

Abstract
Hepatocellular carcinoma is a multifactorial disease which is associated with a background of many causal risk factors. Diabetes mellitus however is one of the most common co-morbid illnesses found in hepatocellular carcinoma patients that are significantly associated with worsening of hepatocellular carcinoma development, patient prognosis and survival. Therefore, efforts have been focused on understanding the mechanisms underlying progression of hepatocellular carcinoma onset and development especially in diabetic patients. To our knowledge, there are no reports which address the impact of tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) along with epigenetic regulations associated with increased risk of hepatocellular carcinoma confounded by diabetes mellitus. Therefore, this mini-review focuses on the possible intermediary mechanisms involved in worsening the onset and progression of hepatocellular carcinoma development confounded by diabetes mellitus. The first approach is to look at the role of inflammatory mediators (TNF-α and IL-6) in apoptosis and inflammation during hepatocarcinogenesis through monitoring levels of apoptotic regulators, B-cell lymphoma 2 protein which is encoded by BCL2 gene and apoptosis regulator BAX known as bcl-2-like protein 4 which is encoded by the BAX gene. The second approach is to focus on the possible epigenomic reprogramming that drives hepatocellular transformation since epigenetic modification of DNA is a key feature in the pathogenesis of hepatocarcinogenesis. Both approaches may suggest role of using Bcl2 and Bax as apoptotic and inflammatory markers for hepatocellular carcinoma detection as well as the importance impact of DNA methylation, hypomethylation or histone modifications as attractive candidates for early-detection biomarkers of hepatocellular carcinoma.
Keywords: Apoptosis and inflammation, Diabetes mellitus, DNA methylation or histone modifications, Epigenomic reprogramming, Hepatocarcinogenesis, Hepatocellular carcinoma
The association between diabetes mellitus and the risk of hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is the fifth most common malignancy in the world with high morbidity and mortality but its pathogenesis remains unclear [1]. Because primary liver cancer is a growing concern showing poor prognosis due to its rapid infiltrating power and complicating liver cirrhosis; more attention were given to address the causal risk factors that could be preventable and/or treatable [2]. Several risk factors have been identified that contribute to the international burden of HCC such as chronic infection with hepatitis B virus (HBV) and hepatitis C virus (HCV), alcoholic liver disease, non-alcoholic steatohepatitis (NASH), diabetes mellitus (DM), obesity, intake of aflatoxins-contaminated food, tobacco smoking, excessive alcohol drinking and genetically inherited disorders (hemochromatosis, α-1 anti-trypsin deficiency, porphyrias); [3]. HCC is phenotypically and genetically heterogeneous tumor, reflecting in part the heterogeneity of etiologic factors involved in (i) the onset of HCC development that is also influenced by age, gender and ethnic differences, (ii) the complexity of hepatocyte functions and epigenome leading to neoplastic transformation and (iii) the late stage of HCC development [2].
Because the liver plays a crucial role in glucose metabolism, it is not surprising that DM is an epiphenomenon of many chronic liver diseases such as chronic hepatitis, fatty liver, liver failure and cirrhosis [4]. DM is a metabolic disorder characterized by hyperglycemia which may predispose the liver to relative insulin resistant due to inadequate secretion or receptor insensitivity to endogenous insulin. DM is one of the leading causes of blindness and the most common cause of end-stage renal disease and cardiovascular complications in developed countries. In recent years, DM has been associated with increase risk for several malignancies including breast, colon, kidney, liver, endometrium and pancreatic cancers [5]. In addition, DM as part of the insulin resistance syndrome, has been implicated as a risk factor for non-alcoholic fatty liver disease (NAFLD), including its most severe form non-alcoholic steatohepatitis (NASH); which has been identified as a cause of both cryptogenic cirrhosis and HCC. DM has been reported to cause a 2.5-fold greater risk of HCC, however this significant association was independent to hepatitis viral infections (HBV and/or HCV) or alcohol consumption as examined from 10 previously reported studies; those reports account for ~90% of HCC cases [1,6]. Many case reports and case reviews of HCC in NASH have showed the association of DM and obesity on the increased risk of HCC and have implicated age and advanced fibrosis as significant risks. Insulin resistance and the resulting inflammatory cascade, which are associated with the development of NASH appears to mediate hepatocarcinogenesis in HCC [1].
The association between DM and HCC has been demonstrated in both case–control and cohort studies, suggesting that DM is independent risk factor for the development of HCC. These several lines of evidence includes; (i) findings that insulin resistance and DM were shown to increase the progression of liver diseases that preceded the development of HCC [6,7]; (ii) findings showing that 2.5 fold increase in the risk of HCC was reported in patients with DM [8,9] attributed with the prolonged DM disease duration [10-12]; (iii) findings demonstrating the synergistic interaction between DM and other HCC risk factors [11,13,14]; (iv) findings showed significant association of DM for the recurrence of HCC after treatment [15,16]; (v) findings suggesting significant biological plausibility underlying the association between DM and HCC [7,17].
Nevertheless, the exact pathophysiological mechanisms of this significant association are still unclear. In this mini-review, we aim to further analyze this relationship by evaluating the role of possible intermediary mechanisms that could be associated with the onset and progression of HCC development in the presence of DM. We will mainly focus on; (i) pro-inflammatory gene products (TNF-α and IL-6) and (ii) the epigenetic pathogenesis of DM on the development of HCC.
Possible intermediary mechanisms involved in worsening the onset and progression of hepatocellular carcinoma confounded by diabetes mellitus
TNF-α/ NF-kβ and IL-6/STAT-3 signaling pathways effect on hepatocellular carcinoma development
Malignant transformation of hepatocytes may occur through a pathway of increased liver cell turnover induced by chronic liver injury and regeneration in the context of inflammation, immune response and oxidative DNA damage [18]. This is evidence to suggest that chronic inflammation in the individuals with diabetes mellitus type 2 (T2DM) may influence certain cancers via cytokines. Cancerous and precancerous tissue show signs of inflammation, which is caused by the infiltration of the immune cells into the tissues. The presence of inflammatory cytokines is important constituents of the local environment of tumors in certain type of cancer including HCC. These cytokines cause oncogenic changes that promote tumor development by blocking apoptosis and increasing the survival of malignant cells [19]. Although an elevation in the circulating levels of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-12 (IL-12), interleukin-1-beta (IL-1β) and transforming growth factor beta (TGF-β-1) has been observed in obese and diabetic individuals [20-24]; plasma levels of interleukin-10 (IL-10) were found elevated in obese individuals thus, decreased in diabetics [25].
Cytokines such as IL-1β, TNF-α, IL-8 and IL-6 have been involved in chronic liver inflammation and pathogenesis of various liver diseases; among which IL-6 and TNF-α, are thought to be most important [26-28]. In this regard, TNF-α and IL-6 were found to positively correlated with the progression of HCC development in humans [29-31].
IL-6 is a pleiotropic cytokine secreted by T cells, macrophages, kupffer cells, and adipose tissue. Similarly, TNF-α is secreted by several inflammatory cell types, including monocyte/macrophages, neutrophils, T-cells, kupffer cells and adipose tissue [26,32]. Increased circulating levels of IL-6 and TNF-α have been found in animal models and patients with liver disease including HCC [33]. Moreover, studies have revealed that the levels of TNF-α and IL-6 are significantly increased in rats bearing hepatoma, which reflects an aggressive inflammatory response correlated with tumor induction [34]. In obesity and DM, tremendous amount of the tumor-promoting cytokines IL-6 and TNF-α are produced, which is thought to promote HCC development [35,31]. Moreover, DM-mediated inflammatory responses lead to the production of cytokines, which act as growth and angiogenic factors for transformed cells. DM has confounding effects on the factors of inflammation, proliferation and anti-apoptosis in HCC, which explains why HCC and DM co-occur twice as frequently as expected. Furthermore, it has been reported that diabetic patients with HCC are suffer more frequently with liver injury and tumor growth as compared to non-diabetics [19].
IL-6 and TNF-α are involved in multiple signalling pathways, which ultimately lead to liver injury, inflammation and HCC development (Figure 1). Chronic inflammation in diabetic patients may promote carcinogenesis through multifaceted processes such as activation of signal transducer and activator of transcription 3 (STAT-3) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kβ) transcription factors following the production of tumor growth factors or induction of angiogenesis. It is known that STAT-3, activated by IL-6 in hypatocytes, promotes HCC cell growth in vitro and in vivo [36]. In addition, activated IL-6/STAT-3 pathways have been observed in liver cancer and are thought to be important factors in the initiation, development, and progression of HCC [37,38]. Furthermore, blockade of STAT-3 may have therapeutic potential in preventing and treating liver cancer [39]. Since serum concentrations of IL-6 are elevated in diabetic patients, it is reasonable to surmise that IL-6-activated STAT-3 signaling pathway plays play a role in HCC development in DM. The oncogenic role of constitutively activated STAT-3 is driven through the up-regulation of cell survival proteins (Bcl-xL, BCL-2) and cell cycle regulators (c-Myc, cyclin D) [38,40-42]. The second main HCC oncogenic pathway is TNF-α-mediated activation of NF-kβ, which is a key regulator of inflammation that provides a mechanistic link between inflammation and apoptosis during carcinogenesis [43]. Constitutively increased NF-kβ activation has been observed in tumor tissues [44]; however, IKK-α, a critical kinase for NF-kβ activation, is necessary to produce malignant properties in liver cancer [45]. NF-kβ activation not only protects tumor cells against cell death but also provides essential growth factors to non-parenchymal cells of the liver, such as Kupffer cells [46,47]. TNF-α-mediated NF-kβ activation has both positive and negative impact on our immune system [48,49]. An imbalance in TNF-α mediated NF-kβ could lead to several types of inflammatory disorders. In DM, a balance shift towards a prolonged activation of IKK-α/NF-kβ, and simultaneous blockade of apoptosis, is essential for the inhibition of tumor growth. TNF-α induced activation of mitogen-activated protein kinases (MAPKs) are also significant factors for tumour growth. TNF-α levels were shown to be higher in obese and diabetic individuals and to correlate with insulin resistance and liver disease [27,32,50]. In addition, increased STAT-3 and NF-kβ were shown to be correlated with impaired insulin sensitivity and more advanced development of T2DM. Furthermore, systemic abnormalities in the activation of STAT-3 and NF-kβ in subjects with DM provide clinically important milieu for increased risk of severe HCC development [51,52]. Since the presence of TNF-α and IL-6 is considered to be a possible risk factor for the development of HCC in diabetic individuals, DM is thought to be a major risk factor for the development of aggressive HCC. In conclusion, these studies suggest that the presence of liver inflammation in the context of DM, leads to the exposure of hepatocytes to increased levels of IL-6 and TNF-α, which promote the activation of JAK/STAT-3 and IKKα/NF-kβ signaling pathways, followed by lack of apoptosis, and consequently uncontrolled proliferation of hepatocytes; this results in initiation and promotion of HCC development [53].
Figure 1.
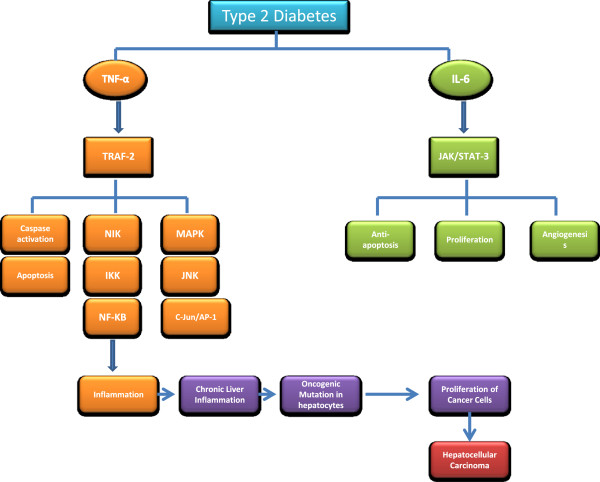
Oncogenic impact of IL-6/STAT-3 and TNF-α/NF-kβ signaling pathways on the development and progression of HCC. Obesity induced type-2 diabetes, produces IL-6 and TNF-α by accessory cells, adipose tissue and Kupffer cells. These inflammatory markers (IL-6 and TNF-α) activate the downstream signaling molecules STAT-3 and NF-kβ in residual liver cells, respectively. This activation of NF-kβ and STAT-3 signaling pathways are likely to be involved in critical processes such as anti-apoptosis, proliferation and angiogenesis, which contribute to hepatocarcinogenesis.
Epigenomic reprogramming that drives hepatocellular transformation
Multifactorial diseases such as DM and HCC are far more complex compared to single gene disorders; whereby multiple genes in addition to non-genetic components dictate the pathogenic manifestations of the disease. Further sophistication layer is added onto these disorders from modifications on the genetic material in cells and tissues. These are referred to as “epigenetics factors”. Epigenetic (meaning “above” genetics) inheritance is essential for the development of critical cellular processes such as gene transcription, differentiation and protection against viral genomes. Aberrant epigenetic states may predispose to genetic changes or vice versa [54-56]. Therefore, epigenetic and genetic mechanisms may work together to silence key cellular genes and destabilize the genome, leading to oncogenic transformation and observed complexity and heterogeneity in human cancers. The main epigenetic modifications that can alter gene expression and tissue-specific cellular changes [57] are DNA methylations, histone modifications, and non-coding RNAs (micro and long non-coding RNAs); that together impact on transcriptional regulation of wide range of genes [58,59]. Thus, unregulated epigenetic events play key role in pathogenic mechanisms affecting the expression of many genes including tumor-suppressor genes and cancer-associated genes as has been observed in broad human cancers [60]. Moreover, a number of studies suggested that a frequent loss of heterozygosity (LOH) in chromosome 8p in HCC cases which leads to inactivation of the Deleted in Liver Cancer 1 gene (DLC-1) may play a pivotal role in HCC development [61]. Thus, in the late stage of HCC development; somatic mutations in several tumor suppressor genes (such as TP53, p16 and RB); oncogenes (such as c-MYC and β-catenin) and other cancer-associated genes including E-cadherin and cyclin D1 have also been observed. However, the significance and sequence of these genetic events remain to be established [61,62].
Recent data suggest that the epigenetic pattern is age-dependent influencing key mitochondrial respiratory chain genes [63,64]. An example is the complex 4 COX7A1 protein (which is a target of age-related DNA methylation) as evidenced from its expression reduction in muscle tissue from diabetic patients [64,65]. In young and elderly twin studies; COX7A1 gene promoter methylation was shown to be increased in skeletal muscles of elderly compared with young twins, and that was reciprocal to expression pattern of COX7A1 gene. The transcript level of COX7A1 gene in skeletal muscle was associated with increased in vivo glucose uptake [64]. Polymorphism can also lead to generation of DNA methylation sites (CpG dinucleotides). In addition, putative transcription factor binding site in the NDUFB6 promoter are associated with increased DNA methylation, decreased gene expression, and decreased in vivo metabolism with increasing age [66]. The impact of aging on T2DM is also evident from data on the hepatic function of glucokinase enzyme glucose utilization, where its activity is decreased in the diabetic patients’ liver [67]. Further evidence for epigenetic role was demonstrated in a recent study in which significant differential DNA methylation profiling was indicated when comparing pancreatic islets from type 2 diabetics with non-diabetic controls; where 276 CpG loci associated with 254 genes were uncovered. These methylation signatures were absent in peripheral blood cells of diabetic individuals, and couldn’t be experimentally induced in non-diabetic islets by exposure to high glucose, suggesting tissue-specific methylation patterns [68]. There is also the potential role for epigenetic control microRNAs (miRNAs) on chromatin-modifying enzymes leading to effects in gene expression. Histone modifications and changes in chromatin structure can affect transcription and expression of miRNAs [69]; which are associated with the regulation of pathogenic pathways critical in insulin secretion, cholesterol synthesis and fat metabolism [70-72]. Increased telomeric activity and hTERT expression in HCC cases were also reported. In one study on 106 hepatic tissues with and without HCC; hTERT expression has correlated inversely with DNA methylation levels in normal tissues compared to tumor tissues in a mechanism thought to be regulated by DNA methylation and histone H3-K9 modifications [73].
Furthermore, genome-wide oncogenes promoter demethylation was detected during HCC progression [74]. This demethylation was accompanied by selective regional hypermethylation in the CpG islands leading to silencing of antitumor genes like tumor suppressor genes, proliferation inhibitor genes, in addition to apoptotic and DNA repair genes [75,76]. A proposed mechanism for HCC is suggested to occur via activating inflammatory response components’ (NF-κB and JAK/STAT) to induce epigenetic changes resulting in switching on the long-term oncogenic memory system in hepatocytes [77]. The epigenetic switch in turn would contribute to a chronic inflammatory response during the course of altered gene expression concordant with a positive feedback loop to aggravate a chronic state of inflammation. These uncontrolled epigenetic-driven transcriptional alterations ultimately promote hepatocytes proliferative and oncogenic transformations.
Conclusions
In this mini-review, we summarized the intermediary mechanisms conveying the association between DM and HCC development. Whereby, in the first section, we highlighted that DM can work as a promoter for HCC development since number of evidence lines showed that it may worsen the HCC progression as evident by co-existence of pro-inflammatory gene products (TNF-α and IL-6) during hepatocarcinogenesis. These inflammatory markers play a role in monitoring levels of apoptotic regulators (Bcl2 and Bax), suggesting their importance as apoptotic and inflammatory markers for HCC. The second section, we have focused on the possible effects of epigenetic mechanisms of HCC upon co-existence of DM. Number of studies showed that DNA methylation, histone modifications, and RNA interference, may lead to activation of pro-inflammatory signaling (NF-κB and JAK/STAT) and deregulation of metabolic pathways. The crosstalk of chromatin-modifying enzymes, microRNAs, signaling pathways and the downstream transcription factors may result in epigenomic reprogramming that drives hepatocellular transformation. Many of the epigenetic events and methylated genes described previously in other studies have been detected in premalignant tissues and in the serum of patients prior to or concurrent with the diagnosis of cancer. For this reason, DNA methylation, hypomethylation or histone modifications are attractive candidates for early-detection biomarkers (Figure 2).
Figure 2.
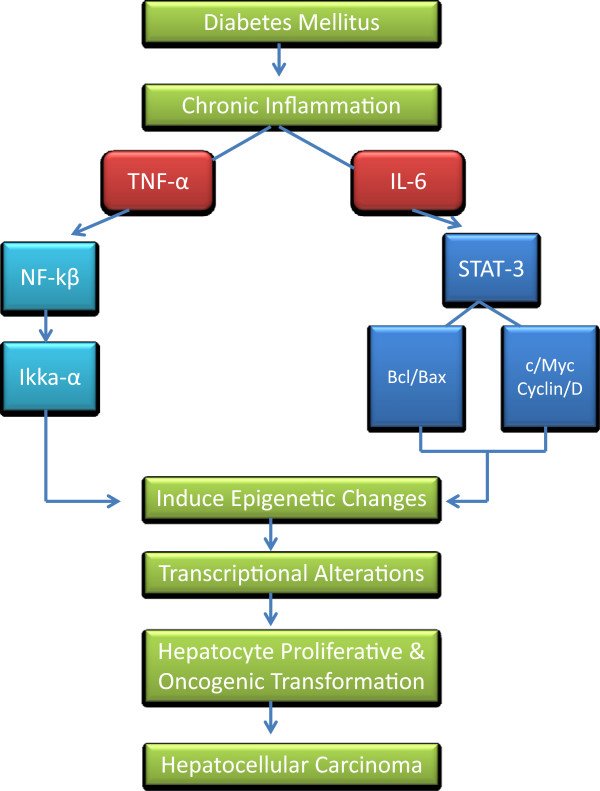
Overview of the possible intermediary mechanisms associated with onset and progression of hepatocellular carcinoma confounded by diabetes mellitus; (i) possible effects of IL-6 and TNF-α as apoptotic and inflammatory markers for the development of HCC and (ii) role of epigenomic reprogramming that drives hepatocellular transformation.
Future trends
Inflammatory markers (TNF-α and IL-6) can be suggested to be used as diagnostic tools for HCC along with alpha-fetoprotein (AFP) that may help to monitor the severity of hepatocellular damage and hepatic function in diabetic patients. These markers can also help to follow-up patients diagnosed with HCC prognostically. On the other hand, epigenetic alterations could be used as biomarkers of early-detection for HCC which may help in prognosticate diabetic patient outcomes or even as novel therapeutic compounds. However, large-scale prospective data is warranted before this approach can be translated into personalized medicine.
Competing interests
All authors have nothing to disclose.
Author’s contribution
MMAK was involved with literature review to interpret the association, drafting of the manuscript and critical revision of the manuscript. RA, OA and KB were involved with drafting of the manuscript and critical revision of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Maisa Mahmoud Ali Kamkar, Email: maisa.mahmoud@dasmaninstitute.org.
Rasheed Ahmad, Email: rasheed.ahmad@dasmaninstitute.org.
Osama Alsmadi, Email: osama.alsmadi@dasmaninstitute.org.
Kazem Behbehani, Email: kazem.behbehani@dasmaninstitute.org.
Acknowledgements
Other members who contributed in reviewing the manuscript are, Dr. Ali Tiss, Mr. Amro Shehabeldin, Dr. Mohammed Abufarha and Dr. Amal Hasan.
References
- Montella M, Crispo A, Giudice A. HCC, diet and metabolic factors. Hepat Mon. 2011;11:159–162. [PMC free article] [PubMed] [Google Scholar]
- Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist. 2010;15:14–22. doi: 10.1634/theoncologist.2010-S4-14. [DOI] [PubMed] [Google Scholar]
- Nordenstedt H, White DL, El-Serag HB. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl 3):S206–S214. doi: 10.1016/S1590-8658(10)60507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffy G. Editorial: hepatocellular carcinoma in type 2 diabetes: more than meets the eye. Am J Gastroenterol. 2012;107:53–55. doi: 10.1038/ajg.2011.390. [DOI] [PubMed] [Google Scholar]
- Herman WH, Zimmet P. Type 2 diabetes: an epidemic requiring global attention and urgent action. Diabetes Care. 2012;35:943–944. doi: 10.2337/dc12-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systemic review of epidemiology evidence. Clin Gastroenterol Hepatol. 2006;4:369–380. doi: 10.1016/j.cgh.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Bell DS, Allbright E. The multifaceted associations of hepatobiliary disease and diabetes. Endocr Pract. 2007;13:300–312. doi: 10.4158/EP.13.3.300. [DOI] [PubMed] [Google Scholar]
- Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serage HB. Diabetes increases the risk of hepatocellular carcinoma in the United States: a population based case control study. Gut. 2005;54:533–539. doi: 10.1136/gut.2004.052167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldt BJ, Chen W, Heathcote EJ, Wedemeyer H, Reichen J, Hofmann WP, Knegt RJ, Zeuzem S, Manns MP, Hansen BE, Schalm SW, Janssen HL. Increased risk of hepatocellular carcinoma among patients with hepatitis C cirrhosis and diabetes mellitus. Hepatology. 2008;47:1856–1862. doi: 10.1002/hep.22251. [DOI] [PubMed] [Google Scholar]
- Hassan MM, Curley SA, Li D, Kaseb A, Davila M, Abdalla EK, Javle M, Moghazy DM, Lozano RD, Abbruzzese JL, Vauthey JN. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer. 2010;116:1938–1946. doi: 10.1002/cncr.24982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JM, Govindarajan S, Arakawa K, Yu MC. Synergism of alcohol, diabetes and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer. 2004;101:1009–1017. doi: 10.1002/cncr.20427. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004;126:460–468. doi: 10.1053/j.gastro.2003.10.065. [DOI] [PubMed] [Google Scholar]
- Chen CL, Yang HI, Yang WS, Liu CJ, Chen PJ, You SL, Wang LY, Sun CA, Lu SN, Chen DS, Chen CJ. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: a follow-up study in Taiwan. Gastroenterology. 2008;135:111–121. doi: 10.1053/j.gastro.2008.03.073. [DOI] [PubMed] [Google Scholar]
- Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Abbruzzese JL, Beasley P, Patt YZ. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;361:1206–1213. doi: 10.1053/jhep.2002.36780. [DOI] [PubMed] [Google Scholar]
- Komura T, Mizukoshi E, Kita Y, Sakurai M, Takata Y, Arai K, Yamashita T, Ohta T, Shimizu K, Nakamoto Y, Honda M, Takamura T, Kaneko S. Impact of diabetes on recurrence of hepatocellular carcinoma after surgical treatment in patients with viral hepatitis. Am J Gastroenterol. 2007;102:1939–1946. doi: 10.1111/j.1572-0241.2007.01354.x. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Shimada M, Hasegawa H, Gion T, Kajiyama K, Shirabe K, Yanaga K, Takenaka K, Sugimachi K. Prognosis of hepatocellular carcinoma with diabetes mellitus after hepatic resection. Hepatology. 1998;27:1567–1571. doi: 10.1002/hep.510270615. [DOI] [PubMed] [Google Scholar]
- Dellon ES, Shaheen NJ. Diabetes and hepatocellular carcinoma: associations, biological plausibility and clinical implications. Gastroenterology. 2005;129:1132–1134. doi: 10.1053/j.gastro.2005.06.079. [DOI] [PubMed] [Google Scholar]
- El-Serag H, Rudolph K. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, Vidal H, Hainque B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab. 2000;58:3338–3342. doi: 10.1210/jcem.85.9.6839. [DOI] [PubMed] [Google Scholar]
- Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J, Kay TW. IL-1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes. 2004;53:113–121. doi: 10.2337/diabetes.53.1.113. [DOI] [PubMed] [Google Scholar]
- Fain JN. Release of inflammatory mediators by humuna adipose tissue is enhanced in obesity and primarily by the nonfat cells: a review. Mediators Inflamm. 2010;2010:513948. doi: 10.1155/2010/513948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal R, Faizy AF, Siddiqui SS, Singhai M. Evaluation of TNF-α and IL-6 levels in obese and Non-obese diabetics: pre- and postinsulin effects. North Am J Med Sci. 2012;4:180–184. doi: 10.4103/1947-2714.94944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Middelberg-Bisping K, Drewes C, Schatz H. Elevated plasma levels of transforming growth factor-beta 1 in NIDDM. Diabetes Care. 1996;19:1113–1117. doi: 10.2337/diacare.19.10.1113. [DOI] [PubMed] [Google Scholar]
- Straczkowski M, Kowalska I, Nikolajuk A, Krukowska A, Gorska M. Plasma interleukin-10 concentration is positively related to insulin sensitivity in young healthy individuals. Diabetes Care. 2005;28:2036–2037. doi: 10.2337/diacare.28.8.2036. [DOI] [PubMed] [Google Scholar]
- Montecucco F, Mach F. Does non-alcoholic fatty liver disease (NAFLD) increase cardiovascular risk? Endocr Metab Immune Disord Drug Targets. 2008;8:301–307. doi: 10.2174/187153008786848268. [DOI] [PubMed] [Google Scholar]
- Lesmana CR, Hasan I, Budihusodo U, Gani RA, Krisnuhoni E, Akbar N, Lesmana LA. Diagnostic value of a group of biochemical markers of liver fibrosis in patients with non-alcoholic steatohepatitis. J Dig Dis. 2009;10:201–206. doi: 10.1111/j.1751-2980.2009.00386.x. [DOI] [PubMed] [Google Scholar]
- Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. 2010;22:347–352. doi: 10.1093/intimm/dxq030. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Maeda S, Yoshida H, Tateishi R, Masuzaki R, Ohki T, Hayakawa Y, Kinoshita H, Yamakado M, Kato N, Shiina S, Omata M. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: an analysis based on gender differences. Int J Cancer. 2009;125:2264–2269. doi: 10.1002/ijc.24720. [DOI] [PubMed] [Google Scholar]
- Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES, Ng EK, Chan FK, Sung JJ, Chan HL. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int J Cancer. 2009;124:2766–2770. doi: 10.1002/ijc.24281. [DOI] [PubMed] [Google Scholar]
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Österreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol. 2012;18:727–735. doi: 10.3748/wjg.v18.i8.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38:413–419. doi: 10.1053/jhep.2003.50316. [DOI] [PubMed] [Google Scholar]
- Abdel-Hamid NM, Nazmy MH, Abdel-Ghany MI, Nazmy WH. Cytokines as important playmakers of experimental hepatocarcinogenesis confounded by diabetes. Ann Hepatol. 2012;11:118–127. [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- Sheweita SA, Tilmisany AK. Cancer and phase II drug-metabolizing enzymes. Curr Drug Metab. 2003;4:45–58. doi: 10.2174/1389200033336919. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F, Kim WH, Tian Z, Jaruga B, Ishac E, Shen X, Gao B. Elevated interleukin-6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: involvement of induction of Bcl-2 and Bcl-x (L) proteins. Oncogene. 2002;21:32–43. doi: 10.1038/sj.onc.1205016. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, Dey S, Sung B. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59–76. doi: 10.1111/j.1749-6632.2009.04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, Sun Z. Interleukin-6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology. 2010;52:2137–2147. doi: 10.1002/hep.23909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link? Biochem Pharmacol. 2006;72:1605–1621. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Jiang R, Xia Y, Li J, Deng L, Zhao L, Shi J, Wang X, Sun B. High expression levels of IKKalpha and IKKbeta are necessary for the malignant properties of liver cancer. Int J Cancer. 2010;126:1263–1274. doi: 10.1002/ijc.24854. [DOI] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullaert A, van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor-kappaB: effects on liver homeostasis and beyond. Endocr Rev. 2007;28:365–386. doi: 10.1210/er.2006-0031. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Szlosarek PW, Balkwill FR. Tumour necrosis factor alpha: a potential target for the therapy of solid tumours. Lancet Oncol. 2003;4:565–573. doi: 10.1016/S1470-2045(03)01196-3. [DOI] [PubMed] [Google Scholar]
- Dandona P, Weinstock R, Thusu K, Abdel-Rahman E, Aljada A, Wadden T. Tumor necrosis factor-alpha in sera of obese patients: fall with weight loss. J Clin Endocrinol Metab. 1998;83:2907–2910. doi: 10.1210/jcem.83.8.5026. [DOI] [PubMed] [Google Scholar]
- Muto Y, Sato S, Watanabe A, Moriwaki H, Suzuki K, Kato A, Kato M, Nakamura T, Higuchi K, Nishiguchi S, Kumada H, Ohashi Y. Long-Term Survival Study (LOTUS) Group. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long-term oral supplementation with branched-chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol Res. 2006;35:204–214. doi: 10.1016/j.hepres.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Dinh W, Füth R, Nickl W, Krahn T, Ellinghaus P, Scheffold T, Bansemir L, Bufe A, Barroso MC, Lankisch M. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol. 2009;8:58. doi: 10.1186/1475-2840-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, Peck-Radosavljevic M, Leffert HL, Karin M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2(Suppl 1):S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- Herceg Z. Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis. 2007;22:91–103. doi: 10.1093/mutage/gel068. [DOI] [PubMed] [Google Scholar]
- Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–48. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Wolffe AP, Guschin D. Review: chromatin structural features and targets that regulate transcription. J Struct Biol. 2000;129:102–122. doi: 10.1006/jsbi.2000.4217. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity. 1998;9:765–775. doi: 10.1016/S1074-7613(00)80642-1. [DOI] [PubMed] [Google Scholar]
- Rao A, Avni O. Molecular aspects of T-cell differentiation. Br Med Bull. 2000;56:969–984. doi: 10.1258/0007142001903634. [DOI] [PubMed] [Google Scholar]
- Pogribny IP. Epigenetic events in tumorigenesis: putting the pieces together. Exp Oncol. 2010;32:132–136. [PubMed] [Google Scholar]
- Herath NI, Leggett BA, MacDonald GA. Review of genetic and epigenetic alterations in hepatocarcinogenesis. J Gastroenterol Hepatol. 2006;21:15–21. doi: 10.1111/j.1440-1746.2005.04043.x. [DOI] [PubMed] [Google Scholar]
- Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA, Schroeder I, Factor VM, Thorgeirsson SS. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007;117:2713–2722. doi: 10.1172/JCI31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu Y-Z, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro¨nn T, Poulsen P, Hansson O, Holmkvist J, Almgren P, Nilsson P, Tuomi T, Isomaa B, Groop L, Vaag A, Ling C. Age influences DNA methylation and gene expression of COX7A1 in human skeletal muscle. Diabetologia. 2008;51:1159–1168. doi: 10.1007/s00125-008-1018-8. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Ling C, Poulsen P, Simonsson S, Ro¨nn T, Holmkvist J, Almgren P, Hagert P, Nilsson E, Mabey AG, Nilsson P, Vaag A, Groop L. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J Clin Invest. 2007;117:3427–3435. doi: 10.1172/JCI30938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro JF, Triester S, Patel VK, Tapscott EB, Frazier NL, Dohm GL. Liver glucokinase: decreased activity in patients with type II diabetes. Horm Metab Res. 1995;27:19–22. doi: 10.1055/s-2007-979899. [DOI] [PubMed] [Google Scholar]
- Volkmar M, Dedeurwaerder S, Cunha DA, Ndlovu MN, Defrance M, Deplus R, Calonne E, Volkmar U, Igoillo-Esteve M, Naamane N, Del Guerra S, Masini M, Bugliani M, Marchetti P, Cnop M, Eizirik DL, Fuks F. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012;31:1405–1426. doi: 10.1038/emboj.2011.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Jothi R, Cuddapah S, Cui K, Roh TY, Schones DE, Zhao K. Chromatin poises miRNA- and protein-coding genes for expression. Genome Res. 2009;19:1742–1751. doi: 10.1101/gr.090951.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneghan HM, Miller N, Kerin MJ. Role of microRNAs in obesity and the metabolic syndrome. Obes Rev. 2010;11:354–361. doi: 10.1111/j.1467-789X.2009.00659.x. [DOI] [PubMed] [Google Scholar]
- Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- Poy MN, Spranger M, Stoffel M. microRNAs and the regulation of glucose and lipid metabolism. Diabetes Obes Metab. 2007;9:67–73. doi: 10.1111/j.1463-1326.2007.00775.x. [DOI] [PubMed] [Google Scholar]
- Papanikolaou V, Iliopoulos D, Dimou I, Dubos S, Tsougos I, Theodorou K, Kitsiou-Tzeli S, Tsezou A. The involvement of HER2 and p53 status in the regulation of telomerase in irradiated breast cancer cells. Int J Oncol. 2009;35:1141–1149. doi: 10.3892/ijo_00000430. [DOI] [PubMed] [Google Scholar]
- Hamilton JP. Epigenetic mechanisms involved in the pathogenesis of hepatobiliary malignancies. Epigenomics. 2010;2:233–243. doi: 10.2217/epi.10.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Wu G, Bu F, Lu B, Liang A, Cao L, Tong X, Lu X, Wu M, Guo Y. Epigenetic silence of ankyrin-repeat-containing, SH3-domain-containing, and proline-rich-region- containing protein 1 (ASPP1) and ASPP2 genes promotes tumor growth in hepatitis B virus-positive hepatocellular carcinoma. Hepatology. 2010;51:142–153. doi: 10.1002/hep.23247. [DOI] [PubMed] [Google Scholar]
- Zhang C, Li H, Wang Y, Liu W, Zhang Q, Zhang T, Zhang X, Han B, Zhou G. Epigenetic inactivation of the tumor suppressor gene RIZ1 in hepatocellular carcinoma involves both DNA methylation and histone modifications. J Hepatol. 2010;53:889–895. doi: 10.1016/j.jhep.2010.05.012. [DOI] [PubMed] [Google Scholar]
- Martin M, Herceg Z. From hepatitis to hepatocellular carcinoma: a proposed model for cross-talk between inflammation and epigenetic mechanisms. Genome Med. 2012;4:8. doi: 10.1186/gm307. [DOI] [PMC free article] [PubMed] [Google Scholar]