lawrence S. M., Ishii M., Abshari M., Washington A. V., Feigenbaum L., Warner A. C., Sims D. J., Li W. Q., Hixon J. A., Gray D. H., Rich B. E., Morrow M., Anver M. R., Cherry J., Naf D., Sternberg L. R., McVicar D. W., Farr A. G., Germain R. N., Rogers K., Jenkins N. A., Copeland N. G., Durum S. K. (2009) Visualization and identification of IL-7 producing cells in reporter mice. PLoS One 4, e7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heufler C., Topar G., Grasseger A., Stanzl U., Koch F., Romani N., Namen A. E., Schuler G. (1993) Interleukin 7 is produced by murine and human keratinocytes. J. Exp. Med. 178, 1109–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Watanabe M., Ueno Y., Yajima T., Iwao Y., Tsuchiya M., Ishikawa H., Aiso S., Hibi T., Ishii H. (1995) Interleukin 7 is produced by human intestinal epithelial cells and regulates the proliferation of intestinal mucosal lymphocytes. J. Clin. Invest. 95, 2945–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma A., Koka R., Burkett P. (2006) Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu. Rev. Immunol. 24, 657–679 [DOI] [PubMed] [Google Scholar]
- 44. Mazzucchelli R., Durum S. K. (2007) Interleukin-7 receptor expression: intelligent design. Nat. Rev. Immunol. 7, 144–154 [DOI] [PubMed] [Google Scholar]
- 45. Shalapour S., Deiser K., Sercan O., Tuckermann J., Minnich K., Willimsky G., Blankenstein T., Hammerling G. J., Arnold B., Schuler T. (2010) Commensal microflora and interferon-γ promote steady-state interleukin-7 production in vivo. Eur. J. Immunol. 40, 2391–2400 [DOI] [PubMed] [Google Scholar]
- 46. Oshima S., Nakamura T., Namiki S., Okada E., Tsuchiya K., Okamoto R., Yamazaki M., Yokota T., Aida M., Yamaguchi Y., Kanai T., Handa H., Watanabe M. (2004) Interferon regulatory factor 1 (IRF-1) and IRF-2 distinctively up-regulate gene expression and production of interleukin-7 in human intestinal epithelial cells. Mol. Cell. Biol. 24, 6298–6310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sawa S., Kamimura D., Jin G. H., Morikawa H., Kamon H., Nishihara M., Ishihara K., Murakami M., Hirano T. (2006) Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J. Exp. Med. 203, 1459–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ritter M., Mennerich D., Weith A., Seither P. (2005) Characterization of Toll-like receptors in primary lung epithelial cells: strong impact of the TLR3 ligand poly(I:C) on the regulation of Toll-like receptors, adaptor proteins and inflammatory response. J. Inflamm. 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., Taniguchi T. (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777 [DOI] [PubMed] [Google Scholar]
- 50. Goldrath A. W., Sivakumar P. V., Glaccum M., Kennedy M. K., Bevan M. J., Benoist C., Mathis D., Butz E. A. (2002) Cytokine requirements for acute and basal homeostatic proliferation of naive and memory CD8+ T cells. J. Exp. Med. 195, 1515–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaech S. M., Tan J. T., Wherry E. J., Konieczny B. T., Surh C. D., Ahmed R. (2003) Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4, 1191–1198 [DOI] [PubMed] [Google Scholar]
- 52. Sudo T., Nishikawa S., Ohno N., Akiyama N., Tamakoshi M., Yoshida H. (1993) Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc. Natl. Acad. Sci. USA 90, 9125–9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deshmane S. L., Kremlev S., Amini S., Sawaya B. E. (2009) Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 29, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bracke K. R., Demedts I. K., Joos G. F., Brusselle G. G. (2007) CC-chemokine receptors in chronic obstructive pulmonary disease. Inflamm. Allergy Drug Targets 6, 75–79 [DOI] [PubMed] [Google Scholar]
- 55. Culley F. J. (2009) Natural killer cells in infection and inflammation of the lung. Immunology 128, 151–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McCartney S., Vermi W., Gilfillan S., Cella M., Murphy T. L., Schreiber R. D., Murphy K. M., Colonna M. (2009) Distinct and complementary functions of MDA5 and TLR3 in poly(I: C)-mediated activation of mouse NK cells. J. Exp. Med. 206, 2967–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim K. D., Zhao J., Auh S., Yang X., Du P., Tang H., Fu Y. X. (2007) Adaptive immune cells temper initial innate responses. Nat. Med. 13, 1248–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schmidt K. N., Leung B., Kwong M., Zarember K. A., Satyal S., Navas T. A., Wang F., Godowski P. J. (2004) APC-independent activation of NK cells by the Toll-like receptor 3 agonist double-stranded RNA. J. Immunol. 172, 138–143 [DOI] [PubMed] [Google Scholar]
- 59. Lauzon N. M., Mian F., MacKenzie R., Ashkar A. A. (2006) The direct effects of Toll-like receptor ligands on human NK cell cytokine production and cytotoxicity. Cell. Immunol. 241, 102–112 [DOI] [PubMed] [Google Scholar]
- 60. Baccala R., Hoebe K., Kono D. H., Beutler B., Theofilopoulos A. N. (2007) TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 13, 543–551 [DOI] [PubMed] [Google Scholar]
- 61. Takeuchi O., Akira S. (2008) MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 20, 17–22 [DOI] [PubMed] [Google Scholar]
- 62. Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K. J., Yamaguchi O., Otsu K., Tsujimura T., Koh C. S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S. (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 [DOI] [PubMed] [Google Scholar]
- 63. Wang Q., Miller D. J., Bowman E. R., Nagarkar D. R., Schneider D., Zhao Y., Linn M. J., Goldsmith A. M., Bentley J. K., Sajjan U. S., Hershenson M. B. (2011) MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 7, e1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Andersson A., Srivastava M. K., Harris-White M., Huang M., Zhu L., Elashoff D., Strieter R. M., Dubinett S. M., Sharma S. (2011) Role of CXCR3 ligands in IL-7/IL-7R α-Fc-mediated antitumor activity in lung cancer. Clin. Cancer Res. 17, 3660–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Andersson A., Yang S. C., Huang M., Zhu L., Kar U. K., Batra R. K., Elashoff D., Strieter R. M., Dubinett S. M., Sharma S. (2009) IL-7 promotes CXCR3 ligand-dependent T cell antitumor reactivity in lung cancer. J. Immunol. 182, 6951–6958 [DOI] [PubMed] [Google Scholar]
- 66. Martinez F. O., Helming L., Gordon S. (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483 [DOI] [PubMed] [Google Scholar]
- 67. Martinez F. O., Gordon S., Locati M., Mantovani A. (2006) Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177, 7303–7311 [DOI] [PubMed] [Google Scholar]
- 68. Sica A., Bronte V. (2007) Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Invest. 117, 1155–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pettersen J. S., Fuentes-Duculan J., Suarez-Farinas M., Pierson K. C., Pitts-Kiefer A., Fan L., Belkin D. A., Wang C. Q., Bhuvanendran S., Johnson-Huang L. M., Bluth M. J., Krueger J. G., Lowes M. A., Carucci J. A. (2011) Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J. Invest. Dermatol. 131, 1322–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhou Z. H., Han Y., Wei T., Aras S., Chaturvedi P., Tyler S., Rani M. R., Ransohoff R. M. (2001) Regulation of monocyte chemoattractant protein (MCP)-1 transcription by interferon-gamma (IFN-γ) in human astrocytoma cells: postinduction refractory state of the gene, governed by its upstream elements. FASEB J. 15, 383–392 [DOI] [PubMed] [Google Scholar]
- 71. Van der Velden V. H., Verheggen M. M., Bernasconi S., Sozzani S., Naber B. A., van der Linden-van Beurden C. A., Hoogsteden H. C., Mantovani A., Versnel M. (1998) Interleukin-1β and interferon-γ differentially regulate release of monocyte chemotactic protein-1 and interleukin-8 by human bronchial epithelial cells. Eur. Cytokine Netw. 9, 269–277 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.