

Significance
Cytokine signaling is essential for cell growth, hematopoiesis, and immune system function. Cytokine-mediated receptor dimerization induces intracellular activation of receptor-bound Janus kinases (JAKs), which then induce downstream transcriptional responses. We have determined a two-domain crystal structure containing the pseudokinase and kinase domains from the JAK family member TYK2, which identifies an inhibitory interaction interface between the two domains. Cancer-associated mutations found in other JAK family members map to this inhibitory interaction site, whereas analogous mutations in TYK2 cause in vitro activation of the kinase. This study identifies a mechanism for pseudokinase-mediated autoinhibition of the TYK2 kinase domain and suggests a means by which cancer-associated JAK mutations induce aberrant kinase activity.
Keywords: JAK1, JAK3
Abstract
Janus kinases (JAKs) are receptor-associated multidomain tyrosine kinases that act downstream of many cytokines and interferons. JAK kinase activity is regulated by the adjacent pseudokinase domain via an unknown mechanism. Here, we report the 2.8-Å structure of the two-domain pseudokinase–kinase module from the JAK family member TYK2 in its autoinhibited form. We find that the pseudokinase and kinase interact near the kinase active site and that most reported mutations in cancer-associated JAK alleles cluster in or near this interface. Mutation of residues near the TYK2 interface that are analogous to those in cancer-associated JAK alleles, including the V617F and “exon 12” JAK2 mutations, results in increased kinase activity in vitro. These data indicate that JAK pseudokinases are autoinhibitory domains that hold the kinase domain inactive until receptor dimerization stimulates transition to an active state.
Helical bundle cytokines of the interleukin and IFN families regulate a wide variety of immune and cellular growth responses (1). These signaling pathways are initiated by cytokine-mediated receptor dimerization and subsequent activation of nonreceptor tyrosine kinases called Janus kinases (JAKs) that are associated with the receptor intracellular domains (2). JAK activation leads to phosphorylation of the receptor and recruitment of STAT family transcription factors, which are then phosphorylated by the JAK and translocated to the nucleus where they induce gene transcription. The JAK family consists of four members (JAK1, JAK2, JAK3, and tyrosine kinase 2 or TYK2), each of which binds a distinct set of cytokine receptor subtypes and hence has a unique gene knockout phenotype, ranging from embryonic lethality due to neuronal and erythropoietic defects (JAK1 and JAK2, respectively) (3–5), to profound immune system deficiencies (JAK3 and TYK2) (6).
The JAK kinases are large (∼1,150-aa) multidomain proteins whose primary sequences were initially organized into seven JAK-homology (JH) domains that reflected distinct regions of high sequence homology (7). Subsequent structure predictions indicated that JAKs contain four distinct domains: N-terminal 4.1, Ezrin, Radixin, Moesin (FERM) and Src homology 2 (SH2) domains, followed by C-terminal pseudokinase and kinase domains (7, 8) (Fig. 1A). The FERM and SH2 domains constitute the receptor-binding module (2), whereas the pseudokinase, which has a canonical kinase fold but lacks key catalytic residues, is thought to regulate kinase activity. Deletion of the pseudokinase in JAK2 and JAK3 increases basal kinase activity and deregulates signaling through cognate receptors (9, 10). Additionally, the JAK2 pseudokinase and kinase domains have been reported to coimmunoprecipitate, and coexpression of the JAK2 pseudokinase domain inhibits activity of the isolated kinase domain (10). Recent work on the JAK2 pseudokinase domain indicates that it has weak catalytic activity and autophosphorylates two inhibitory sites within the SH2-pseudokinase linker and pseudokinase N-lobe (11, 12). However, these JAK2 phosphorylation sites are not universally conserved in the other JAK family members (13), suggesting this may be a JAK2-specific regulatory pathway. Moreover, the in vitro activity of the isolated JAK2 pseudokinase–kinase protein is reduced and the Km value for ATP increased compared with the kinase domain alone (14), suggesting there may also be a more direct mechanism by which the pseudokinase regulates enzymatic activity.
Fig. 1.

Crystal structure of the TYK2 pseudokinase–kinase. (A) Simplified domain structure of TYK2 indicating the boundaries of the pseudokinase–kinase construct used in this study. (B) Diagram of the pseudokinase (blue) and kinase (tan) complex. From this view, the N and C termini of the crystallized construct are visible. Protein backbone is shown with the all-atom surface of the kinase domain displayed to aid consideration of the interaction interface. The kinase inhibitor molecule compound 7012 is shown in the active site as spheres (carbon atoms colored purple). (C) The diagram from B is rotated 180°, with pseudokinase and kinase represented in the same color, but the surface of the pseudokinase is shown. In this view, the region of the interdomain linker between the pseudokinase and kinase are visible, with a distance measurement and a dashed line shown to illustrate the connection made by the unobserved peptide linkage. Simplified diagrams are shown beneath each panel for orientation.
Although JAK kinase inhibition has proven to be an attractive drug target for a number of immune indications (15), the discovery of the JAK2 V617F mutation as causative for polycythemia vera and other myeloproliferative neoplasms (MPNs) has prompted the investigation of all JAK family members in a number of malignancies (16). Recent cancer genome sequencing studies have shown that assorted mutations in JAK1, JAK2, and JAK3 can also induce myeloid neoplasias as well as a smaller number of solid tumors (16, 17). Most of these mutations cluster in and around the N-lobe of the pseudokinase domain near the JAK2 V617F mutation, with a smaller subset in the N-lobe of the kinase domain. Functional screening for constitutively active JAK2 alleles has also identified several point mutations in the pseudokinase N-lobe (18). Importantly, JAK2 V617F is a kinase-activating mutation, increasing JAK2 activity in both cell-based assays (17) and in vitro in the context of the isolated JAK2 pseudokinase–kinase module (14). Analogous mutations made in both JAK1 (L653F) and TYK2 (V678F) also activate their respective kinase domains (19). These data suggest that cancer-associated pseudokinase mutations relieve inhibition of JAK kinase activity, and therefore JAK pseudokinase domains have a tumor-suppressing function.
Although recent work has provided valuable structural information about the JAK kinase (20–22) and pseudokinase (11, 13) domains, the molecular mechanism by which JAK kinase activity is regulated by the pseudokinase is poorly understood. A recent structure of the JAK1 pseudokinase (13) provides tantalizing evidence that its N terminus may act as a switch controlling JAK1 kinase activation. This N-terminal segment corresponds to exon 12 in JAK2, which contains a number of MPN-associated mutations (23). We build on this finding by presenting a 2.8-Å crystal structure of the pseudokinase and kinase domains of TYK2, which shows that the TYK2 pseudokinase interacts with the kinase via a composite interface formed by the pseudokinase N-lobe and the N-lobe “exon 12” segment. Mapping of tumor-derived, activating JAK mutations onto our structure, along with kinase activity measurements of interface mutants, indicates this is an autoinhibitory interface that, when disrupted, relieves kinase inhibition and can initiate JAK-mediated malignancies.
Results
Architecture of the TYK2 Pseudokinase–Kinase Module.
To initiate structural studies of the TYK2 pseudokinase–kinase protein, we used insect cells to express a kinase-inactive (D1023N) version of human TYK2 residues 566–1187. Despite high expression levels and good solution behavior, this fragment of TYK2 failed to crystallize in its apo-state. Addition of a TYK2-specific ATP-competitive inhibitor (24) to the protein sample enabled crystallization, and we obtained a structure of the pseudokinase–kinase module at a resolution of 2.8 Å (Table S1 and Fig. S1). The structure contains one pseudokinase and one kinase domain in the asymmetric unit. After model refinement, a 17-aa pseudokinase–kinase interdomain linker remained unresolved in electron density maps. To determine the proper linkage for a pseudokinase–kinase monomer within the crystal lattice, we sought symmetry-related pseudokinase domains that had crystal contacts with the kinase domain. We found three possible pseudokinase–kinase combinations, and measured distances between the observed C terminus of the pseudokinase (Pro871) and observed N terminus of the kinase domain (Pro889) for each complex (Fig. S2). One of the three possible combinations showed a distance of 26 Å, which was less than one-half the second shortest distance (54 Å). Given that a 17-aa linker could extend to an absolute maximum distance of 63 Å (at 3.7 Å per amino acid residue), we hypothesized that the pair with shortest linker distance likely is the covalently linked combination and therefore might represent a physiologically relevant arrangement. The interface between this pseudokinase–kinase pair is generated by the back side of the pseudokinase N-lobe (opposite the active site cleft) as well the N-terminal exon 12 segment, tucked up underneath the kinase N-lobe sheet against the hinge region next to the active site (Fig. 1 B and C). Using this model, we analyzed of the overall structure and JAK family mutations, allowing us to validate this as a bona fide inhibitory conformation.
Structural Analysis of the TYK2 Kinase and Pseudokinase Domains.
The TYK2 kinase domain adopts the characteristic bilobate protein kinase fold and has high similarity to previously reported structures (21, 25) (Fig. S3A). Superposition with TYK2 [Protein Data Bank (PDB) ID code 3NZ0] shows a Cα rms deviation (rmsd) of 0.9 Å. There are three loops (residues 933–939, 1035–1036, and 1114–1118) without discernable electron density and presumed to be disordered. The TYK2 inhibitor (compound 7012) interacts with the kinase hinge via both the amide nitrogen and carbonyl oxygen atoms of residue Val981 as has been shown previously for this series of compounds (24) (Fig. S4B). Interestingly, the kinase domain activation loop (A-loop) resides in an active-like conformation, with minor backbone rearrangement relative to 3NZ0 likely due to the presence of a crystal contact with a neighboring symmetry-related molecule. As for the structure of TYK2 kinase domain (25), we do not see evidence of A-loop phosphorylation in the electron density or in mass-spectrometric analysis (discussed below).
The TYK2 pseudokinase domain also adopts the bilobate kinase fold, and has high secondary structure similarity to the published JAK1 and JAK2 pseudokinase domains (11, 13) (Fig. 2A), as well as to an unpublished TYK2 pseudokinase in PDB (PDB ID code 3ZON) (Cα rmsd of 0.5 Å) (Fig. S3B). We also find compound 7012 bound within the active site of the pseudokinase. The overall hinge-binding mode is similar to that seen for the kinase domain, with the pyridyl-amide moiety interacting with residue Val690 (Fig. 2B and Fig. S4C). At the same time, compound 7012 is rotated about 30° away from the pseudo-DFG motif (DPG 759–761) and catalytic Lys (Lys642). A single residue change at the bottom of the binding pocket (Gly1040 in kinase, Ser758 in pseudokinase) likely forces the chloro-phenyl ring upward, and this shift can be accommodated due to the more open nature of the pseudokinase active site cleft (Fig. S4C).
Fig. 2.
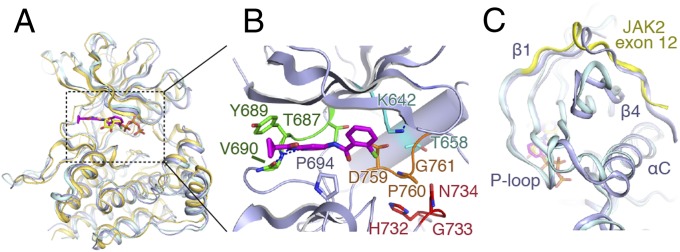
Comparison of the TYK2, JAK1, and JAK2 pseudokinases. (A) Overlay of the TYK2 pseudokinase domain (blue) with the pseudokinase from JAK1 (PDB ID code 4L00; gold) and JAK2 (PDB ID code 4FVQ; cyan). Protein chain is shown as a cartoon model, with ligands shown as sticks (TYK2-bound compound 7012 in magenta, JAK2-bound ATP in yellow). (B) Close-up view of the TYK2 pseudokinase active site. Compound 7012 is shown in magenta, and the positions of canonical kinase features colored [hinge: green; HGN (∼“HRD”) motif: red; DPG (∼“DFG”) motif: orange; αC-helix salt bridge residues: cyan]. (C) View of the exon 12 N-terminal segment of JAK2 compared with TYK2. Protein chains are colored as in A, with the JAK2 exon 12 segment colored yellow.
As in the other JAK pseudokinases, the TYK2 pseudokinase is lacking a number of the structural hallmarks of an active kinase. Specifically, three key active site motifs, the αC helix Glu-β3 Lys salt bridge, DFG, and HRD motifs are altered (Fig. 2B). The canonical Glu-Lys salt bridge between the αC-helix and the β3-strand is replaced by a Thr658–Lys642 pair that does not make contact. The Asp/Phe/Gly (DFG) motif is replaced by Asp/Pro/Gly motif (residues 759–761), with the catalytic Asp residue deeply recessed inside the kinase cleft as seen in JAK2 (11). The His/Arg/Asp (HRD) motif is replaced by His/Gly/Asn sequence (residues 732–734), with Asn734 replacing the canonical phosphate-binding aspartate and Gly733 replacing the activation loop-stabilizing arginine from HRD. Additionally, the pseudokinase A-loop is 7 aa shorter than that of the kinase domain (Fig. S4A) and contains no potential tyrosine phosphorylation sites. This shortened A-loop does not extend out of the pseudokinase active site pocket, and instead forms a two-turn α helix near its C-terminal end and packs against the C-lobe helices.
Another unique feature of the JAK family pseudokinases is an extended N-terminal segment that starts on the back of the N-lobe packed against the αC helix, crosses perpendicular to the β4 strand, makes a single α-helical turn and enters the β1 strand (Fig. 2C). This N-terminal conformation is conserved in JAK1, JAK2, and TYK2 pseudokinase structures, and is encoded by the frequently mutated exon 12 in JAK2 (23). Comparison of the exon 12 segment (residues 536–547) from JAK2 pseudokinase structure (11) and our TYK2 structure (residues 579–591) shows significant structural homology between the regions (Fig. 2C). Importantly, this exon 12 segment in TYK2 forms a portion of the pseudokinase–kinase interface (discussed below).
The Pseudokinase–Kinase Interface.
The interaction between the TYK2 pseudokinase and kinase domains is predominantly mediated by the N-lobes of each domain, with the back of the pseudokinase N-lobe interacting at a near 90° angle with the underside of the kinase domain N-lobe β1–β3 sheet and the β2–β3 loop, as well as the hinge region and subsequent αD helix (Fig. 3A). The pseudokinase interface surface is primarily composed of residues in the αC helix–β4 loop, the extended β7–β8 loop, and the N-terminal exon 12 segment. Approximately 1,460 Å2 of surface area is buried between the two domains in a long stripe of interactions, which is anchored by a hydrophobic center and ringed by several points of polar contacts (Fig. 3 B and C). The core of the interface is represented by pseudokinase residue Phe672, which interacts with a patch of hydrophobic residues (Ile900, Leu913, Met926, and Tyr980) on the kinase side. Nearby, αC helix residue Gln664 as well as Val665 and Ser666 interact with Tyr980, Pro982, and Leu983 in the kinase hinge. Several residues in the extended pseudokinase β7–β8 loop also make contact with the kinase, including a salt bridge between Arg744 and Asp921 in the kinase β2–β3 loop, and a hydrogen bond between Lys756 and the backbone carbonyl of Gly922 (Fig. 3A). Contacts between the pseudokinase exon 12 segment (TYK2 residues 579–591) and the kinase domain include His582 with Arg901, as well as Leu579 and Ser580 contacts with Leu983, Tyr989, Arg992, and His993 (Fig. 3A). This contact interface, in which the pseudokinase binds the kinase N-lobe and hinge directly adjacent to the active site, may limit the conformational mobility of the active site required for catalysis.
Fig. 3.

The TYK2 pseudokinase–kinase interaction. (A) (Upper) Overview of the pseudokinase–kinase complex. The pseudokinase is colored in blue, and kinase is colored in tan. (Lower) Zoomed-in view of the pseudokinase–kinase interface. The pseudokinase is shown facing up as a blue semitransparent molecular surface, with the kinase in front shown as tan worm and stick side chains. Pseudokinase residues within 4.5 Å of the kinase domain have side chains displayed as dark blue sticks (with tan surface). (B and C) Open-book view of the pseudokinase–kinase interface. Surface representations of the pseudokinase (B) and kinase (C) domains, colored as in A with surfaces within 4.5 Å of the partner domain outlined and highlighted the opposite color. Residues found in the interface are labeled.
Tumor-Derived JAK Mutations Cluster Near the Pseudokinase–Kinase Interface.
To investigate the possibility that cancer-associated, activating JAK mutations are concentrated in the pseudokinase–kinase interface, we compiled a comprehensive list of human JAK mutations from the literature for mapping onto our structure (Table S2). These include mutations in JAK1, JAK2, and JAK3 that are primarily associated with MPNs and hematologic malignancies, but include some solid tumors. Highlighting these mutations on a sequence alignment of the four human JAKs (Fig. 4A) confirms that the bulk of the point mutations (green) and deletions (orange) map to the N-lobe of the pseudokinase. A second subset can be found in the N-lobe of the kinase domain (magenta), whereas a third subset includes the JAK2 exon 12 deletions near the N terminus of the pseudokinase domain (yellow). A fourth subset of mutations (cyan) reside outside these regions in the pseudokinase C-lobe, pseudokinase–kinase linker, or kinase C-lobe.
Fig. 4.

Cancer-associated JAK alleles map to the TYK2 pseudokinase–kinase interface. (A) Sequence alignment of the four human JAKs, with cancer-associated JAK alleles colored according to the type of mutation. For the pseudokinase, point mutants are shown in green, deletions are shown in orange, and the exon 12 segment is shown in yellow. For the kinase, point mutants are shown in magenta. Mutations outside the N-lobes of the pseudokinase or kinase are shown in cyan. Residues found within the pseudokinase and kinase interface are boxed and shaded in tan and blue, respectively. Protein secondary structure is also shown above the sequences, with α-helices shown as cylinders and β-sheets shown as rectangular arrows. (B and C) Open-book surface views of the pseudokinase (B) and kinase (C) domains, with select JAK alleles mapped onto the models. Mutations are colored as in A. The perspective shown is identical to that displayed in Fig. 3 B and C.
We then mapped a representative set of these mutations onto the corresponding residues in our TYK2 pseudokinase–kinase structure (Fig. 4 B and C). Remarkably, nearly all of these pseudokinase N-lobe mutations were found in or close to the interface with the kinase (Fig. 4B). Many cluster on the back side of the N-lobe, including a number of JAK1 and JAK2 point mutations that are found C-terminal to the pseudokinase αC helix (JAK1 S646F, H647Y, V651M, Y652H, L653F; JAK2 R564L and L611S), a JAK2 deletion/insertion region (JAK2 del682–686), as well as homologous Arg point mutants in JAK1 (R724H) and JAK3 (R657Q). The JAK2 exon 12 deletions map to the N-terminal segment of the pseudokinase in and near the interface with the kinase domain. Interestingly, a number of mutations also occur adjacent to the interface, including JAK2 V617F, JAK2 D620E, JAK3 A593T, a JAK1 deletion (624–629insW), and internal point mutants that occur in the αC helix of JAK1 (A634D) and JAK3 (A572V). On the kinase side, mutations are found in the region near the “left side” of the active site cleft (Fig. 4C). This includes a number of mutations in the first three strands of the kinase domain N-lobe (JAK1 R879/S/C/H; JAK2 R867Q, D873N, T875N) and a proline-to-arginine (JAK2 P933R) mutation in the kinase hinge. This striking result suggests these cancer-associated mutations could disrupt this pseudokinase–kinase interface, leading to the constitutive JAK activity associated with these diseases.
Tumor-Derived TYK2 Interface Mutants Are Active in Vitro.
To investigate the effect of disruption of the pseudokinase–kinase interface on kinase activity in vitro, we introduced several representative interface mutations found in JAK-driven malignancies into the TYK2 pseudokinase–kinase construct (residues 566–1187) and tested their enzymatic activity. These mutations included V678F, a mimic of the JAK2 V617F mutation (19); R744G, which falls in a region mutated in JAK1, JAK2, and JAK3-driven malignancies (26–31); R901S, a mimic of the R879S/C/H mutation found in the JAK1 kinase N-lobe (26); and a deletion of residues Q586 and K587, designed to mimic a JAK2 exon 12 deletion (23). As a positive control, we also generated a TYK2 kinase domain construct (residues 885–1176) and compared the activity of the isolated kinase domain with the wild-type pseudokinase–kinase protein (Fig. 5). The wild-type pseudokinase–kinase construct was ∼100-fold less active than the kinase domain alone, recapitulating a similar effect reported for JAK2 (14) and indicating that the pseudokinase has a direct role in inhibiting TYK2 enzymatic activity. Comparison of the four TYK2 analogs that mimic JAK-associated mutations showed that all of the mutants were more active than wild type pseudokinase–kinase, with the R744G and R901S mutants approaching levels of activity seen for the isolated kinase domain (Fig. 5). These data indicate a higher level of enzymatic activity is induced by mutations in the interface between the pseudokinase and kinase domains of TYK2.
Fig. 5.

TYK2 pseudokinase–kinase mutants analogous to cancer-associated JAK alleles are active in vitro. The activity of the kinase domain of wild-type TYK2 (KD, residues 885–1176) was compared with the pseudokinase and kinase domains of wild-type TYK2 (wt, residues 566–1187) as well as a number of pseudokinase–kinase interface mutants (V678F, R744G, R901S, and delQ586/K587). Specific activity was measured in an assay monitoring phosphorylation of a synthetic peptide derived from the JAK3 sequence and calculated based on the percentage conversion to phosphorylated product over time and the concentration of TYK2 used. Values shown have units of nanomolar concentration of product formed per minute per nanomolar concentration of TYK2 and are the mean of more than five measurements ± SD.
We also assessed the phosphorylation state of the pseudokinase–kinase constructs by mass spectrometry after purification from insect cells. The TYK2 kinase domain contains two sites of tyrosine phosphorylation, Y1054 and Y1055, which reside in the activation loop and are phosphorylated during insect cell expression of the isolated kinase domain (32). We found that the interface mutants and isolated kinase domain all showed significant fractions of singly and doubly phosphorylated species, whereas the wild-type pseudokinase–kinase was completely unphosphorylated, as we see in the crystal structure (Fig. S5). A likely explanation for the presence of phosphorylation is that the pseudokinase–kinase mutants are active and transautophosphorylate during expression in the insect cells, whereas the wild-type pseudokinase–kinase is held inactive and unable to autophosphorylate. We then purified the unphosphorylated fraction of the V678F and R744G mutant proteins by anion exchange chromatography (Fig. S6) and found the nonphosphorylated mutants to be inactive (Fig. S7), agreeing with a published study showing phosphorylation is required for TYK2 kinase activity in vitro (32). These phosphorylation-state studies show the role of the pseudokinase is to inhibit activity of the kinase domain, with the kinase able to transautophosphorylate and become activated when interface mutations disrupt the autoinhibited state.
Discussion
In the past decade, mutant alleles of the JAK kinases (in particular JAK1, JAK2, and JAK3) have been linked to the development and progression of myeloproliferative diseases and hematological cancers. Here, we have described the structure of the human TYK2 pseudokinase–kinase module and show that the pseudokinase and kinase participate in an interdomain interaction between their respective N-lobes. Most clinically relevant N-lobe pseudokinase and kinase mutations cluster in or near this interdomain interface, strongly suggesting this complex represents a physiological autoinhibitory conformation. Although oncogenic mutations have so far been found exclusively in JAK1, JAK2, and JAK3 (Table S2), our data and others (19) clearly demonstrate that homologous mutations can also convert TYK2 into a constitutively active kinase. The high frequency of MPN-inducing mutations found in JAK2 (and to a lesser extent JAK1) likely speaks to the oncogenic potential of specific STAT signaling pathways these kinases activate. However, the evidence that homologous mutations can confer activity to the TYK2 pseudokinase–kinase module suggests that TYK2 can be used as a surrogate system for understanding pseudokinase-mediated regulation of the JAK family kinases.
Although a number of JAK mutations lie directly in the pseudokinase–kinase interface, it is important to note that many lie adjacent to the interface or within the hydrophobic core of the N-lobe. This “second shell” of mutations includes the most clinically relevant changes, such as JAK2 V617F and many of the exon 12 mutations. However, structural analysis suggests that these mutations could still have a significant effect on the ability of the pseudokinase and kinase to form a stable, autoinhibitory interaction. For example, insertion of an aromatic residue at the V617 location (V678 in TYK2) would likely disrupt the structure of the exon 12 residues at the pseudokinase–kinase interface (Fig. 6). Indeed, a recent structure of the JAK1 pseudokinase suggests that insertion of a phenylalanine at the analogous JAK1 position (V658) induces remodeling of a highly conserved Phe residue (F575) in the N-terminal exon 12 segment (13). It has also been shown that any number of large hydrophobic residues in place of V617 can activate JAK2 (33), whereas replacement of the V617-adjacent aromatic residue F595 with a smaller aliphatic residue can reverse the increased activity conferred by V617F (34), suggesting that simple steric disruption of the packing near JAK2 V617 and exon 12 can relieve inhibition. Bulky substitutions in the pseudokinase αC-helix (e.g., JAK1 A634D, JAK3 A572V or A573V) or exon 12 deletions and insertions may also relieve autoinhibition in a similar manner to the V617F mutation (Fig. 6). Interestingly, our activity data suggest the V678F and exon 12 (delQ586/K587) mutants have lower activity than the interface mutants R744G and R901S (Fig. 5), suggesting mutations peripheral to the interface may have lower activating potential than interface mutants. Taken as a whole, these data imply that V617 and other “second-shell” alleles may disrupt the autoinhibited dimer by remodeling N-terminal pseudokinase residues found in the kinase interface.
Fig. 6.

V617 and αC-helix alleles carry “second-shell” mutations that may disrupt the exon 12 segment. A close-up view of the TYK2 N terminus, which corresponds to the JAK2 exon 12 segment that is frequently mutated in MPN disorders. The TYK2 N-terminal segment is highlighted in yellow with side chains shown as sticks. Point mutations in this region are highlighted in green, with a JAK1 deletion highlighted in orange.
On the other side of the interface, all known kinase N-lobe mutations participate directly in interdomain interactions. Save the JAK2 R867Q position (C915 in TYK2), the residues on the kinase side that are mutated in JAK-associated cancers are conserved in TYK2. Interestingly, two residues found mutated in JAK2 (D921 and T923 in TYK2) reside in the kinase β2–β3 loop and come in direct contact with the often-mutated arginine at position 744 in the TYK2 pseudokinase. Although there are fewer cancer-associated mutations found on the kinase side, this may reflect the relative importance of kinase N-lobe residues for substrate recognition or efficient catalysis.
A common theme in kinase regulation has been the role of protein–protein interactions in modulating kinase activity, with pseudokinases often playing a role (35). Interestingly, we find that the structure of the TYK2 kinase domain in complex with the pseudokinase is very similar to the structure of the kinase domain alone, with the A-loop of the kinase domain in an “active-like” conformation even in the absence of phosphorylation at tyrosine residues 1054 and 1055. This conformation has previously been identified in crystal structures of the nonphosphorylated TYK2 kinase domain (25). Moreover, the kinase A-loop forms a crystal contact with the active site cleft of an adjacent pseudokinase molecule, suggesting this active-like conformation may be an artifact of crystal packing. Regardless, the presence of an active-like kinase conformation in our structure suggests that the pseudokinase regulates the kinase domain by steric inhibition of ATP binding and/or a reduction in flexibility of the kinase active site required for catalysis, rather than by blocking substrate access, as found for SOCS3 (36).
Currently, it is unclear how the dimer we have characterized is disrupted to activate the kinase domain. Because the TYK2 kinase activation loop is found in an active conformation in our structure, activation loop phosphorylation likely does not have a role in regulating pseudokinase-mediated inhibition. Given that isolated JAK kinase domains are significantly active (14, 32, 37) and pseudokinase–kinase interface mutations increase kinase activity, we speculate that, in the context of receptor-induced JAK dimerization, a displacement event, direct or perhaps allosteric, relieves the inhibitory influence of the pseudokinase domain, allowing the kinase domain to autophosphorylate and become the active phospho-transfer catalyst required for downstream signaling.
Methods
Human TYK2 constructs containing the wild-type or mutant pseudokinase and kinase domain (residues 566–1187) or kinase domain alone (residues 885–1176) were expressed in insect cells and purified by Ni affinity followed by size exclusion chromatography. For crystallization, TYK2 pseudokinase–kinase protein was concentrated to 10 mg/mL and crystallization drops set in the presence of 1 mM compound 7012. Data were collected at 110 K at Stanford Synchrotron Radiation Lightsource and Advance Light Source, and the structure solved by molecular replacement using the coordinates of the TYK2 kinase domain. Coordinates and structure factors have been deposited in the PDB under ID code 4OLI.
TYK2 activity was measured by monitoring phosphorylation of a fluorescently labeled synthetic peptide derived from the JAK3 protein sequence. The peptide substrate in each reaction was electrophoretically separated from phosphorylated product using a LabChip 3000 instrument and peak heights detected and recorded. The specific activity of each TYK2 protein (i.e., nanomolar concentration of phosphorylated product formed per minute per nanomolar concentration of TYK2) was calculated based on the percentage conversion and the concentration of TYK2 used. See SI Methods for full crystallization and assay details.
Supplementary Material
Acknowledgments
We thank the Structural Biology expression group at Genentech for their technical support. The Advanced Light Source and Stanford Synchrotron Radiation Lightsource are supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the National Institute of General Medical Sciences and the National Center for Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4OLI).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1401180111/-/DCSupplemental.
References
- 1.Leonard WJ, O’Shea JJ. Jaks and STATs: Biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 2.Haan C, Kreis S, Margue C, Behrmann I. Jaks and cytokine receptors—an intimate relationship. Biochem Pharmacol. 2006;72(11):1538–1546. doi: 10.1016/j.bcp.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Rodig SJ, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93(3):373–383. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 4.Parganas E, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93(3):385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 5.Neubauer H, et al. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93(3):397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 6.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36(4):515–528. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilks AF, et al. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11(4):2057–2065. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girault JA, Labesse G, Mornon JP, Callebaut I. The N-termini of FAK and JAKs contain divergent band 4.1 domains. Trends Biochem Sci. 1999;24(2):54–57. doi: 10.1016/s0968-0004(98)01331-0. [DOI] [PubMed] [Google Scholar]
- 9.Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277(49):47954–47963. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- 10.Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20(10):3387–3395. doi: 10.1128/mcb.20.10.3387-3395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bandaranayake RM, et al. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat Struct Mol Biol. 2012;19(8):754–759. doi: 10.1038/nsmb.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ungureanu D, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18(9):971–976. doi: 10.1038/nsmb.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toms AV, et al. Structure of a pseudokinase-domain switch that controls oncogenic activation of Jak kinases. Nat Struct Mol Biol. 2013;20(10):1221–1223. doi: 10.1038/nsmb.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanz A, et al. Analysis of Jak2 catalytic function by peptide microarrays: The role of the JH2 domain and V617F mutation. PLoS One. 2011;6(4):e18522. doi: 10.1371/journal.pone.0018522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kontzias A, Kotlyar A, Laurence A, Changelian P, O’Shea JJ. Jakinibs: A new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol. 2012;12(4):464–470. doi: 10.1016/j.coph.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen E, Staudt LM, Green AR. Janus kinase deregulation in leukemia and lymphoma. Immunity. 2012;36(4):529–541. doi: 10.1016/j.immuni.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32(21):2601–2613. doi: 10.1038/onc.2012.347. [DOI] [PubMed] [Google Scholar]
- 18.Zhao L, et al. A JAK2 interdomain linker relays Epo receptor engagement signals to kinase activation. J Biol Chem. 2009;284(39):26988–26998. doi: 10.1074/jbc.M109.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Staerk J, Kallin A, Demoulin JB, Vainchenker W, Constantinescu SN. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: Cross-talk with IGF1 receptor. J Biol Chem. 2005;280(51):41893–41899. doi: 10.1074/jbc.C500358200. [DOI] [PubMed] [Google Scholar]
- 20.Lucet IS, et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107(1):176–183. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 21.Chrencik JE, et al. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6. J Mol Biol. 2010;400(3):413–433. doi: 10.1016/j.jmb.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 22.Boggon TJ, Li Y, Manley PW, Eck MJ. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106(3):996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott LM. The JAK2 exon 12 mutations: A comprehensive review. Am J Hematol. 2011;86(8):668–676. doi: 10.1002/ajh.22063. [DOI] [PubMed] [Google Scholar]
- 24.Liang J, et al. Lead optimization of a 4-aminopyridine benzamide scaffold to identify potent, selective, and orally bioavailable TYK2 inhibitors. J Med Chem. 2013;56(11):4521–4536. doi: 10.1021/jm400266t. [DOI] [PubMed] [Google Scholar]
- 25.Tsui V, et al. A new regulatory switch in a JAK protein kinase. Proteins. 2011;79(2):393–401. doi: 10.1002/prot.22889. [DOI] [PubMed] [Google Scholar]
- 26.Flex E, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205(4):751–758. doi: 10.1084/jem.20072182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato T, et al. Functional analysis of JAK3 mutations in transient myeloproliferative disorder and acute megakaryoblastic leukaemia accompanying Down syndrome. Br J Haematol. 2008;141(5):681–688. doi: 10.1111/j.1365-2141.2008.07081.x. [DOI] [PubMed] [Google Scholar]
- 28.Bercovich D, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372(9648):1484–1492. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 29.Mullighan CG, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2009;106(23):9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearney L, et al. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009;113(3):646–648. doi: 10.1182/blood-2008-08-170928. [DOI] [PubMed] [Google Scholar]
- 31.Gaikwad A, et al. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol. 2009;144(6):930–932. doi: 10.1111/j.1365-2141.2008.07552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korniski B, et al. Expression, purification, and characterization of TYK-2 kinase domain, a member of the Janus kinase family. Biochem Biophys Res Commun. 2010;396(2):543–548. doi: 10.1016/j.bbrc.2010.04.141. [DOI] [PubMed] [Google Scholar]
- 33.Dusa A, et al. Substitution of pseudokinase domain residue Val-617 by large non-polar amino acids causes activation of JAK2. J Biol Chem. 2008;283(19):12941–12948. doi: 10.1074/jbc.M709302200. [DOI] [PubMed] [Google Scholar]
- 34.Dusa A, Mouton C, Pecquet C, Herman M, Constantinescu SN. JAK2 V617F constitutive activation requires JH2 residue F595: A pseudokinase domain target for specific inhibitors. PLoS One. 2010;5(6):e11157. doi: 10.1371/journal.pone.0011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16(9):443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Kershaw NJ, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. 2013;20(4):469–476. doi: 10.1038/nsmb.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall T, et al. Expression, purification, characterization and crystallization of non- and phosphorylated states of JAK2 and JAK3 kinase domain. Protein Expr Purif. 2010;69(1):54–63. doi: 10.1016/j.pep.2009.09.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.