

Significance
Sharks are among the evolutionary oldest living organisms with an immune system that possesses a number of elements similar to ours, including antibodies. In this article, we present structural insights into one of the most ancient antibodies, shedding light on the molecular evolution of the immune system and the structural features of heavy chain-only antibodies. Sharks enrich urea in their blood to prevent osmotic loss of water in the marine environment. Urea, however, denatures proteins if they are not sufficiently stable. Indeed, we find that shark antibodies are particularly stable. We pinpointed specific features responsible for their high stability and found that transplanting them into a human antibody increased its secretion.
Keywords: protein evolution, antibody structure, protein folding, protein engineering
Abstract
Sharks and other cartilaginous fish are the phylogenetically oldest living organisms that rely on antibodies as part of their adaptive immune system. They produce the immunoglobulin new antigen receptor (IgNAR), a homodimeric heavy chain-only antibody, as a major part of their humoral adaptive immune response. Here, we report the atomic resolution structure of the IgNAR constant domains and a structural model of this heavy chain-only antibody. We find that despite low sequence conservation, the basic Ig fold of modern antibodies is already present in the evolutionary ancient shark IgNAR domains, highlighting key structural determinants of the ubiquitous Ig fold. In contrast, structural differences between human and shark antibody domains explain the high stability of several IgNAR domains and allowed us to engineer human antibodies for increased stability and secretion efficiency. We identified two constant domains, C1 and C3, that act as dimerization modules within IgNAR. Together with the individual domain structures and small-angle X-ray scattering, this allowed us to develop a structural model of the complete IgNAR molecule. Its constant region exhibits an elongated shape with flexibility and a characteristic kink in the middle. Despite the lack of a canonical hinge region, the variable domains are spaced appropriately wide for binding to multiple antigens. Thus, the shark IgNAR domains already display the well-known Ig fold, but apart from that, this heavy chain-only antibody employs unique ways for dimerization and positioning of functional modules.
The phylogenetically oldest living organisms identified that possess most major components of a vertebrate adaptive immune system are cartilaginous fish (Chondrichthyes) such as sharks, skates, and rays (1, 2). They shared the last common ancestor with other jawed vertebrates roughly 500 million years ago (2, 3). Accordingly, shark antibodies can provide unique insights into the molecular evolution of the immune system. Furthermore, shark antibodies have evolved under challenging conditions; for example, the high osmolarity of shark blood is partially sustained by the protein denaturant urea (4, 5). Even though it is partially counteracted by other osmolytes (6), shark antibodies are believed to be particularly stable (7). Insights into the structural features that provide this increased stability may provide attractive applications for biotechnology (8). Sharks and other Elasmobranchs have two conventional antibodies, IgM and IgW, but the structurally simplest antibody molecule in sharks is the so-called Ig new antigen receptor (IgNAR) (9). In its secreted form, it consists of two identical heavy chains (HCs) composed of one variable domain (V) and five constant domains (C1–C5) each (4, 9) (Fig. 1A). Similar to camelid antibodies, IgNARs are devoid of light chains (LCs) (9, 10), an example of convergent evolution (11). The variable domain of IgNAR, whose structure had been solved (12, 13), shows similarity to the variable domains of evolutionarily more recent immunoglobulins (12, 13). In contrast, its constant domains (C1–C5; Fig. 1A) are most homologous to the primordial IgW of sharks (14). Of the five human antibody classes, IgA, IgD, IgE, IgG, and IgM, IgW is most closely related to IgD, which, along with IgM, are the oldest Ig isotypes (14–17). Except for low-resolution electron microscopic images (18), no structural data are available for any of the constant IgNAR domains.
Fig. 1.
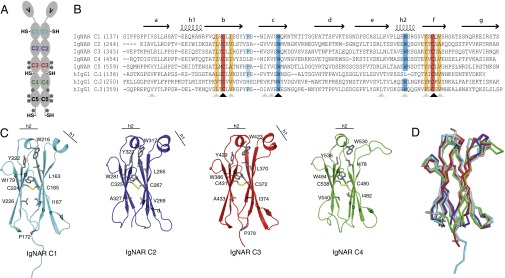
Sequence and structure of IgNAR domains C1–C4. (A) Schematic of the secreted dimeric IgNAR molecule, comprising one variable (V) and five constant (C1–C5) domains. Predicted glycosylation sites are shown as gray hexagons. Cysteines that are not part of the intradomain disulfide bridges are indicated (–SH). The secretory tail is C terminally of the C5 domain. (B) Sequence alignment of IgNAR C1–C5 with the human IgG1 HC domains CH1–CH3. Conserved cysteines are highlighted in red, and conserved hydrophobic residues of a YxCxY (Y, hydrophobic residue) motif around the disulfide bridge are highlighted in orange. Conserved tryptophans in strand c and the second helix are highlighted in blue, and the cis-proline residue in the loop between strand b and c is depicted in cyan. Secondary structure elements are indicated above the alignment. Black arrows indicate strictly conserved residues, and gray arrows homologous residues. (C) Ribbon diagram of the isolated constant IgNAR domains C1–C4 (C1, cyan; C2, blue; C3, red; C4, green; colors like in A). Residues marked in the alignment are shown in stick representation, the small helices are indicated. (D) Superposition of the IgNAR C1-4 domains (C1, cyan; C2, blue; C3, red; C4, green) on a human IgG CH3 domain (gray, Protein Data Bank ID code 1HZH).
Here, we determined the structures of the four N-terminal IgNAR constant domains (C1–C4) at atomic resolution and present a model for the complete IgNAR molecule that reveals key adaptations of HC-only antibodies. We identified structural elements that contribute to the high stability of some IgNAR domains and transferred these to human antibodies to improve their stability and secretion.
Results
The Structures of the IgNAR Constant Domains Reveal Key Elements of the Ig Fold and Differences to Modern Antibody Domains.
To determine the structure of IgNAR, we expressed the full-length protein, different truncation constructs, and individual domains in Escherichia coli. As we could not obtain crystals for the complete C1–C5 fragment or several multidomain subfragments, we proceeded with the individual domains. The structures of the IgNAR C1, C2, and C3 domains were determined by X-ray crystallography. The 3D structure of the C2 domain was solved by Patterson search techniques, with a resolution of 1.5 Å (Rfree = 21.6%), using the structure of the mouse λLC constant domain (CL; Protein Data Bank ID code 1IND) as starting point. The structure of C2 was used to determine the structures of the C1 and C3 domains by molecular replacement. For C1 and C3, resolutions of 2.7 Å (Rfree = 28.4%) and 2.8 Å (Rfree = 28.7%), respectively, were obtained. C4 was not amenable to crystallization, and thus the structure of the C4 domain was solved by NMR spectroscopy. Its structure is well-defined on the basis of 1,265 NOE-derived distance restraints, orientational restraints from residual dipolar coupling data, and dihedral angle restraints. Details can be found in the SI Appendix, Materials and Methods and SI Appendix, Tables S1 and S2. Recombinant C5 was unstructured (see following for details), and thus no high-resolution structure could be obtained.
The IgNAR domains have only limited sequence conservation in comparison with human Ig domains (Fig. 1B and SI Appendix, Fig. S1). However, C1–C4 all show a typical constant domain Ig fold (C1-type). Similar to most C1-type domains, these IgNAR domains consist of a two-layer sandwich structure, with strands b, c, e, and f forming the common core (Fig. 1C and SI Appendix, Fig. S1) (19, 20). In each case, the two layers of the β-sandwich are covalently linked by a buried disulfide bridge that is orientated roughly perpendicular to the sheets (Fig. 1 B and C and SI Appendix, Fig. S1) (19). Around this disulfide bridge, several hydrophobic residues form a tight core with a highly conserved tryptophan at its center, which is present in all IgNAR domains (Fig. 1 B and C and SI Appendix, Fig. S1). Essentially all known modern antibody C-type domain structures exhibit this core. Thus, most likely, this feature developed very early and remained one of its defining characteristics. In the C1–C3 domains of IgNAR, the β-sandwich layers are intercalated by two short α-helices, which are found in most antibody domains but are not generally present in other proteins with the Ig fold. In the C4 domain, only the second helix between strand e and f is formed (Fig. 1C). C1–C4 each contain a tryptophan in this second helix, which is highly conserved within Chordata (Fig. 1B and SI Appendix, Fig. S1). This tryptophan interacts with an almost equally conserved tyrosine or phenylalanine located C terminally (Fig. 1 B and C and SI Appendix, Fig. S1). The evolutionary conservation of this motif is in agreement with an important structural role in the antibody domain folding process (21) and suggests that, together with the core of the fold, it developed early in the evolution of antibody domains. Of note, in C2 and C4, a salt bridge exists between the second helix and the loop connecting strand c and d that is likely to stabilize the helix and this loop.
Despite the overall similarities, several differences exist between mammalian and IgNAR domains. One is the absence of a proline residue between strand b and c in some of the IgNAR domains (Fig. 1 B and C). In many mammalian antibody domains, this proline is in the cis state in the native structure, and thus, because of its slow isomerization, it leads to the greater population of folding intermediates, decisively influencing the domain folding reaction (22, 23). In the C1 and C3 domain, a cis-proline residue is located in this conserved position; however, no proline is found at the corresponding position in C2 or C4 (Fig. 1 B and C), suggesting that rate-limiting steps in the folding pathway for these IgNAR domains might differ. A further difference is that the first helix between strands a and b is not formed in the C4 domain (Fig. 1C). Instead, this region is unstructured and flexible (SI Appendix, Fig. S2) and contains two consecutive charged residues that are flanked by three hydrophobic residues, two of which are aromatic residues (F467-E-E-I-W471; Fig. 1B). These characteristics might suggest a role in interactions with other domains (e.g., the C5 domain) or Ig receptors that might be further modulated by a glycosylation site between the C4 and C5 domains (Fig. 1 A and C). Except for this unstructured loop in C4, the topologies of the shark domains are very similar to the ones observed for human antibody constant domains (Fig. 1D). The isolated C5 domain was an exception, as circular dichroism (CD) and fluorescence spectroscopy revealed the characteristics of an unfolded protein (SI Appendix, Fig. S3), which was also observed for a glycosylated C5 domain expressed in insect cells and in the context of a C4–C5 construct (SI Appendix, Fig. S3). In agreement with this notion, the isolated C5 domain did not form its internal disulfide bridge (SI Appendix, Fig. S3). Taken together, these data suggest that isolated IgNAR C5 is unfolded and that its folding depends on further factors.
A Structural Analysis of the Complete IgNAR Molecule Reveals Adaptations of Heavy Chain-Only Antibodies.
Even though IgNAR is known to be a covalent dimer (9), to date it has been unclear which of the IgNAR domains contribute to dimerization of the antibody molecule and how they interact. This cannot be easily deduced from the sequence, and different modes of dimerization are realized, for example, for IgM antibody domains (24). For IgNAR, we find that the isolated C1 and C3 domains each form a dimer, both in the crystal structure (Fig. 2 A and B) and in solution (SI Appendix, Fig. S4). The Kd for the C1 domain interaction was 595 ± 43 nM, whereas the C3 domain had a significantly higher dissociation constant of Kd = 188 ±16 μM (SI Appendix, Fig. S4), suggesting C1 dimerization drives dimer formation of the C3 domain. Contrary to the strong polar interactions, which are responsible for the domain pairing of, for example, the IgG CH3 domains (Fig. 2C), the IgNAR C1 dimer interface is dominated by hydrophobic interactions and contains only two hydrogen bonds (Fig. 2 B and C).
Fig. 2.

Characterization of the IgNAR C1 and C3 dimerization interfaces. (A) Ribbon and surface diagram of the IgNAR C1 and C3 dimers. The two subunits are colored in cyan and gray (C1) or red and gray (C3), respectively. (B) Hydrophobic residues within the C1 and C3 dimerization interfaces are shown in orange. One monomer is in surface representation; its counterpart is shown as a mesh surface. (C) Comparison of the dimerization interfaces of IgNAR C1 and C3 and different human and murine dimeric domains, as determined by the PISA server (48) (Protein Data Bank ID codes IgG CH3, 3HKF; IgA CH3, 1OW0; IgE CH4, 1O0V; IgM CH2, 4JVU; and IgM CH4, 4JVW).
Combining our results on domain interactions with the structural information available for the IgNAR variable domain (12, 13), and with the reported interchain disulfide bonds of the IgNAR molecule (9) allowed us to obtain a model for the complete IgNAR molecule (Fig. 3A). Of note, the wide angle of the C1 dimerization interface induces a significant distance and flexibility between the variable domains, which might be important for efficient antigen binding, whereas the small angle between the C3 domains leads to the formation of a narrow stalk for the IgNAR molecule. A flexible, disulfide-bridged linker connects the C3 and the C4 domains, restricting their relative positions but at the same time providing flexibility within the stalk (Fig. 3A). To assess our structural model, we performed small-angle X-ray scattering (SAXS) experiments on complete IgNAR molecules purified from shark serum (SI Appendix, Fig. S5). The SAXS measurements are consistent with the overall shape of our model (Fig. 3B and SI Appendix, Table S3) and suggested that in solution, a major fraction of IgNAR is kinked approximately in the middle of the molecule, where the flexible linker between C3 and C4 is located (Fig. 3 A and C), in agreement with previously published electron microscopic images (18). In structural models of IgNAR calculated using HADDOCK (25, 26), kinked clusters agree best when scored against the experimental SAXS data (see SI Appendix, Materials and Methods for details). An independent ab initio analysis of our SAXS data using DAMMIF (27) further supports the kinked conformation (Fig. 3C). The radius of gyration (Rg) derived from our SAXS data (67 Å) is slightly larger than the one calculated from our model (59 Å), suggesting that full-length IgNAR occupies a larger conformational space then a single model would imply, further supporting the idea that full-length IgNAR is flexible (Fig. 3A).
Fig. 3.

A model of the complete IgNAR antibody. (A) A structural model of full-length IgNAR based on restraints from the C1 and C3 dimer structures and intermolecular disulfide bonds, derived by docking with HADDOCK (26). (Left) Crystal structures of the variable domain (Protein Data Bank ID code 1SQ2) and constant domains C1–C4. Because of the lack of structural information for C5, it is drawn as a gray oval. Cysteines forming interchain disulfide bonds are highlighted in orange and predicted glycosylation sites in red, respectively. The arced arrows indicate flexibility within the molecule. (Right) The same model is shown in surface view, including a possible conformation of the flexible C4–C5, as adopted in the lowest energy structure of the best cluster, assessed by the HADDOCK score and SAXS χ2. (B) SAXS data of full-length IgNAR (cyan dots). The Q range above 0.25 Å−1 has been removed because of noise. The blue line corresponds to the back-calculated curve from the HADDOCK-derived structure with the best fit to the SAXS data (shown in A; χ2 = 1.43). (C) An ab initio bead model derived from the SAXS data, using DAMMIF (27), superimposed with the lowest-energy structure of the HADDOCK cluster with the best fit to the SAXS data, using SUPCOMB (49).
Of note, we find all predicted glycosylation sites in IgNAR to be surface-exposed (Fig. 3A); thus, their further modification would be in agreement with our model.
Defined Structural Elements Contribute to the High Stability of IgNAR Domains and Can Be Transferred to Human Antibodies.
Shark blood contains urea at concentrations of several hundred millimolar to prevent osmotic-driven water loss in a marine environment (4, 5). Urea is a protein denaturant, and thus shark antibodies are likely to be more stable than those from organisms that do not enrich urea in their blood (7, 8), even though sharks also accumulate the protein-stabilizing agent trimethylamine-N-oxide in their blood (6). When we individually analyzed the stability of the constant domains C1–C4, we found the C2 and C4 domains to be very resistant to chemical denaturation (Fig. 4 and SI Appendix, Fig. S6). The C1 and C3 domains, however, did not show a particularly high stability under the conditions tested (Fig. 4). C3 has two predicted glycosylation sites (Fig. 1A), and their modification would likely increase C3 stability. In addition, because of the high dissociation constant for C3 dimerization (Fig. 4 and SI Appendix, Fig. S4), isolated C3 is essentially monomeric under our experimental conditions, whereas in the context of the full-length IgNAR molecule, it will be a dimer resulting from induced proximity driven by C1 dimerization and interchain disulfide bridges (Fig. 4 and SI Appendix, Fig. S5).
Fig. 4.

Stability and oligomerization state of the constant IgNAR domains. C1–C4 were reversibly unfolded by guanidinium chloride (GdmCl) (C1, cyan; C2, blue; C3, red; C4, green; unfolding, closed circles; refolding, open circles). Midpoints of thermal transitions and thermodynamic parameters obtained by GdmCl-induced unfolding transitions are listed in the table (Tmelt, melting temperature; ΔG0unf, free energy of unfolding; meq, cooperativity parameter). The association state and dissociation constant Kd of the domains in solution were obtained by analytical ultracentrifugation. All data are shown ± SD.
In agreement with their high chemical stability, C2 and C4 also showed superior stabilities against elevated temperatures (Fig. 4 and SI Appendix, Fig. S6), which were higher than most monomeric mammalian antibody domains, which typically have melting temperatures ranging from about 40–60 °C (28–32). We hypothesized that these high stabilities were a result of well-defined structural differences between human antibody domains and C2/C4, and thus searched for motifs that are present in C2 and C4 but absent in human antibody domains. We identified two structural elements: the aforementioned salt bridge between the loop separating strand c and d and the second helix connecting strand e and f, and an extended hydrophobic core in C2 and C4 resulting from an additional Val residue in strand d (SI Appendix, Fig. S7). With a view to test whether these elements could improve the properties of evolutionary more-recent Ig domains, we transplanted these motifs into the monomeric human Ig κLC CL domain and assessed their effect on its stability. When both the additional salt bridge (mutant M1) and the extended hydrophobic core (mutant M2) were introduced together into the CL domain (mutant M1+2), a significant stabilization was achieved. The melting point of M1+2 was almost 10 °C higher (67.0 ± 0.4 °C) than that of the wild-type (wt) CL domain (58.3 ± 0.3 °C) (Fig. 5A and SI Appendix, Fig. S8), and its stability against urea was markedly increased (23.1 ± 0.8 kJ/mol for M1+2 compared with 15.0 ± 0.2 kJ/mol for the wt CL domain) (Fig. 5A). Separately, M1 and M2 each had only a modest effect on the stability of the CL domain (Fig. 5A and SI Appendix, Fig. S8). Importantly, all CL mutants tested still associated with the HC CH1 domain, its authentic partner domain, and induced its folding (SI Appendix, Fig. S8), which is a critical test for their structural integrity (33).
Fig. 5.

The effect of IgNAR-based mutations of a human CL domain on domain stability and antibody secretion. (A) Thermal stabilities and thermodynamic parameters derived from urea melts are shown for the wt CL domain and mutants M1, M2, and M1+2. Urea melts are shown on top (wt, gray; M1, red; M2, blue; M1+2, black; unfolding, closed circles; refolding, open circles). (B) LCWT, LCM1, LCM2, and LCM1+2 or empty vector (pSVL) were coexpressed with γHCs, as indicated. Cells were metabolically labeled for 1 h and either immediately lysed (lysate) or subsequently chased for 24 h before analysis of the medium (medium). Lysates were immunoprecipitated with anti-FLAG antibody to capture the LCs together with protein A to isolate the HCs; media were immunoprecipitated with protein A only to detect LC-induced HC secretion. (C) HC secretion data were quantified by phosphorimager analysis [n = 7 ± SD; data that are statistically significantly different from the wt (P ≤ 0.05) are marked with an asterisk].
On the basis of these improved biophysical characteristics, our mutants were engineered into the CL domain in the context of a full-length κLC and assessed for their ability to exert their advantageous characteristics in mammalian cells, where the accuracy of antibody folding and assembly are monitored by quality control processes in the endoplasmic reticulum (ER) (34). We expressed the different LC variants, comprising mutants M1, M2, or M1+2, either alone or together with IgG HCs (γHCs) in COS-1 cells (35), and compared them with wild-type κLC. When the various LCs were expressed alone, we found less expression of LCM2 in the cell lysate, which was not a result of enhanced secretion (SI Appendix, Fig. S9), even though the CL domain possessing this mutation was more stable in vitro. The other two κLC mutants were expressed at similar or slightly higher levels than LCWT in the cell lysate, and there was a significant increase in secretion of the combined mutant LCM1+2 into the culture medium compared with LCwt (SI Appendix, Fig. S9).
Importantly, when the different LCs were coexpressed with a γHC, we found a significant increase in the secretion of completely assembled IgG antibody molecules with the LCM1+2 mutant compared with LCWT (Fig. 5 B and C). Of note, directly after the metabolic labeling pulse, we reproducibly detected higher levels of γHC upon coexpression of LCM1+2 than when the γHC was expressed alone or with any of the other LC constructs (Fig. 5B). LCs can already associate cotranslationally with HCs (36–38). It is thus possible that the LCM1+2 mutant, which also shows enhanced properties in vivo in isolation (SI Appendix, Fig. S9), is already exerting beneficial effects on γHCs during chain synthesis, leading to an increased yield of full-length HCs. In keeping with the reduced secretion of free LCM2 (SI Appendix, Fig. S9), it showed a decreased ability to induce transport of γHCs to the media (Fig. 5 B and C). Together, our data demonstrate that the IgNAR-based optimization of a human CL domain, the scaffold on which the HC CH1 domain has to fold for antibodies to pass ER quality control (33, 35), can increase domain stability and positively influences limiting steps in antibody biosynthesis in the cell.
Discussion
The atomic resolution structures of four IgNAR constant domains show that the Ig fold has been conserved for ca. 500 million years, since the last common ancestor of shark and men. Comparison of Ig domains from both species, which are not well conserved at the sequence level, allowed the identification of highly conserved structural features of the Ig fold in antibodies. These consist of the hydrophobic core surrounding the internal disulfide bridge, including an evolutionary conserved tryptophan residue. In addition, we found the small helix between strands e and f of evolutionary more recent antibody domains to also be present in C1–C4. A highly conserved tryptophan in this helix interacts with an almost equally conserved aromatic residue located C terminally, arguing that this motif is evolutionary ancient and contributes to the stability and robust folding pathway of antibody domains, as opposed to some other Ig domains, which usually do not possess these elements (23, 39, 40).
A peculiarity of IgNAR was that we found C5 to be unfolded by all methods we used to measure structure. Similarly, the CH1 domain of IgG, which does not possess the characteristic sequence elements of intrinsically disordered proteins (41), is unfolded in isolation (33). It plays a critical role in IgG assembly control (33, 35, 42), and analogously, C5 folding might depend on assembly of the complete IgNAR chains. Alternatively, authentic glycosylation or extrinsic cellular factors might be required for C5 folding.
Despite similarities to more modern antibodies, IgNAR also shows major evolutionary adjustments as a HC-only antibody. The complementarity-determining region 3 (CDR3) loop of its variable domain is particularly extended, which allows more sequence diversity to be accommodated and the binding of cryptic antigens to occur. Furthermore, the variable domain is stabilized by additional internal disulfide bridges compared with variable domains that undergo dimerization in typical HC-LC antibodies (12, 13). Interestingly, CDR3 extension combined with disulfide bridge stabilization seems to have evolved multiple times in nature to extend the antibody-binding repertoire of either HC-only antibodies (10, 12, 13, 43) or in species with a limited repertoire of variable domains (44).
To perform their biological functions, antibodies often bind spatially separated epitopes on antigens. Camelid antibodies have lost the ability to pair with LCs, yet retained a hinge region that allows orientational flexibility of their variable domains (10). For IgNAR, it has been less clear what allows the observed orientational flexibility of their variable domains (18) and adequate spacing to bind multiple antigens. Our structural model of the complete IgNAR molecule explains how, despite the absence of a canonical hinge region, IgNAR might still be able to bind spatially separated epitopes: the wide angle of C1 dimerization positions the IgNAR variable domains appropriately. These are followed by a narrow stalk with intrinsic flexibility. In solution, a major fraction of IgNAR seems to be kinked in the stalk, and the flexibility of IgNAR might be of relevance for binding to cellular receptors or accessing confined sites of antigen presentation.
Given the very different solvent environments in which antibodies from sharks and men must function, it was possible that IgNAR domains contain structural elements that could influence their folding pathway and enhance their stability. Indeed, we could identify two well-defined structural features, which contributed to the high stability of some IgNAR domains. Individually, these elements each led to a slight stabilization of a human CL domain in vitro, which became quite pronounced when these two elements were combined. Such a simple additive effect was not observed in vivo. Only the combination of both mutations led to a significant increase in both the secretion of free LC and assembled IgG antibody, whereas one mutant (M2) actually decreased both. This might be a result of the lower folding cooperativity observed for M2 in comparison with the other two mutants, which, together with its extended hydrophobic core, could possibly account for less-efficient folding in vivo in the presence of the ER quality control machinery. Alternatively, it is possible that elements in the particular VL domain of the antibody we investigated abrogate the positive effects of this mutation on the stability of the CL domain. The significantly increased stability and secretion of the combination mutant make it tempting to speculate that it might be useful for future applications in diagnostic and therapeutic antibodies, in particular, as the stabilizing structural motif we have identified could also be transplanted to other domains.
In conclusion, the first structures of antibody constant domains from sharks reveal key structural elements of the Ig fold that have been conserved for hundreds of millions of years. The results of this study also show that although the oldest antibody domains known already exhibit a “modern” overall fold, the IgNAR molecule shows structural adaptations to specifically organize its functional modules.
Materials and Methods
Protein Expression and Purification.
Genes were cloned using either the IgNAR cDNA or synthetic, E. coli codon-usage optimized templates. Proteins were expressed in E. coli or insect cells, and authentic IgNAR was purified from shark serum. Details can be found in the SI Appendix, Materials and Methods.
Optical Spectroscopy and Protein Stability Measurements.
CD and fluorescence spectra were measured as described in the SI Appendix, Materials and Methods. GdmCl/urea-induced unfolding and temperature-induced unfolding transitions were followed by far-UV CD spectroscopy or fluorescence spectroscopy and analyzed as described in the SI Appendix, Materials and Methods. Association-induced folding of a human CH1 domain by the different CL constructs was analyzed as published (33).
Analytical Ultracentrifugation.
Experiments were performed as described in the SI Appendix, Materials and Methods. For C1 and C3, the Kd was determined using a monomer-dimer self-association model.
Structure Determination.
Crystallization conditions were identified using different crystallization suites. Data sets were collected either using synchrotron radiation (SLS) or on a Bruker Microstar/X8 Proteum (Bruker AXS). Data sets were processed and models completed as described in the SI Appendix, Materials and Methods. Molecular illustrations were prepared in PyMOL (DeLano Scientific).
For NMR backbone assignments, triple resonance experiments were performed, where magnetization is transferred between the nuclei of interest [CBCA(CO)NH and CBCANH]. For side chain assignments, total correlation spectroscopy was used (HCCH-TOCSY). Distance restraints were obtained from 15N- and 13C-edited NOESY spectra. Residual dipolar couplings were recorded using an IPAP-HSQC (45) and HNCO-based NMR experiments (46). NMR spectra were processed and analyzed, and structures were calculated as described in the SI Appendix, Materials and Methods.
Docking.
A full-length dimeric IgNAR model was built using MODELLER 9.4 (47). Docking of the identical protein chains was performed using the HADDOCK Web server (25, 26), with structural restraints based on C1 and C3 dimerization and the intermolecular disulfide bridges. Details can be found in the SI Appendix, Materials and Methods.
SAXS.
SAXS measurements were performed on a Rigaku BioSAXS1000, using a Pilatus detector, as described in the SI Appendix, Materials and Methods.
Cell Culture, Metabolic Labeling, and Western Blots.
LC constructs were amplified from synthetic genes optimized for human expression. Mutants were generated by site-directed mutagenesis. Experiments were performed in COS-1 cells, using a previously described γHC construct (35). Experiments were performed as described in the SI Appendix, Materials and Methods.
Statistical Analysis.
Results are shown as means ± SD. Where indicated, a two-tailed Student t test was used to analyze the data.
Supplementary Material
Acknowledgments
We thank the staff of the SLS (Villigen, Switzerland) for their assistance in X-ray data collection. Initial model building of C2 was carried out at the CCP4 workshop, supported by the National Cancer Institute (Grant Y1-CO-1020) and the National Institute of General Medical Sciences (Grant Y1-GM-1104). We thank the Bavarian NMR Center and the TUM SFB1035 for NMR and SAXS measurement time, respectively, Dr. Ralf Stehle for assisting the SAXS measurements, and Helmut Krause for performing mass spectrometry. We are grateful to Muralidhar Reddivari and the protein production facility staff of St. Jude Children’s Research Hospital for protein production in insect cells. This work was supported by grants from the National Institutes of Health [R01 OD010549-21 (to M.F.); R01 GM54068 (to L.M.H.)] and the Deutsche Forschungsgemeinschaft (to M.S., M.G., and J. Buchner). Funding was also received from the Leopoldina-National Academy of Sciences [Grant LPDS 2009-32 (to M.J.F.)], the Paul-Barrett endowed fellowship of St. Jude Children’s Research Hospital (to M.J.F.), the Peter und Traudl Engelhornstiftung (to M.A.G.), the Studienstiftung des Deutschen Volkes (to M.M. and J.P.), the European Molecular Biology Organization (ALTF-276-2010) and the Swedish Research Council (Vetenskapsrådet) to J.H. and the Boehringer Ingelheim Fonds (to J. Behnke).
Footnotes
Conflict of interest statement: A patent for optimized antibodies based on the results presented in this study has been filed.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 4Q97 (C1), 4Q9B (C2), 4Q9C (C3), and 2MKL (C4)]. The NMR chemical shifts have been deposited in the BioMagResBank, www.bmrb.wisc.edu (accession no. 19783).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321502111/-/DCSupplemental.
References
- 1.Cooper MD, Alder MN. The evolution of adaptive immune systems. Cell. 2006;124(4):815–822. doi: 10.1016/j.cell.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: Genetic events and selective pressures. Nat Rev Genet. 2010;11(1):47–59. doi: 10.1038/nrg2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blair JE, Hedges SB. Molecular phylogeny and divergence times of deuterostome animals. Mol Biol Evol. 2005;22(11):2275–2284. doi: 10.1093/molbev/msi225. [DOI] [PubMed] [Google Scholar]
- 4.Dooley H, Flajnik MF. Antibody repertoire development in cartilaginous fish. Dev Comp Immunol. 2006;30(1-2):43–56. doi: 10.1016/j.dci.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 5.England JL, Haran G. Role of solvation effects in protein denaturation: From thermodynamics to single molecules and back. Annu Rev Phys Chem. 2011;62:257–277. doi: 10.1146/annurev-physchem-032210-103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yancey PH, Somero GN. Counteraction of urea destabilization of protein structure by methylamine osmoregulatory compounds of elasmobranch fishes. Biochem J. 1979;183(2):317–323. doi: 10.1042/bj1830317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frommel D, Litman GW, Finstad J, Good RA. The evolution of the immune response. XI. The immunoglobulins of the horned shark, Heterodontus francisci: Purification, characterization and structural requirement for antibody activity. J Immunol. 1971;106(5):1234–1243. [PubMed] [Google Scholar]
- 8.Saerens D, Ghassabeh GH, Muyldermans S. Single-domain antibodies as building blocks for novel therapeutics. Curr Opin Pharmacol. 2008;8(5):600–608. doi: 10.1016/j.coph.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Greenberg AS, et al. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature. 1995;374(6518):168–173. doi: 10.1038/374168a0. [DOI] [PubMed] [Google Scholar]
- 10.Hamers-Casterman C, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363(6428):446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 11.Flajnik MF, Deschacht N, Muyldermans S. A case of convergence: Why did a simple alternative to canonical antibodies arise in sharks and camels? PLoS Biol. 2011;9(8):e1001120. doi: 10.1371/journal.pbio.1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanfield RL, Dooley H, Flajnik MF, Wilson IA. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science. 2004;305(5691):1770–1773. doi: 10.1126/science.1101148. [DOI] [PubMed] [Google Scholar]
- 13.Streltsov VA, et al. Structural evidence for evolution of shark Ig new antigen receptor variable domain antibodies from a cell-surface receptor. Proc Natl Acad Sci USA. 2004;101(34):12444–12449. doi: 10.1073/pnas.0403509101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berstein RM, Schluter SF, Shen S, Marchalonis JJ. A new high molecular weight immunoglobulin class from the carcharhine shark: Implications for the properties of the primordial immunoglobulin. Proc Natl Acad Sci USA. 1996;93(8):3289–3293. doi: 10.1073/pnas.93.8.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohta Y, Flajnik M. IgD, like IgM, is a primordial immunoglobulin class perpetuated in most jawed vertebrates. Proc Natl Acad Sci USA. 2006;103(28):10723–10728. doi: 10.1073/pnas.0601407103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenberg AS, et al. A novel “chimeric” antibody class in cartilaginous fish: IgM may not be the primordial immunoglobulin. Eur J Immunol. 1996;26(5):1123–1129. doi: 10.1002/eji.1830260525. [DOI] [PubMed] [Google Scholar]
- 17.Hsu E, Pulham N, Rumfelt LL, Flajnik MF. The plasticity of immunoglobulin gene systems in evolution. Immunol Rev. 2006;210:8–26. doi: 10.1111/j.0105-2896.2006.00366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roux KH, et al. Structural analysis of the nurse shark (new) antigen receptor (NAR): Molecular convergence of NAR and unusual mammalian immunoglobulins. Proc Natl Acad Sci USA. 1998;95(20):11804–11809. doi: 10.1073/pnas.95.20.11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bork P, Holm L, Sander C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol. 1994;242(4):309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- 20.Hamill SJ, Steward A, Clarke J. The folding of an immunoglobulin-like Greek key protein is defined by a common-core nucleus and regions constrained by topology. J Mol Biol. 2000;297(1):165–178. doi: 10.1006/jmbi.2000.3517. [DOI] [PubMed] [Google Scholar]
- 21.Feige MJ, et al. The structure of a folding intermediate provides insight into differences in immunoglobulin amyloidogenicity. Proc Natl Acad Sci USA. 2008;105(36):13373–13378. doi: 10.1073/pnas.0802809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goto Y, Hamaguchi K. Unfolding and refolding of the constant fragment of the immunoglobulin light chain. J Mol Biol. 1982;156(4):891–910. doi: 10.1016/0022-2836(82)90146-2. [DOI] [PubMed] [Google Scholar]
- 23.Feige MJ, Hendershot LM, Buchner J. How antibodies fold. Trends Biochem Sci. 2010;35(4):189–198. doi: 10.1016/j.tibs.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Müller R, et al. High-resolution structures of the IgM Fc domains reveal principles of its hexamer formation. Proc Natl Acad Sci USA. 2013;110(25):10183–10188. doi: 10.1073/pnas.1300547110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5(5):883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 26.Dominguez C, Boelens R, Bonvin AM. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125(7):1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 27.Franke D, Svergun DI. DAMMIF, a program for rapid ab-inito shape determination in small-angle scattering. J Appl Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feige MJ, et al. Dissecting the alternatively folded state of the antibody Fab fragment. J Mol Biol. 2010;399(5):719–730. doi: 10.1016/j.jmb.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 29.Feige MJ, Walter S, Buchner J. Folding mechanism of the CH2 antibody domain. J Mol Biol. 2004;344(1):107–118. doi: 10.1016/j.jmb.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 30.Hellman R, Vanhove M, Lejeune A, Stevens FJ, Hendershot LM. The in vivo association of BiP with newly synthesized proteins is dependent on the rate and stability of folding and not simply on the presence of sequences that can bind to BiP. J Cell Biol. 1999;144(1):21–30. doi: 10.1083/jcb.144.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goto Y, Hamaguchi K. Role of amino-terminal residues in the folding of the constant fragment of the immunoglobulin light chain. Biochemistry. 1987;26(7):1879–1884. doi: 10.1021/bi00381a014. [DOI] [PubMed] [Google Scholar]
- 32.Gong R, et al. Engineered human antibody constant domains with increased stability. J Biol Chem. 2009;284(21):14203–14210. doi: 10.1074/jbc.M900769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feige MJ, et al. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell. 2009;34(5):569–579. doi: 10.1016/j.molcel.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 35.Lee YK, Brewer JW, Hellman R, Hendershot LM. BiP and immunoglobulin light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Mol Biol Cell. 1999;10(7):2209–2219. doi: 10.1091/mbc.10.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shapiro AL, Scharff MD, Maizel JV, Jr, Uhr JW. Polyribosomal synthesis and assembly of the H and L chains of gamma globulin. Proc Natl Acad Sci USA. 1966;56(1):216–221. doi: 10.1073/pnas.56.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bole DG, Hendershot LM, Kearney JF. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J Cell Biol. 1986;102(5):1558–1566. doi: 10.1083/jcb.102.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergman LW, Kuehl WM. Formation of intermolecular disulfide bonds on nascent immunoglobulin polypeptides. J Biol Chem. 1979;254(13):5690–5694. [PubMed] [Google Scholar]
- 39.Goto Y, Yagi H, Yamaguchi K, Chatani E, Ban T. Structure, formation and propagation of amyloid fibrils. Curr Pharm Des. 2008;14(30):3205–3218. doi: 10.2174/138161208786404146. [DOI] [PubMed] [Google Scholar]
- 40.Eichner T, Radford SE. Understanding the complex mechanisms of β2-microglobulin amyloid assembly. FEBS J. 2011;278(20):3868–3883. doi: 10.1111/j.1742-4658.2011.08186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He B, et al. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009;19(8):929–949. doi: 10.1038/cr.2009.87. [DOI] [PubMed] [Google Scholar]
- 42.Hendershot L, Bole D, Köhler G, Kearney JF. Assembly and secretion of heavy chains that do not associate posttranslationally with immunoglobulin heavy chain-binding protein. J Cell Biol. 1987;104(3):761–767. doi: 10.1083/jcb.104.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muyldermans S, Atarhouch T, Saldanha J, Barbosa JA, Hamers R. Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 1994;7(9):1129–1135. doi: 10.1093/protein/7.9.1129. [DOI] [PubMed] [Google Scholar]
- 44.Wang F, et al. Reshaping antibody diversity. Cell. 2013;153(6):1379–1393. doi: 10.1016/j.cell.2013.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cordier F, Dingley AJ, Grzesiek S. A doublet-separated sensitivity-enhanced HSQC for the determination of scalar and dipolar one-bond J-couplings. J Biomol NMR. 1999;13(2):175–180. doi: 10.1023/a:1008301415843. [DOI] [PubMed] [Google Scholar]
- 46.Yang D, Kay LE. Improved 1HN-detected triple resonance TROSY-based experiments. J Biomol NMR. 1999;13(1):3–10. doi: 10.1023/A:1008329230975. [DOI] [PubMed] [Google Scholar]
- 47.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 48.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 49.Kozin M, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Cryst. 2001;34:33–41. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.