

Abstract
Long-term potentiation (LTP) of synaptic strength occurs during learning and can last for long periods, making it a probable mechanism for memory storage. LTP induction results in calcium entry, which activates calcium–calmodulin-dependent protein kinase II (CaMKII). CaMKII subsequently translocates to the synapse, where it binds to the NMDA-type glutamate receptors and produces potentiation by phosphorylating principal and auxiliary subunits of AMPA-type glutamate receptors. These processes all are localized to stimulated spines and account for the synapse specificity of LTP. In the later stages of LTP, CaMKII has a structural role in enlarging and strengthening the synapse.
Introduction
Long-term potentiation (LTP) is a process whereby brief periods of synaptic activity can produce a long-lasting increase in the strength of a synapse, as shown by an increase in size of the excitatory postsynaptic current (EPSC). LTP, in vivo, can last for at least a month1, and similar changes in synaptic strength occur during learning2, 3. When LTP is prevented by various mutations, memory is impaired4. Taken together, these findings indicate that LTP has an important role in learning and memory, and a major effort has been therefore made to understand the mechanisms underlying this process.
The CA1 region of the hippocampus, a brain region that is vital for long-term memory formation, has been used as the primary model system for understanding LTP5. The process of LTP at hippocampal CA1 synapses is initiated by brief periods of high-frequency presynaptic stimulation. Glutamate, the neurotransmitter that is released at these synapses, activates postsynaptic AMPA- and NMDA-type glutamate receptors (AMPARs and NMDARs). The opening of NMDARs during the induction of LTP leads to calcium entry that triggers a biochemical cascade, the end product of which is the long-lasting potentiation of the AMPAR-mediated EPSC.
Strong evidence now exists that calcium elevation initiates this biochemical cascade through activation of calcium–calmodulin-dependent protein kinase II (CaMKII), a major synaptic protein6. This kinase is a large holoenzyme consisting of 12 subunits. There are two types of subunits (alpha and beta) both of which are capable of catalysis. Recent work has led to crystallization of the holoenzyme and the elucidation of how the enzyme’s structure relates to its function (BOX 1)7. The kinase can act as a protein switch8; once activated by calcium–calmodulin, the enzyme can be autophosphorylated at T286 (on the alpha subunits; 287 on the beta subunits), an event that makes CaMKII activity persist even after the calcium concentration falls to baseline levels9 (the T286 autophosphorylated state of CaMKII is termed the ‘autonomous’ state). If active CaMKII subunits or holoenzymes are introduced into CA1 neurons, the EPSC becomes large and LTP can no longer be induced by synaptic stimulation10, 11. If persistent activation of CaMKII is prevented by a knock-in mutation encoding T286A in CaMKII, LTP induction is prevented and mice show profound memory impairments, notably after one-trial learning 12, 13. In many mutants with enhanced learning, CaMKII activation is enhanced14. Taken together, these results indicate that activated CaMKII is sufficient to trigger LTP and that CaMKII activation is necessary for LTP induction and certain forms of learning.
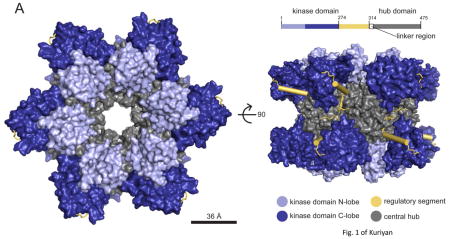
Box 1. Structural and functional properties of CaMKII.
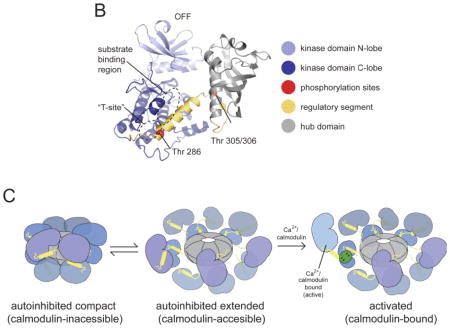
The architecture of the CaMKII holoenzyme has been determined on the basis of its crystal structure (see the figure, part a) 7. The holoenzyme has 12 subunits, each with a carboxy-terminal association domain that forms a central organizing hub (grey) on to which the catalytic kinase domains (blue) fold into two rings of 6. These domains have a bi-lobed structure with the ATP binding site located between the two lobes. A regulatory segment (yellow) regulates the activity of the kinase domain (the S-site). A linker region connects the regulatory domain to the hub, which has a central pore of unknown function 146. In the autoinhibited state, the regulatory segment forms an α-helix that blocks the catalytic S-site in a pseudosubstrate manner (see the figure, part b). This inactive state is further stabilized by sequestering the T286 phosphorylation site in a hydrophobic groove (the T-site) 147. The crystal structure reveals a ‘compact’ state in which the portion of the regulatory domain that is recognized by calmodulin is sequestered into the association domain, making it impossible for calmodulin to bind. Activation is only possible because the compact state is in equilibrium with an extended state to which calmodulin can bind (see the figure, part c) 7, 148, 149. The linker region varies in different CaMKII isoforms and determines the set-point of this equilibrium and, secondarily, the calcium level required for activation. Upon calmodulin binding, the regulatory segment is stripped from the S-site 7, 150, allowing substrate phosphorylation and starting the slower process of T286 intersubunit autophosphorylation. If T286 phosphorylation has not occurred, lowering calcium rapidly turns off activity; if T286 phosphorylation has occurred, activity remains (this is termed the ‘autonomous’ state) and the T-site is open for binding (for example, to the NR2B subunit of NMDA-type glutamate receptors). T286 phosphorylation dramatically strengthens the binding of calmodulin, a process called ‘trapping’. The autonomous activity of CaMKII for phosphorylating various substrates is up to five times higher with calmodulin bound than it is when calmodulin is not bound126. Formation of autonomous CaMKII has a Hill coefficient of approximately eight with respect to calcium, indicating a virtual threshold 151. Various cooperative reactions underlie this ultrasensitivity. Four calcium binding events are required to fully activate calmodulin, which displays cooperativity when binding to the holoenzyme 152. Calmodulin can activate a CaMKII subunit, but the subunit can only phosphorylate a neighbour if that subunit also has calmodulin molecule bound (as required to expose T286).
In this Review, we will describe recent progress in understanding the role of CaMKII in LTP. It has been shown that LTP occurs specifically at synapses that are stimulated (inactive synapses that are only microns away from a stimulated synapse remain unpotentiated). We will review recent data showing that activation of CaMKII following synaptic stimulation is synapse-specific and elucidating how this specificity is achieved. We will also describe experiments that have identified the downstream process by which activated CaMKII causes EPSC enhancement during the early stages of LTP. Finally, we will discuss experiments suggesting that CaMKII can contribute to the later stages of LTP, when structural changes in the synapse become important for long-term memory. As a result of space limitations, we will not discuss the presynaptic functions of CaMKII in LTP15, the role of other kinases and second messengers in LTP16, or the phosphatase-dependent processes that limit or reverse CaMKII-dependent activation17.
Synapse-specific activation
Early experimental data suggested that LTP is a synapse-specific event18, 19, and optical methods have now made it possible to demonstrate directly that this is correct. An identified spine can be stimulated using two-photon glutamate uncaging. Such uncaging gives rise to a glutamate pulse with submicron dimensions that directly targets the identified spine and induces LTP at the synapse on that spine. Nearby synapses that are only microns away are not potentiated (FIG. 1)20. Given the complex biochemical cascade that is involved in LTP and the small distances that separate different synapses, it is remarkable that synapse-specific LTP can be achieved.
Figure 1. Synapse-specificity of the processes involved in LTP induction.

a A transient elevation of calcium (detected by the calcium-sensitive dye Fluo4-FF; green signal) in a spine can be observed in response to glutamate uncaging at that spine (arrowhead). The calcium-insensitive dye Alexa-594 outlines the spine volume (red). 35. b Local glutamate uncaging can potentiate AMPA receptor-mediated excitatory postsynaptic currents (EPSCs) in the stimulated spine but not in an adjacent spine154. c After LTP induction by local glutamate uncaging (arrowhead), transient activation of CaMKII can be observed (red denotes high activation; level of activation measured by the FRET sensor Camui). 22. d Stimulation of a spine (arrowhead) in this fashion can cause a persistent increase in spine volume and translocation of CaMKII (here shown by translocation of green fluorescent protein-labelled αCaMKII; red denotes high levels of this kinase)… From 41.
High-resolution imaging of CaMKII activation during LTP induction has been made possible by the development of the Camui activity sensor21, 22. This sensor comprises two different fluorophores — monomeric enhanced green fluorescent protein (mEGFP) and resonance energy-accepting chromoprotein (REAch) — that are attached to different ends of CaMKII subunits. The level of fluorescence resonance energy transfer (FRET) that occurs between the two fluorophores is an indicator of the distance between them: when the enzyme is activated, conformational changes increase the distance between the fluorophores and thus decrease the amount of FRET. Through use of this sensor and two-photon fluorescence lifetime imaging (2pFLIM), which allows quantification of FRET, the conformational change in CaMKII that is associated with activation can be monitored at the level of a single spine in real time. Moreover, such monitoring can be conducted while LTP is induced at the same spine using a second two-photon laser to uncage glutamate, which activates NMDARs on that spine. Using this approach, it can be seen that CaMKII is activated within seconds after the start of synaptic stimulation and that the activation is restricted to the stimulated spine (FIG. 1)22. At the end of stimulation, CaMKII activity decays to baseline levels within ~1 min. By contrast, when using a mutant variant of CaMKII (T286A) that cannot be autophosphorylated, the activity of the enzyme decays within seconds. Taken together, these results show that CaMKII activation has the localization that is required to account the for synapse specificity of LTP. The results also show autophosphorylation makes CaMKII activity persist for much longer than the duration of calcium elevation. The slow rate at which active CaMKII turns off (~1 min) means that during this period, the enzyme acts as a biochemical integrator of the multiple calcium pulses that generally occur during LTP induction. This integration is important for the full activation of CaMKII that is required to produce LTP.
Investigations have examined the mechanisms of CaMKII activation, and the results of these studies provide a framework for understanding why activation of the kinase is localized to the stimulated spine. The primary source of calcium entry into spines is through NMDARs. The opening of these channels requires both the presynaptic release of glutamate and strong postsynaptic depolarization, which relieves the magnesium blockade of these receptors23. These channel properties allow NMDARs to compute the Hebbian condition that is crucial for associative memory storage. Calcium indicators have been used to study calcium entry through NMDARs into spines, and such studies have confirmed that calcium elevation is localized to stimulated spines (FIG. 1)22, 24–27.
Spines largely contain two types of NMDARs: those containing the NMDAR subtype 2A (NR2A) subunit and those containing the NR2B subunit28. NR2A-containing NMDARs open briefly but reliably following glutamate release, whereas NR2B-containing receptors have a lower probability of opening but continue to have many brief openings for several hundred milliseconds after glutamate release29, 30. When calcium enters through a NMDAR, the concentration of calcium becomes high near the inner mouth of the channel, a region called a calcium nanodomain31. Calcium then diffuses into the bulk of the spine head within microseconds.
Experiments have been conducted to determine whether CaMKII activation depends on events that occur in the nanodomain or in the bulk of the spine head; these were based on the calcium-buffering capabilities of BAPTA, a fast calcium buffer, and EGTA, a slow calcium buffer (the steady-state dissociation constant is similar for both of these buffers). As calcium traverses the nanodomain quickly, only a fast buffer can block calcium elevation in the nanodomain, whereas either a fast or a slow buffer can block calcium elevation in the bulk of the spine head. The aforementioned experiments showed that CaMKII activation can be inhibited by either BAPTA or EGTA, implying that activation of this enzyme is dependent on an elevation of calcium in the bulk of the spine head22. However, a nanodomain process may also be required for such activation, as a bulk calcium signal, when produced by depolarization of voltage-dependent calcium channels, is not sufficient to activate CaMKII.
The actual calcium sensor for CaMKII activation is not the enzyme itself, but the small diffusible molecule calmodulin. A new view of calmodulin activation has been recently presented32. This view is based on measurements showing that the on-rate constant for calcium binding to calmodulin is significantly higher than it is for calcium binding to calbindin, the other major calcium buffer in pyramidal cells. Reconstitution experiments and computer simulations indicate that because of the high intracellular concentration of calmodulin (~100 μM) and its high on-rate constant for calcium binding, this protein can account for much of what has been termed the ‘fast calcium buffer’27, 33. From a signaling perspective, these conditions make sense: a substantial fraction of the calcium that enters the spine becomes bound to calmodulin, making it an excellent detector of calcium and an efficient activator of CaMKII subunits, which are present in spines at a concentration of ~100μM34.
The ability of local activation of NMDARs to lead to local activation of CaMKII is non-trivial because calcium, calmodulin and CaMKII can all diffuse. For a small diffusible molecule, the diffusion length scale is of the order of 1μm per ms. A spine can be as close as 1μm to another, yet somehow LTP remains localized to the stimulated spine. A rather satisfactory biophysical explanation can now be given for how the synapse specificity of CaMKII activation is achieved (BOX 2). In brief, the narrow spine neck slows diffusion out of the spine, and the lifetime of the activated state of these molecules is short compared with the time that would be required for diffusion to carry the signal to other spines35, 36.
Box 2. Biophysical basis of the synapse specificity of long-term potentiation.
Calcium, calmodulin and CaMKII are all diffusible (albeit not freely), but the narrow spine neck that connects the spine head to the dendrite helps the compartmentalization of these molecules35, 36. In general, a signal will be localized to the spine if the decay rate of its activity is faster than the spine–neck diffusion coupling. Early experiments using two-photon imaging of caged fluorescein showed that the spine–neck diffusion coupling time can be estimated as:
where V is the spine volume, l is the spine neck length, s is the cross sectional area of the spine neck, and D is the diffusion constant 153. Assuming V~0.2 μm3, l~1 μm and s~0.052π, τ can be calculated as ~0.1 s for small molecules, with D of the order of 100 μm/s2. Thus, the active state of a signaling molecule will be localized to the spine if the lifetime of this state is <100 msec.
This condition is met by calcium. The spine–neck diffusion coupling of calcium (D~200 μm/s2) is ~100 ms 27 and the clearance of calcium by extrusion or binding to a slow calcium buffer is ~20 ms 27, much faster than the spine–neck diffusion coupling. Thus, a calcium signal will stay largely within the activated spine.
The next step in the long-term potentiation cascade is the binding of calcium to the four binding sites on calmodulin. Calcium-saturated calmodulin keeps calcium bound for ~1 ms, a long enough time for calmodulin to diffuse within the spine and to bind CaMKII, but too short a period of time to diffuse out of the spine . Size considerations indicate that the upper bound for the calmodulin diffusion constant is similar to that of green fluorescent protein (~20 μm/s2), yielding a diffusion coupling time of ~0.5 s. Thus, the decay time of activated calmodulin is considerably shorter than the time required for diffusion of calmodulin out of spines, ensuring localization.
The time constant for CaMKII inactivation is 45 s22, but as CaMKII diffuses at a very slow rate (for CaMKII to exit spines may take many minutes), activated CaMKII is mostly localized to the activated spine.
Translocation and binding to the NMDA receptor
Once activated by calcium entry, CaMKII moves from the cytoplasm to the synapse, a phenomenon called translocation37. The drivers of this translocation are simple diffusion and the binding of activated CaMKII to synaptic proteins, most importantly NMDARs (see below). However, translocation is also due to spine enlargement, the resulting increase in F-actin in spines, and the resulting increase in CaMKII-beta/CaMKII-alpha heteromers bound to F-actin 38, 39,40.
An important issue is whether translocation is synapse specific. This was demonstrated to be the case in experiments using two-photon glutamate uncaging to induce LTP at an identified spine (FIG. 1). A second method, using synaptically evoked LTP, showed similar results: in this case, active spines were identified by synaptically induced calcium elevation41.
Electron microscopy immunolabeling42 has been used to determine whether some of the CaMKII that translocates into the spine binds to the postsynaptic density (PSD). In this study, LTP induction, which was chemically induced, produced a two-fold increase in the amount of CaMKII per unit length of the PSD, as measured one hour after LTP induction (FIG. 2). Other work has revealed the molecular processes that contribute to CaMKII binding to the PSD. Of note, activated CaMKII has been shown to bind NMDARs, most strongly to NR2B43, 44. Several binding sites involved in the formation of CaMKII–NMDAR complexes have been identified. The region near S1303 in NR2B binds activated CaMKII in a way that does not require T286 phosphorylation; a second binding site in NR2B between amino acids 839 and 1,120 does require T286 phosphorylation45. A I205K-encoding mutation in CaMKII, which affects the so-called T-site of the enzyme (BOX 1), blocks complex formation and strongly attenuates CaMKII translocation in HEK293 cells expressing CaMKII and NMDARs46. Activation of NMDARs in hippocampal slices leads to a two-fold increase in the amount of the CaMKII–NMDAR complex (FIG. 4D), as determined by coimmunoprecipitation experiments43. In experiments in which CaMKII translocation was caused by ionophore-mediated calcium entry, once CaMKII become bound to an NMDAR via the T-site, the complex persisted for more than 30 min after the removal of calcium47. Importantly, the binding of CaMKII to NR2B locks the enzyme in an active state45 (but see 48).
Figure 2. Role of CaMKII–NMDAR complex in LTP maintenance.

Electron microscopy immunolabelling has been used to measure CaMKII levels in postsynaptic densities (PSDs) a before and b one hour after chemically induced LTP. c The histogram shows that there is more CaMKII per unit length of the PSD after LTP induction than before induction of LTD 42. d The CaMKII–NMDAR complex can be immunoprecipitated with an antibody that detects CaMKII. Synaptic stimulation with NMDA increases the amount of NR2B and NR1 in complexes with CaMKII. These increases can be blocked by the NMDAR antagonist MK801 or by the CaMK inhibitor KN62 43. e Overexpression of a mutant form of NR2B (RS/QD) blocks LTP (LTP induction denoted by the arrow) 49. f LTP (induced at arrow) can be reversed by tatCN21, a peptide that interferes with CaMKII binding to NR2B. In this example, LTP was induced by 4 tetani (T), and an additional tetanus had no effect, indicating that LTP saturation had been reached (left-hand side). Subsequent application of tatCN21 reversed LTP. After 30 mins of tatCN21 application, the peptide was removed and only partial recovery of LTP was observed (note that there was an acute inhibitory effect that was reversed when the peptide was removed). Additional LTP could then be induced using a 4T protocol (right-hand panel). 107
Figure 4. Mechanisms of expression processes by which CaMKII enhances AMPA receptor-mediated transmission.

a CaMKII enhances AMPA receptor (AMPAR) conductance in isolated CA1 pyramidal cells of wild-type mice but not in knockin mice in which phosphorylation of the AMPAR subunit glutamate receptor 1 (GluR1) on S831 or both S831 and S845 is prevented by mutation of the serine residues to alanine residues. Pseudophosphorylation of these sites (that is, expression of S831D or S845D GluR1) enhances AMPAR conductance and prevents further enhancement by CaMKII 78. b In addition to affecting AMPAR conductance, CaMKII also affects the mobility of these receptors. Single-particle tracking experiments have shown that in neurons under control conditions (left-hand panel), GluR2 subunits exhibit rapid diffusion that follows a broad path (three examples of particle paths are shown at the top and a histogram of diffusion constants for GluR2-labelled particles is shown at the bottom). In neurons overexpressing an active (truncated) form of CaMKII (tCaMKII; right-hand panel), GluR2 molecules show limited diffusion paths (top) and a lower average diffusion constant (bottom), as AMPARs are captured at the synapse70 c The influence of CaMKII on AMPA receptor-mediated transmission can be observed in LTP experiments. LTP can be induced by a pairing protocol (arrow denotes the point of protocol initiation) in wild-type CA1 pyramidal cells but is prevented in cells expressing a mutant variant of CaMKII (T286A) that cannot become persistently active 12. d Block of exocytosis of AMPAR containing vesicles by postsynaptic injection of botulinum toxin (left-hand panel) did not affect earliest phase of LTP; control is shown after injection of inactivated botulinum toxin (right-hand panel) 91. e Stargazin has an important role in CaMKII-enhanced AMPA receptor-mediated transmission. LTP is blocked in CA1 pyramidal cells expressing a mutant variant of stargazin in which the nine potential CaMKII/PKC serine phosphorylation sites are mutated to alanine residues (S9A). LTP can be induced in cells expressing pseudophosphorylated stargazing (S9D). The control condition is without LTP induction, and the arrow denotes the start of the induction protocol 69.
Formation of the CaMKII–NMDAR complex seems to have a key role in LTP induction and learning. LTP is greatly reduced by overexpression of mutant variants of NR2B (variants with mutations affecting residues near S1303) that block CaMKII binding to the NMDAR49 (FIG. 2). The results of this study are in agreement with other experiments showing that total deletion of the cytoplasmic tail of NR2B can block LTP despite having no effect on NMDAR-mediated currents50. Transgenic mice have been generated that overexpress the cytoplasmic tail of NR2B, which interferes with the formation of the CaMKII–NMDAR complex and reduces the level of this complex by 20–50%51. These animals have a reduction in LTP of ~50% and exhibit learning deficits.
Although the NMDAR is probably of primary importance in initiating the binding of CaMKII to the PSD, other CaMKII-binding proteins may also have a role in this process. PSDs in the average spine contain about 80 CaMKII holoenzymes and the molar ratio of PSD-95 (a synaptic scaffolding protein) to NR1 is about 7:152, 53. Thus, assuming that each NMDAR has two NR1 subunits, the PSD-95:NMDAR molar ratio is 14:1. The absolute number of PSD-95 molecules is ~30052, 54, so a rough estimate of NMDARs per synapse is 20–25. Thus, many of the CaMKII holoenzymes may be held in the PSD by other binding partners, including densin-180 (also known as leucine-rich repeat-containing protein 7)55, α-actinin55, synapse-associated protein 97 (also known as disks large homologue 1)56, and multiple PDZ domain protein57. Also, CaMKII is known to associate with other channels and receptors, including L-, T-, and P/Q-type voltage-gated calcium channels58–60 and dopamine receptors61. Many of these channels and receptors are associated with the PSD and could therefore contribute to CaMKII binding. CaMKII holoenzymes can also bind to other holoenzymes in a self-association process62. Such binding serves to amplify the amount of CaMKII that is bound in the PSD and probably gives rise to the CaMKII towers that are observed in the PSD63. Although the CaMKII in the PSD probably has special importance in the control of synaptic strength (see below), most of the CaMKII in the spine is not in the PSD34.
In summary, the available data point to the following model: activated CaMKII diffuses to the synapse and binds to NR2B, an event that is necessary for LTP (FIG. 3). Other binding partners may further enhance the amount of CaMKII in the PSD and spine head. Interestingly, the number of NR2B-containing NMDARs is independent of synapse size53. Thus, one possibility is that the amount of the CaMKII–NMDAR complex is graded; with increasing rounds of stimulation, NR2Bs eventually become saturated with CaMKII. This scenario could potentially explain why the stronger a synapse becomes, the less it can be potentiated64. Similarly, this scenario would explain why the higher the fraction of CaMKII that is bound in the spine, the fewer the number of CaMKII molecules that can be added to the spine by further LTP42. The enzymatic and structural processes by which CaMKII enhances AMPAR-mediated transmission are discussed below.
Figure 3. Role of CaMKII is in AMPA receptor-mediated transmission during early LTP.

Calcium entry through the NMDAR activates calmodulin both in the bulk cytoplasm and in the channel nanodomain. Calmodulin activates CaMKII, which then translocates to the PSD where it enhances AMPAR-mediated transmission in two ways: 1) by phosphorylation of GluR1 and S831, which leads to increased average conductance of the channel and 2) phosphorylation of stargazin, which leads to its binding to PSD-95, thereby increasing the number of AMPAR at the synapse. CaMKII, perhaps with other calcium dependent processes, stimulates fusion of vesicles containing AMPAR with the plasma membrane, increasing the extrasynaptic channel concentration. This increase is not necessary for the earliest stage of LTP, but is necessary for subsequent stages.
Potentiation of AMPA receptor function
The enhancement of AMPAR-mediated transmission during LTP occurs through two processes (FIG. 3). One process increases the average single channel conductance of AMPARs, which can be measured by non-stationary noise analysis65. A similar increase occurs after overexpression of an active truncated form of CaMKII66. The second process involves an increase in the number of AMPARs at the synapse67, 68. One of the studies revealing this process showed that LTP produced an increase in the rectification of the EPSC of the type expected from insertion into the membrane of AMPAR glutamate receptor 1 (GluR1) subunit homomers (GluR1 was overexpressed in this study). Further analysis revealed that extrasynaptic AMPARs that diffused into the synapse could be phosphorylated by CaMKII68. This phosphorylation occurs on the AMPAR auxiliary protein stargazin and allows the receptors to bind to PSD-9569, 70. In this way, AMPARs become captured by the synapse. We will now review the evidence for these processes.
GluR1 contains several important phosphorylation sites: S831, which is phosphorylated by PKC or CaMKII71, 72; S567, which is phosphorylated by CaMKII73; S845, which is phosphorylated by PKA74; and S818, which is a substrate for PKC75. Evidence now suggests that the increase in AMPAR conductance that is observed during the early stages of LTP is due to S831 phosphorylation. Of note, the phosphorylation of this site increases during LTP76, 77. Moreover, S831 phosphorylation blocks the increase in AMPAR conductance that is observed when active CaMKII is expressed (FIG. 4)78. Finally, a knockin mutation that encodes S831 pseudophosphorylated GluR1 increases channel conductance. Single-channel recordings show that S831 phosphorylation does not change the magnitude of any of the conductance states of AMPARs but, rather, enhances the probability that high conductance states will be activated by intermediate glutamate concentrations78. Interestingly, for AMPARs containing both GluR1 and GluR2, the ability of S831 phosphorylation to increase channel conductance only occurs if stargazin is present 78, 79. Thus, understanding the interactions of subunits within the AMPAR is likely to provide mechanistic insight into channel regulation. Given that S831 phosphorylation occurs during LTP and that such phosphorylation increases the average channel conductance, one would expect that a knockin mutation encoding GluR1 S831A by itself would decrease the magnitude of LTP. Such an effect, however, is not observed unless GluR1 S845 phosphorylation is also prevented80. Thus, the increase in AMPAR conductance during LTP may depend on interactions between the two sites.
The CaMKII-dependent phosphorylation of GluR1 probably occurs at the synapse itself. This scenario is suggested by the fact that extrasynaptic membrane patches that are excised after LTP induction have a higher number of AMPARs than do such patches prior to LTP induction (see below) but show no change in single-channel conductance81. The possibility that phosphorylation occurs at the synapse itself is further supported by the observation that activation of CaMKII in PSD preparations leads to strong phosphorylation of S83182 (but see 83).
As stated, the second mechanism leading to enhancement of AMPAR-mediated currents during the early phases of LTP is the insertion of AMPARs into the synapse. One step in this process could involve the exocytosis of AMPAR-containing vesicles into the plasma membrane, thereby supplying a pool of receptors that could be inserted into the synapse84–86. This step has been shown to occur by imaging of pHluorin-tagged AMPARs that only exhibit fluorescence when they are exposed to the high pH extracellular environment. After induction of LTP at single spines, a rapid increase occurs in the rate of exocytotic events, a rise that lasts for about a minute. These events occur in the stimulated spine and also in dendrites within 3 μm from the stimulated spine. Exocytosis depends on the RAS–extracellular signal-regulated kinase (ERK) pathway 86, RAB-GTPase proteins, soluble NSF attachment protein receptors (SNARE proteins), syntaxin 4 and 13, and the motor protein myosin V. However, inhibition of any of these proteins does not affect the first 20 minutes of LTP, although it does strongly reduce potentiation at later times (Fig. 4)87–94. The fact that inhibition of exocytosis does not affect the first 20 minutes of potentiation suggests that this early phase is due to the capture of AMPARs that are present extrasynaptically before LTP induction85.
The calcium sensor that triggers the exocytosis pathway is unclear. The RAS– ERK pathway can be stimulated by CaMKII94, but exocytosis is only partially blocked by the CaMK inhibitor KN6286. This finding raises the possibility that there are other calcium sensors for exocytosis, perhaps one of the calcium-sensitive RAS activators (guanine nucleotide exchange factors or GEFs) or inactivators (GTPase-accelerating proteins or GAPs)95.
Insertion of AMPARs into the extrasynaptic membrane is not sufficient to enhance transmission; translocation of these channels into the synapse requires the phosphorylation of stargazin, probably by CaMKII69, 83. This requirement was demonstrated by mutating the putative CaMKII phosphorylation sites of stargazin to preclude phosphorylation. Although these mutations did not affect the surface expression of AMPARs, they did prevent these channels from enhancing synaptic transmission after LTP induction (Fig. 4). Conversely, pseudophosphorylation of stargazin led to enhanced transmission that occluded LTP. These findings suggest that LTP-induced phosphorylation of stargazin is required for the capture of extrasynaptic AMPARs by the synapse.
This capturing process has now been directly observed using quantum-dot labeling of AMPARs and single-particle tracking70. Under basal conditions, AMPARs diffused freely in the extrasynaptic space while pausing only briefly at synaptic locations. Upon activation of CaMKII by NMDA application or synaptic input, diffusing AMPARs were captured at synapses (Fig. 4). The arrest of AMPARs at stimulated synapses is dependent on the phosphorylation of stargazin, as receptors with non-phosphorylatable stargazin did not exhibit immobilization at synapses. Immobilization of AMPARs could also be blocked by inhibition of CaMKII. Thus, although stargazin can be phosphorylated by either CaMKII or PKC, CaMKII seems to have the dominant role in stimulating the capture process. Capture was also prevented by expressing a mutant variant of CaMKII that is unable to bind NMDARs but still allows CaMKII activation. Given that NR2B is at the synapse, these results suggest that the stargazin phosphorylation that triggers AMPAR capture occurs at the synapse rather than extrasynaptically.
It was initially thought that AMPARs were bound in the synapse through the association of GluR1 subunits with PSD proteins having PDZ, but this seems now not to be the case96. Rather, the synaptic localization of AMPARs depends on the binding of stargazin to PSD-9597, 98. Synaptic clustering of AMPARs can be inhibited by mutating the binding sites on stargazin that bind to PSD-95 or by mutating the binding site on PSD-95 that binds stargazin 98. As noted earlier, the early phase of LTP can be blocked by deleting the CaMKII phosphorylation sites on stargazin. If the role of this phosphorylation is to allow stargazin to bind to PSD-95, then early LTP should also be blocked by deleting the PDZ-1 and PDZ-2 domainson PSD-95 to which stargazin binds. Consistent with this prediction, overexpression of PSD-95 lacking these domains prevents LTP induction99.
Additional evidence for the importance of the stargazin–PSD-95 interaction comes from the study of basal transmission in CA1100. Application of a ligand that disrupts both PDZ-1 and PDZ-2 interactions produced a rapid reduction in EPSCs and miniature EPSCs (mEPSCs). The fact that the saturating concentration of the ligand produced only a 45% reduction in these currents indicates that only a component of AMPAR-mediated transmission is dependent on the stargazin–PSD-95 interaction. This notion is consistent with previous work showing that mutliple mechanisms underlie basal AMPAR-mediated transmission at CA1 synapses101, 102.
A structural picture for how phosphorylation of stargazin leads to anchoring of receptors at the synapse is beginning to emerge. The PDZ-binding site on stargazin is normally situated close to the membrane, where it can be held by electrostatic interactions with positively charged lipids103. Synaptic PSD-95 has been visualized by electron microscope tomography and is oriented perpendicular to the membrane with the PDZ-1 and -2 domains that bind stargazin situated quite far (15 nm) from the membrane 104. CaMKII phosphorylation of stargazin may interfere with the electrostatic interactions at the membrane, allowing the PDZ ligand sites on stargazin to extend from the membrane and bind to PSD-95.
Thus, there is now rather strong support for a relatively simple model of early LTP (FIG. 3). The process begins with CaMKII activation. CaMKII then binds to the NMDAR, putting the kinase in close juxtaposition to GluR1 and to the carboxy-terminal region of stargazin83. The resulting phosphorylation of GluR1 enhances AMPAR conductance, while the resulting phosphorylation of stargazin leads to the anchoring of additional AMPARs at the synapse. Together, these processes enhance synaptic transmission by an enzymatic process. The duration of this enzymatic action may be only several minutes105, but the consequences of CaMKII enzymatic activity may last longer because it takes time for the substrates to become dephosphorylated.
Maintenance of long-term potentiation
We now turn our attention to the later phases of LTP and the processes that maintain synaptic strength. Less is known about these processes and the specific role of CaMKII. Most of the CaMKII in spines is active only for about a minute after synaptic stimulation22, suggesting that CaMKII may exert its enzymatic effects for only this brief period. Thus, one view is that the involvement of CaMKII in LTP is only transient and that different downstream processes are required to maintain LTP106. Support exists, however, for an alternative scenario in which a small subset of CaMKII molecules has a persistent role in LTP, a role that may be structural rather than catalytic. Several lines of evidence point to a long-lasting role. As mentioned above, formation of CaMKII–NMDAR complexes is required for LTP induction49 (Fig. 2). In a neuronal cell-culture model of LTP, these complexes, once formed, can be persistent47. Furthermore, the LTP-induced association of CaMKII with the PSD can persist for at least 60 min after induction (Fig. 2). Finally, large pools of CaMKII–NMDAR complexes exist in the hippocampus under basal conditions43, perhaps resulting from previous learning (Fig. 2). Thus, one hypothesis is that synaptic strength is controlled by the amount of CaMKII–NMDAR complex that is present in the synapse.
A key test of any model of synaptic memory mechanisms is to inhibit or disrupt the putative memory molecule after LTP induction and show that saturated LTP can be reversed. However, this result might simply indicate interference with LTP expression, i.e. processes that couple the memory mechanism to the change in AMPAR properties. To show that a maintenance mechanism has been affected, two further tests are required. First, the attacking molecule must be subsequently removed; if an expression process was affected, LTP will recover, whereas if a maintenance process was affected, LTP will not recover. Second, to exclude the possibility that the manipulations have damaged a cell, it must be shown that LTP can be re-induced.
Tests of this kind have been conducted to test the role of the CaMKII–NMDAR complexe in the maintenance of LTP. These tests used CN compounds, a class of CaMKII inhibitor known to interfere with the binding of CaMKII to NR2B in vitro. As shown in (Fig. 2), transient treatment of a hippocampal slice with CN compound reversed LTP, allowing additional LTP to be induced107. In parallel experiments, it was shown that CN inhibitor reduced the amount of the CaMKII–NMDAR complex in the slice.
A further conclusion of these experiments is that CN inhibitors reverse LTP by interfering with the binding reactions of CaMKII rather than catalytic actions of the kinase. CN compounds bind strongly to the T-site on CaMKII to which the NMDAR binds108 whereas other CaMKII inhibitors bind mainly to the catalytic site of the enzyme (the S-site) and only weakly to the T-site. The latter type of inhibitor does not reverse LTP109, 110. These findings also explain why low concentrations of CN compounds that are sufficient to block CaMKII activity111, but are not sufficient to disrupt the CaMKII–NMDAR complex, do not reverse LTP107. These results taken together, suggest that in addition to the catalytic action of CaMKII important for synaptic strengthening during early LTP, there is a structural process by which CaMKII strengthens transmission during late LTP.
Not much can be said with certainty about what this structural process might be, but there are several lines of relevant evidence. LTP is accompanied by a rapid and persistent increase in spine volume (Fig. 1), an increase that can be mimicked by overexpression of active CaMKII holoenzymes11. In addition to the increase in spine volume, there appears to be a slow increase in the size of the synapse itself (both pre- and postsynaptically)112. The late phase of spine growth is dependent on protein synthesis113, which is stimulated by brain-derived growth factor and dopamine and involves the selective targeting of newly synthesized proteins to the synapses that have undergone LTP114–116. Changes in protein synthesis are due in part to local control mechanisms117 and in part to transcriptional changes118. Changes in DNA methylation control some of the long-term transcriptional changes119. Ultimately, there is change in the synthesis of many proteins and these include CaMKII. A mutation in CaMKII that specifically interferes with its dendritic synthesis blocks late LTP and greatly reduces the CaMKII content of the PSD120.
Another perspective on the structural changes in late LTP comes from consideration of the changes in quantal transmission. According to one class of models122, 123, the synapse is a modular structure, with each module capable of anchoring ~20 AMPARs (the total number of AMPARs at the synapse can be in the hundreds). During LTP, the addition of AMPARs causes these modules to become functional while coordinated presynaptic processes increase the size of the active zone and enhance the full-fusion mode of vesicle release. Thus, in the late phase of LTP, a synapse is stronger both because of enhanced glutamate release and because there are additional AMPAR-containing postsynaptic modules. The mechanisms by which these structural changes are triggered and the role of CaMKII in these processes is not known.
Conclusions and perspectives
Although there has been progress in understanding LTP, many important questions remain unanswered. The ability to optically measure LTP-induced calcium elevation and CaMKII activation in single spines raises the prospect of understanding the initial processes of LTP at a quantitative level. Models of increasing sophistication121, 122 have been developed for how CaMKII is activated by calcium and calmodulin in vitro. Using these models to understand how CaMKII is activated in the living cell requires knowledge of the state of calmodulin, about which less is known. The abundant protein neurogranin binds calmodulin and releases it when Ca2+ binds to calmodulin. Mutations in the neurogranin gene have strong effects on synaptic plasticity123, some of which can be accounted for by computational models124. Direct measurements of the state of free and bound calmodulin will be important in providing the information necessary to quantitatively accounting for CaMKII activation.
The focus of this Review has been on T286 phosphorylation, but there are other phosphorylation sites on CaMKII about which less is known. A short time after T286 phosphorylation, T305 and T306 in the calmodulin-binding region become phosphorylated125. This delayed phosphorylation strongly reduces autonomous kinase activity126, 127. The phosphorylation of T305 and T306 has physiological consequences: overexpression of T286D CaMKII (to mimic the phosphorylated state) actually leads to synaptic weakening (and occlusion with long-term depression (LTD)) unless T305 and T306 phosphorylation is prevented by additional mutations128. Phosphorylation of these sites prevents binding of CaMKII to the PSD and produces learning deficits129. Understanding the conditions that lead to T305 and T306 phosphorylation will have medical implications because Angleman’s disease, a form of intellectual disability, is associated with enhanced phosphorylation at these sites that prevents normal LTP130. CaMKII contains other phosphorylation sites, for example T253, the functional role of which is unclear131, 132. Recent work shows that stimuli that induce moderate levels of calcium elevation cause translocation of CaMKII to inhibitory synapses, leading to enhancement of GABAA receptor insertion133. It will be interesting to determine the properties of CaMKII that target this protein to inhibitory versus excitatory synapses.
There has been substantial progress in understanding the role of CaMKII in LTP expression, but there remain uncertainties. Experiments suggest that the different expression processes (that is, enhancement of channel conductance versus an increase in channel number) may dominate at different synapses on the same cell65. Why this should be is completely unclear. The role of different GluR1 phosphorylation sites and their interactions remain unclear, particularly the role of CaMKII-dependent phosphorylation of S567. Aspects of the role of stargazin in LTP also remain uncertain. It is likely that stargazin phosphorylation is a required step for transition of AMPA receptors from the plasma membrane to the synapse, but the kinetics of this phosphorylation step are not known. Furthermore, if the binding of stargazin to PSD-95 leads to AMPAR-mediated transmission, then the effects on LTP of deleting the PDZ l binding domain of stargazin should be equivalent to the expression of a nonphosphorylatable mutant of this protein, but they are dramatically different, suggesting that the stargazin-mediated mechanism involves additional complexity.
The mechanisms of LTP maintenance remain controversial. Evidence has been presented that protein kinase M ζ (PKM-ζ) is responsible for the maintenance of LTP. Moreover, various forms of memory, as measured at the behavioural level, can be erased by PKM-ζ inhibitors134, 135. However, questions remain regarding the specificity of ZIP115, 136, the main pharmacological agent used to inhibit PKM-ζ, and it will be important to test the PKM-ζ hypothesis of LTP maintenance by knockout methods. A preliminary report indicates that late LTP is not affected by knocking out PKM-ζ expression and that the effects of ZIP still occur in knockout animals137.
An alternative to the PKM-ζ hypothesis is that LTP is maintained by the CaMKII–NMDAR complex. This model is supported by the ability of CN compounds to reverse LTP maintenance107 (Fig. 2). Although it is known that formation of this complex is necessary for LTP and that the complex forms during chemical forms of LTP, methods need to be developed to monitor CaMKII–NMDAR complexes during synaptically-induced LTP. Such methods would make it possible to determine whether the complexes that form have the stability to underlie long-term memory. The fraction of total spine CaMKII subunits that are bound to the NMDAR is probably too small (< 1%)34 to be monitored by optical indicators of CaMKII activity, so the monitoring of such complexes will thus require more specific methods, such as those based on FRET between CaMKII and the NMDAR.
One of the most fundamental problems regarding information storage by synapses is how stable storage can be achieved despite the constant turnover of molecules at the synapse. Whatever the underlying mechanism, they must yield information storage that is stable over many years. One model attributes long-term storage to phosphorylated CaMKII in the PSD and suggests a specific mechanism whereby this phosphorylated state is maintained when unphosphorylated CaMKII proteins replace older phosphorylated ones during protein turnover138. According to this model, the high density of phosphorylated CaMKII molecules in the PSD saturates the level of phosphatase activity in the PSD; thus, a newly inserted CaMKII will become spontaneously phosphorylated because there is a basal rate of calcium-dependent autophosphorylation that is not effectively counteracted by the saturated phosphatases. Models of this kind seem to contradict the evidence for a structural rather than catalytic role of CaMKII in LTP maintenance. That evidence shows that peptides that interfere with the CaMKII–NMDAR complex reverse LTP maintenance within 30 min, whereas peptides whose action is primarily at the catalytic site do not. However, these data do not exclude the possibility that maintenance, in addition to depending on a structural readout of the CaMKII–NMDAR complex, requires CaMKII-dependent autophosphoryhlation over a longer time scale. Indeed, biochemical work strongly hints that keeping PSD CaMKII phosphosphorylated is important. This work shows that although protein phosphatase-1 at the PSD dephosphorylates some sites on CaMKII, it cannot dephosphorylate T286139. Recent work shows that T286 sites cannot be dephosphorylated because the NMDAR itself shields these sites140. On the other hand, there are also hints that long-term memory storage may not depend on autophosphorylation of T286. Whereas long-term memory for one-trial learning is strongly attenuated in T286A CaMKIIα mice, memory after massed training persists141, suggesting that synapses have other mechanisms for memory storage. Whether this is due to very different molecules or to CaMKII-β activity remains unclear142.
Development of an understanding of how informational stability can be achieved during protein turnover will require information about how synaptic proteins are actually synthesized and degraded. Experiments indicate that CaMKII is involved in stimulating both protein synthesis and ubiquitin-dependent protein degradation 143–145. Further work will be required to understand these slow reactions and the mechanisms by which the stability of stored information is ensured.
In summary, our knowledge about LTP is advancing, but major questions remain. The greatest clarity is about the earliest steps involving activation of the NMDAR, the accumulation of calcium, and the resulting activation of calmodulin and CaMKII. Furthermore, there is impressive evidence indicating that early LTP results from CaMKII-dependent phosphorylation of GluR1 and stargazin. Further work will be required before this mechanism can be considered definitive, however. The late stages of LTP, the stages most important for understanding long-term memory, remain much more mysterious. The late stages are clearly more complex than early ones because they involve changes in gene expression, protein synthesis, and synapse structure and number.
Acknowledgments
We thank P. De Koninck, W. Ross and N. Otmakhov for comments on this Review. We especially thank L. Chao for discussion about CaMKII structure and for preparing the figures for Box 1. We gratefully acknowledge the support of the Ellison Foundation and the NIH (grant R01 DA027807), and the 2011 MBL Research Award.
Footnotes
No competing interest for any of the authors Competing interests statem
References
- 1.Abraham WC. How long will long-term potentiation last? Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2003;358:735–44. doi: 10.1098/rstb.2002.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–7. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 3.Gruart A, Munoz MD, Delgado-Garcia JM. Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J Neurosci. 2006;26:1077–87. doi: 10.1523/JNEUROSCI.2834-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grant SG, Silva AJ. Targeting learning. Trends in neurosciences. 1994;17:71–5. doi: 10.1016/0166-2236(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 5.Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nature reviews. Neuroscience. 2008;9:813–25. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nature Reviews Neuroscience. 2002;3:175–90. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 7.Chao LH, et al. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin- dependent kinase II holoenzyme. Cell. 2011;146:732–45. doi: 10.1016/j.cell.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lisman JE. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3055–7. doi: 10.1073/pnas.82.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–70. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- 10.Lledo PM, et al. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:11175–9. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pi HJ, et al. CaMKII control of spine size and synaptic strength: role of phosphorylation states and nonenzymatic action. Proc Natl Acad Sci U S A. 2010;107:14437–42. doi: 10.1073/pnas.1009268107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–3. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 13.Lucchesi W, Mizuno K, Giese KP. Novel insights into CaMKII function and regulation during memory formation. Brain research bulletin. 2011;85:2–8. doi: 10.1016/j.brainresbull.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nature reviews. Neuroscience. 2009;10:126–40. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hojjati MR, et al. Kinase activity is not required for alphaCaMKII-dependent presynaptic plasticity at CA3-CA1 synapses. Nature neuroscience. 2007;10:1125–7. doi: 10.1038/nn1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanderson JL, Dell’Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2011;17:321–36. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blitzer RD, et al. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–2. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 18.Andersen P, Sundberg SH, Sveen O, Wigstrom H. Specific long-lasting potentiation of synaptic transmission in hippocampal slices. Nature. 1977;266:736–7. doi: 10.1038/266736a0. [DOI] [PubMed] [Google Scholar]
- 19.Lynch GS, Dunwiddie T, Gribkoff V. Heterosynaptic depression: a postsynaptic correlate of long-term potentiation. Nature. 1977;266:737–9. doi: 10.1038/266737a0. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takao K, et al. Visualization of synaptic Ca2+ /calmodulin-dependent protein kinase II activity in living neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:3107–12. doi: 10.1523/JNEUROSCI.0085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ascher P, Nowak L. Early biophysics of the NMDA receptor channel. The Journal of physiology. 2009;587:4563–4. doi: 10.1113/jphysiol.2009.178640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mainen ZF, et al. Two-photon imaging in living brain slices. Methods. 1999;18:231–9. 181. doi: 10.1006/meth.1999.0776. [DOI] [PubMed] [Google Scholar]
- 25.Emptage NJ, Reid CA, Fine A, Bliss TV. Optical quantal analysis reveals a presynaptic component of LTP at hippocampal Schaffer-associational synapses. Neuron. 2003;38:797–804. doi: 10.1016/s0896-6273(03)00325-8. [DOI] [PubMed] [Google Scholar]
- 26.Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca(2+) signals in spines of hippocampal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:1791–9. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca(2+) ions in dendritic spines. Neuron. 2002;33:439–52. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- 28.Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:6037–46. doi: 10.1523/JNEUROSCI.1221-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santucci DM, Raghavachari S. The effects of NR2 subunit-dependent NMDA receptor kinetics on synaptic transmission and CaMKII activation. PLoS computational biology. 2008;4:e1000208. doi: 10.1371/journal.pcbi.1000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345–58. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Current opinion in neurobiology. 2005;15:266–74. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Faas GC, Raghavachari S, Lisman JE, Mody I. Calmodulin as a direct detector of Ca2+ signals. Nature neuroscience. 2011;14:301–4. doi: 10.1038/nn.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng B, Raghavachari S, Lisman J. Quantitative estimates of the cytoplasmic, PSD, and NMDAR-bound pools of CaMKII in dendritic spines. Brain Research. 2011;1419:46–52. doi: 10.1016/j.brainres.2011.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SJ, Yasuda R. Spatiotemporal Regulation of Signaling in and out of Dendritic Spines: CaMKII and Ras. Open Neurosci J. 2009;3:117–127. doi: 10.2174/1874082000903020117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasuda R, Murakoshi H. The mechanisms underlying the spatial spreading of signaling activity. Curr Opin Neurobiol. 2011;21:313–21. doi: 10.1016/j.conb.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–6. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 38.Lin B, et al. Theta stimulation polymerizes actin in dendritic spines of hippocampus. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:2062–9. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmed R, Zha XM, Green SH, Dailey ME. Synaptic activity and F-actin coordinately regulate CaMKIIalpha localization to dendritic postsynaptic sites in developing hippocampal slices. Molecular and cellular neurosciences. 2006;31:37–51. doi: 10.1016/j.mcn.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 40.Shen K, Teruel MN, Subramanian K, Meyer T. CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron. 1998;21:593–606. doi: 10.1016/s0896-6273(00)80569-3. [DOI] [PubMed] [Google Scholar]
- 41.Zhang YP, Holbro N, Oertner TG. Optical induction of plasticity at single synapses reveals input-specific accumulation of alphaCaMKII. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12039–44. doi: 10.1073/pnas.0802940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otmakhov N, et al. Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci. 2004;24:9324–31. doi: 10.1523/JNEUROSCI.2350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N- methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–44. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J Biol Chem. 1998;273:20689–92. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- 45.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–5. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 46.Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275:23798–806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- 47.Bayer KU, et al. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–74. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robison AJ, Bartlett RK, Bass MA, Colbran RJ. Differential modulation of Ca2+/calmodulin-dependent protein kinase II activity by regulated interactions with N-methyl-D-aspartate receptor NR2B subunits and alpha-actinin. The Journal of biological chemistry. 2005;280:39316–23. doi: 10.1074/jbc.M508189200. [DOI] [PubMed] [Google Scholar]
- 49.Barria A, Malinow R. NMDA Receptor Subunit Composition Controls Synaptic Plasticity by Regulating Binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 50.Foster KA, et al. Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:2676–85. doi: 10.1523/JNEUROSCI.4022-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Y, et al. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:13843–53. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X, et al. Mass of the postsynaptic density and enumeration of three key molecules. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11551–6. doi: 10.1073/pnas.0505359102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shinohara Y, et al. Left-right asymmetry of the hippocampal synapses with differential subunit allocation of glutamate receptors. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19498–503. doi: 10.1073/pnas.0807461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugiyama Y, Kawabata I, Sobue K, Okabe S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nature methods. 2005;2:677–84. doi: 10.1038/nmeth783. [DOI] [PubMed] [Google Scholar]
- 55.Walikonis RS, et al. Densin-180 forms a ternary complex with the (alpha)-subunit of Ca2+/calmodulin-dependent protein kinase II and (alpha)-actinin. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:423–33. doi: 10.1523/JNEUROSCI.21-02-00423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nikandrova YA, Jiao Y, Baucum AJ, Tavalin SJ, Colbran RJ. Ca2+/calmodulin-dependent protein kinase II binds to and phosphorylates a specific SAP97 splice variant to disrupt association with AKAP79/150 and modulate alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor (AMPAR) activity. J Biol Chem. 2010;285:923–34. doi: 10.1074/jbc.M109.033985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron. 2004;43:563–74. doi: 10.1016/j.neuron.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 58.Hudmon A, et al. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–47. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Welsby PJ, et al. A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:10116–21. doi: 10.1523/JNEUROSCI.23-31-10116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang X, et al. Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc Natl Acad Sci U S A. 2008;105:341–6. doi: 10.1073/pnas.0710213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu XY, et al. Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron. 2009;61:425–38. doi: 10.1016/j.neuron.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hudmon A, et al. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:6971–83. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petersen JD, et al. Distribution of postsynaptic density (PSD)-95 and Ca2+/calmodulin-dependent protein kinase II at the PSD. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:11270–8. doi: 10.1523/JNEUROSCI.23-35-11270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao D, Jones A, Malinow R. Direct measurement of quantal changes underlying long-term potentiation in CA1 hippocampus. Neuron. 1992;9:1089–97. doi: 10.1016/0896-6273(92)90068-o. [DOI] [PubMed] [Google Scholar]
- 65.Luthi A, et al. Bi-directional modulation of AMPA receptor unitary conductance by synaptic activity. BMC neuroscience. 2004;5:44. doi: 10.1186/1471-2202-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poncer JC, Esteban JA, Malinow R. Multiple mechanisms for the potentiation of AMPA receptor-mediated transmission by alpha-Ca2+/calmodulin-dependent protein kinase II. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:4406–11. doi: 10.1523/JNEUROSCI.22-11-04406.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi SH, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–6. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 68.Opazo P, Choquet D. A three-step model for the synaptic recruitment of AMPA receptors. Molecular and cellular neurosciences. 2011;46:1–8. doi: 10.1016/j.mcn.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 69.Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45:269–77. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 70.Opazo P, et al. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–52. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 71.Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. The Journal of biological chemistry. 1997;272:32727–30. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- 72.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–88. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 73.Lu W, Isozaki K, Roche KW, Nicoll RA. Synaptic targeting of AMPA receptors is regulated by a CaMKII site in the first intracellular loop of GluA1. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:22266–71. doi: 10.1073/pnas.1016289107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Banke TG, et al. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boehm J, et al. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–25. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 76.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–9. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 77.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–5. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 78.Kristensen AS, et al. Mechanism of Ca(2)/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nature neuroscience. 2011;14:727–35. doi: 10.1038/nn.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nature reviews. Neuroscience. 2007;8:101–13. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- 80.Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. Journal of neurophysiology. 2010;103:479–89. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andrasfalvy BK, Magee JC. Changes in AMPA receptor currents following LTP induction on rat CA1 pyramidal neurones. The Journal of physiology. 2004;559:543–54. doi: 10.1113/jphysiol.2004.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vinade L, Dosemeci A. Regulation of the phosphorylation state of the AMPA receptor GluR1 subunit in the postsynaptic density. Cellular and molecular neurobiology. 2000;20:451–63. doi: 10.1023/A:1007019030595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsui J, Malenka RC. Substrate localization creates specificity in calcium/calmodulin-dependent protein kinase II signaling at synapses. The Journal of biological chemistry. 2006;281:13794–804. doi: 10.1074/jbc.M600966200. [DOI] [PubMed] [Google Scholar]
- 84.Yudowski GA, Puthenveedu MA, von Zastrow M. Distinct modes of regulated receptor insertion to the somatodendritic plasma membrane. Nat Neurosci. 2006;9:622–7. doi: 10.1038/nn1679. [DOI] [PubMed] [Google Scholar]
- 85.Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–90. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patterson MA, Szatmari EM, Yasuda R. AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc Natl Acad Sci U S A. 2010;107:15951–6. doi: 10.1073/pnas.0913875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling Endosomes Supply AMPA Receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 88.Park M, et al. Plasticity-Induced Growth of Dendritic Spines by Exocytic Trafficking from Recycling Endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Z, et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–48. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–35. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- 92.Yang Y, Wang XB, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:5740–51. doi: 10.1523/JNEUROSCI.3998-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–6. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 94.Zhu J, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap Control AMPA Receptor Trafficking during Synaptic Plasticity. Cell. 2002;110:443. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 95.Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol. 2002;3:339–48. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 96.Kim CH, et al. Persistent hippocampal CA1 LTP in mice lacking the C-terminal PDZ ligand of GluR1. Nat Neurosci. 2005;8:985–7. doi: 10.1038/nn1432. [DOI] [PubMed] [Google Scholar]
- 97.Schnell E, et al. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–7. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bats C, Groc L, Choquet D. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–34. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 99.Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–27. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sainlos M, et al. Biomimetic divalent ligands for the acute disruption of synaptic AMPAR stabilization. Nature chemical biology. 2011;7:81–91. doi: 10.1038/nchembio.498. [DOI] [PubMed] [Google Scholar]
- 101.Kim CH, Lisman JE. A labile component of AMPA receptor-mediated synaptic transmission is dependent on microtubule motors, actin, and N-ethylmaleimide-sensitive factor. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:4188–94. doi: 10.1523/JNEUROSCI.21-12-04188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sanhueza M, McIntyre CC, Lisman JE. Reversal of synaptic memory by Ca2+/calmodulin-dependent protein kinase II inhibitor. J Neurosci. 2007;27:5190–9. doi: 10.1523/JNEUROSCI.5049-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen X, et al. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J Neurosci. 2011;31:6329–38. doi: 10.1523/JNEUROSCI.5968-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–56. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lengyel I, et al. Autonomous activity of CaMKII is only transiently increased following the induction of long-term potentiation in the rat hippocampus. The European journal of neuroscience. 2004;20:3063–72. doi: 10.1111/j.1460-9568.2004.03748.x. [DOI] [PubMed] [Google Scholar]
- 106.Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011 doi: 10.1038/nature09823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sanhueza M, et al. Role of the CaMKII/NMDA Receptor Complex in the Maintenance of Synaptic Strength. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:9170–9178. doi: 10.1523/JNEUROSCI.1250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vest RS, Davies KD, O’Leary H, Port JD, Bayer KU. Dual Mechanism of a Natural CaMKII Inhibitor. Mol Biol Cell. 2007;18:5024–33. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:5357–65. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tsien RW, Schulman H, Malinow R. Peptide inhibitors of PKC and CaMK block induction but not expression of long-term potentiation. Advances in second messenger and phosphoprotein research. 1990;24:101–7. [PubMed] [Google Scholar]
- 111.Buard I, et al. CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:8214–20. doi: 10.1523/JNEUROSCI.1469-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–45. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- 113.Tanaka JI, et al. Protein Synthesis and Neurotrophin-Dependent Structural Plasticity of Single Dendritic Spines. Science. 2008 doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Smith WB, Starck SR, Roberts RW, Schuman EM. Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron. 2005;45:765–79. doi: 10.1016/j.neuron.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 115.Lisman J. Memory erasure by very high concentrations of ZIP may not be due to PKM-zeta. Hippocampus. 2011 doi: 10.1002/hipo.20980. [DOI] [PubMed] [Google Scholar]
- 116.Frey U, Morris RG. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends in neurosciences. 1998;21:181–8. doi: 10.1016/s0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- 117.Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:7143–6. doi: 10.1523/JNEUROSCI.1796-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:11760–7. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Day JJ, Sweatt JD. Cognitive neuroepigenetics: a role for epigenetic mechanisms in learning and memory. Neurobiology of learning and memory. 2011;96:2–12. doi: 10.1016/j.nlm.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miller S, et al. Disruption of dendritic translation of CaMKIIalpha impairs stabilization of synaptic plasticity and memory consolidation. Neuron. 2002;36:507–19. doi: 10.1016/s0896-6273(02)00978-9. [DOI] [PubMed] [Google Scholar]
- 121.Pepke S, Kinzer-Ursem T, Mihalas S, Kennedy MB. A dynamic model of interactions of Ca2+, calmodulin, and catalytic subunits of Ca2+/calmodulin-dependent protein kinase II. PLoS computational biology. 2010;6:e1000675. doi: 10.1371/journal.pcbi.1000675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Byrne MJ, Putkey JA, Waxham MN, Kubota Y. Dissecting cooperative calmodulin binding to CaM kinase II: a detailed stochastic model. Journal of computational neuroscience. 2009;27:621–38. doi: 10.1007/s10827-009-0173-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huang KP, et al. Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:10660–9. doi: 10.1523/JNEUROSCI.2213-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhabotinsky AM, Camp RN, Epstein IR, Lisman JE. Role of the neurogranin concentrated in spines in the induction of long-term potentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:7337–47. doi: 10.1523/JNEUROSCI.0729-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hanson PI, Schulman H. Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J Biol Chem. 1992;267:17216–24. [PubMed] [Google Scholar]
- 126.Coultrap SJ, Buard I, Kulbe JR, Dell’Acqua ML, Bayer KU. CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. The Journal of biological chemistry. 2010;285:17930–7. doi: 10.1074/jbc.M109.069351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jama AM, Fenton J, Robertson SD, Torok K. Time-dependent autoinactivation of phospho-Thr286-alphaCa2+/calmodulin-dependent protein kinase II. The Journal of biological chemistry. 2009;284:28146–55. doi: 10.1074/jbc.M109.005900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pi HJ, Otmakhov N, Lemelin D, De Koninck P, Lisman J. Autonomous CaMKII can promote either long-term potentiation or long-term depression, depending on the state of T305/T306 phosphorylation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:8704–9. doi: 10.1523/JNEUROSCI.0133-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Elgersma Y, et al. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron. 2002;36:493–505. doi: 10.1016/s0896-6273(02)01007-3. [DOI] [PubMed] [Google Scholar]
- 130.Weeber EJ, et al. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:2634–44. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Skelding KA, et al. Regulation of CaMKII by phospho-Thr253 or phospho-Thr286 sensitive targeting alters cellular function. Cellular signalling. 2010;22:759–69. doi: 10.1016/j.cellsig.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 132.Gurd JW, et al. Ischemia and status epilepitcus result in enhanced phosphorylation of calcium and calmodulin-stimulated protein kinase II on threonine 253. Brain research. 2008;1218:158–65. doi: 10.1016/j.brainres.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 133.Marsden KC, Shemesh A, Bayer KU, Carroll RC. Selective translocation of Ca2+/calmodulin protein kinase IIalpha (CaMKIIalpha) to inhibitory synapses. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20559–64. doi: 10.1073/pnas.1010346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sacktor TC. How does PKMzeta maintain long-term memory? Nature reviews. Neuroscience. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- 135.Shema R, et al. Enhancement of consolidated long-term memory by overexpression of protein kinase Mzeta in the neocortex. Science. 2011;331:1207–10. doi: 10.1126/science.1200215. [DOI] [PubMed] [Google Scholar]
- 136.Sacktor TC, Fenton AA. Appropriate application of ZIP for PKMzeta inhibition, LTP reversal, and memory erasure. Hippocampus. 2011 doi: 10.1002/hipo.20978. [DOI] [PubMed] [Google Scholar]
- 137.Volk LJ, Bachman J, Johnson RC, Yu Y, Huganir RL. Annual Society for Neuroscience Meeting. Society for Neuroscience; Washington D.C: 2011. [Google Scholar]
- 138.Miller P, Zhabotinsky AM, Lisman JE, Wang XJ. The stability of a stochastic CaMKII switch: dependence on the number of enzyme molecules and protein turnover. PLoS biology. 2005;3:e107. doi: 10.1371/journal.pbio.0030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mullasseril P, Dosemeci A, Lisman JE, Griffith LC. A structural mechanism for maintaining the ‘on-state’ of the CaMKII memory switch in the post-synaptic density. Journal of neurochemistry. 2007;103:357–64. doi: 10.1111/j.1471-4159.2007.04744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cheriyan J, Kumar P, Mayadevi M, Surolia A, Omkumar RV. Calcium/calmodulin dependent protein kinase II bound to NMDA receptor 2B subunit exhibits increased ATP affinity and attenuated dephosphorylation. PloS one. 2011;6:e16495. doi: 10.1371/journal.pone.0016495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Irvine EE, et al. Properties of contextual memory formed in the absence of alphaCaMKII autophosphorylation. Molecular brain. 2011;4:8. doi: 10.1186/1756-6606-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Radwanska K, et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18471–5. doi: 10.1073/pnas.1109680108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bingol B, et al. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–78. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 144.Atkins CM, Nozaki N, Shigeri Y, Soderling TR. Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:5193–201. doi: 10.1523/JNEUROSCI.0854-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nagerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006;52:239–45. doi: 10.1016/j.neuron.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 146.Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Molecular cell. 2003;11:1241–51. doi: 10.1016/s1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- 147.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–60. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 148.Kolodziej SJ, Hudmon A, Waxham MN, Stoops JK. Three-dimensional reconstructions of calcium/calmodulin-dependent (CaM) kinase IIalpha and truncated CaM kinase IIalpha reveal a unique organization for its structural core and functional domains. The Journal of biological chemistry. 2000;275:14354–9. doi: 10.1074/jbc.275.19.14354. [DOI] [PubMed] [Google Scholar]
- 149.Morris EP, Torok K. Oligomeric structure of alpha-calmodulin-dependent protein kinase II. Journal of molecular biology. 2001;308:1–8. doi: 10.1006/jmbi.2001.4584. [DOI] [PubMed] [Google Scholar]
- 150.Rellos P, et al. Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS biology. 2010;8:e1000426. doi: 10.1371/journal.pbio.1000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bradshaw JM, Kubota Y, Meyer T, Schulman H. An ultrasensitive Ca2+/calmodulin-dependent protein kinase II-protein phosphatase 1 switch facilitates specificity in postsynaptic calcium signaling. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10512–7. doi: 10.1073/pnas.1932759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gaertner TR, et al. Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. The Journal of biological chemistry. 2004;279:12484–94. doi: 10.1074/jbc.M313597200. [DOI] [PubMed] [Google Scholar]
- 153.Svoboda K, Tank DW, Denk W. Direct measurement of coupling between dendritic spines and shafts. Science. 1996;272:716–9. doi: 10.1126/science.272.5262.716. [DOI] [PubMed] [Google Scholar]
- 154.Harvey CD, Yasuda R, Zhong H, Svoboda K. The spread of Ras activity triggered by activation of a single dendritic spine. Science. 2008;321:136–40. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]