

Abstract
Introduction
Myeloproliferative neoplasms (MPNs) are a group of stem cell diseases, including polycythemia vera, essential thrombocythemia and primary myelofibrosis. Currently, there is no curative therapy for these diseases other than bone marrow transplant; therefore there is an apparent need for palliative treatment. MPNs are frequently associated with activating mutations in Janus Kinase 2 (JAK2); small molecule drugs targeting this molecule have entered clinical trials.
Areas covered
In this review novel JAK2 inhibitors will be discussed and alternative approaches to inhibiting their transforming potential will be highlighted.
Expert opinion
Current clinical approaches do not only aim at blocking JAK2 activity, but also at reducing its stability and expression. Inhibition of heat shock protein 90 (HSP90) and deacetylase inhibitors (DACi) have the potential to significantly enhance the efficacy of JAK2 inhibitors. Preliminary results from clinical trials indicate the feasibility and efficacy of JAK2 targeted approaches. However, JAK2 inhibitor treatment is limited by dose-dependent toxicity and combination treatment might be required. The discovery of JAK2 mutations that cause secondary resistance in vitro would further highlight the need for the development of next generation JAK2 inhibitors and novel synergistic approaches.
Keywords: Myeloproliferative neoplasms, JAK2, signaling, metabolism, targeted therapy
1. Introduction
In Philadelphia chromosome negative (Ph-) myeloproliferative neoplasms (MPNs), the non-receptor tyrosine kinase Janus kinase 2 (JAK2), is frequently found to contain an activating V617F mutation [1-5]. This mutation is present at a high frequency in patients with polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (MF) as well as infrequently in other MPNs, myelodysplastic syndromes and acute myeloid leukemia (see for review [6]). Even though the V617F mutation can occur spontaneously, there appears to be a three- to fourfold greater risk for carriers with the so called ‘46/1’ haplotype, encoded by a region in the 3′ of JAK2 [7-9]. Additional genetic abnormalities have been found in the JAK2 gene (exon12-15) in a variety of hematologic malignancies but with lower frequencies [10]. The carboxy-terminal kinase domain in JAK2 can also be activated as part of an oncogenic fusion, involving breakpoints in the JH2-JH5 domain. For example, a t(9;12)(p24;p13) or variant translocations in patients with a chronic myeloproliferative disease or acute lymphoblastic leukemia fuses the ETV6 to the JAK2 gene [11,12]. There are additional rare translocations that involve JAK2 and lead to the formation of a constitutive activation of the kinase (see for review [10]) (Figure 1). JAK2 is also directly involved in the transformation by oncogenic receptors. In MPNs, the thrombopoietin (TPO) receptor MPL, which requires JAK2 for signaling, is an infrequent target of activating mutation, in particular at amino acid W515 [13,14]. Also, in acute lymphoblastic leukemias (ALL), activating CRFL2 (cytokine receptor-like factor 2) mutations and rearrangements and activating JAK2 mutations are frequently found [15], suggesting that this pathway is important for the disease process. Thus, JAK2 targeted approaches may not only be beneficial for the treatment of MPNs, but may also help in the treatment of other malignancies with a constitutively active JAK2 signaling pathway.
Figure 1. Schematic structure of JAK2.
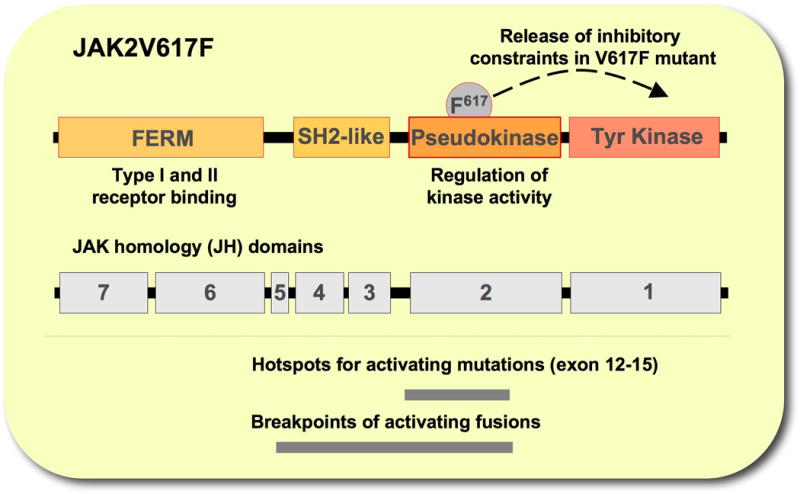
Represented are domains within JAK2, including the FERM (4.1 protein, ezrin, radixin, moesin) domain, SH2 (Scr homology 2) like domain, the pseudokinase domain and the kinase domain (top), the JAK homology (JH) domains (middle) as well as regions that include hotspots for activating mutations and breakpoints for activating fusions (bottom).
2. JAK2 - structure and function
JAK2 belongs to the family of related non-receptor Janus tyrosine kinases, including JAK1-3 and TYK2 [16]. There is a considerable degree of homology between these kinases that can be defined to specific JAK homology (JH) domains. The carboxy terminus contains the kinase domain (JH1) and the related pseudokinase domain (JH2) (Figure 1). The latter is structurally similar to the JH1 domain except for a DFG motif in the activation loop, which results in lack of kinase activity [17]. This distinct architecture of JAKs gave them their name, according to the two-faced Roman god Janus. The JH2 domain plays an important role in regulatory functions of Janus kinases [18,19]. This domain is thought to negatively regulate the kinase activity through interaction with the JH1 domain and the V617F mutation in the JH2 domain found in MPNs has been suggested to overcome these inhibitory constraints. [2,3]. A Src homology 2 (SH2)-like domain (JH3-4) is adjacent to the pseudokinase domain and the amino-terminal region (JH6-7) contains the FERM (4.1 protein, ezrin, radixin, moesin) domain [16]. This domain together with the SH2-like domain form the amino-terminus of JAK2 that is essential for upregulation of surface expression of cytokine receptors such as EpoR [20]. A proline rich eight amino acid motif (box1) in the cytoplasmic portion of membrane-associated receptors typically recruits the FERM domain [21]. Disruption of this interaction, such as in the case of a Y114A substitution in the FERM domain, results in loss of JAK2 activation, independent of the JAK2V617F activating mutation [22,23]. Thus, an intact FERM domain is necessary for phosphorylation and activation of JAK2 signaling pathway [23]. This domain may also promote cell surface localization of the thrombopoietin receptor and thus upregulation of the downstream signaling of JAK2 [22]. However, erythroid progenitors in PV show hypersensitivity to erythropoietin or factor independent growth [24,25], suggesting that in vivo JAK2V617F may, at least in part, require ligand stimulation for signaling.
3. Regulation of cellular functions by JAK2 signaling pathways
JAK2 acts as a kinase for cytokine receptors that lack an intracellular tyrosine kinase domain. Mice with JAK2 gene disruption are embryonically lethal, due in part to ineffective erythropoiesis. JAK2 is also indispensable for functions of various cytokines, such as interleukin 3, thrombopoietin and erythropoietin [26,27]. JAK mediated tyrosine phosphorylation of receptors forms docking sites for intracellular effectors, such as STATs (signal transducers and activators of transcription). STAT proteins are phosphorylated at the receptor and translocate in its active form to the nucleus to initiate transcription of their target genes [28]. In JAK2V617F expressing cells, STAT5 is constitutively phosphorylated [2]. Constitutive activation of STAT5 was shown to be sufficient to induce growth factor independent cell proliferation [29]. Also, disruption of the STAT5 genes demonstrated a role in anti-apoptotic signaling, immature hematopoiesis and fetal erythropoiesis in vivo [30,31]. Signaling and activation of STAT5 by JAK2V617F is crucial for JAK2V617F-dependent growth factor independent proliferation and transformation in vivo and required the functional expression of a cytokine receptor such as the erythropoietin receptor [23,32,33]. Recently, a high throughput chemical screen for small molecule inhibitors identified pimozide as a potential STAT5 targeting drug [34]. Treatment of CML cell lines with pimozide resulted in decreased phosphorylation and expression of STAT5 and was associated with increased apoptosis and cell-cycle arrest. However, the exact mode of action for pimozide is not clear. Furthermore, active STAT5 can induce the expression of the serine/threonine kinases PIM1 and PIM2, which are required for optimal growth in JAK2V617F transformed cells [23]. Early clinical trials with the PIM inhibitor SGI-1776 (SuperGen) [35] for the treatment of refractory leukemias, lymphomas and prostate cancer were terminated due to toxicity and there is a need for safer compounds. In addition to cellular targets, JAK2 was also shown to phosphorylate nuclear histone H3 on tyrosine 41 (H3Y41) in the nucleus of hematopoietic cells [36]. Phosphorylation of H3Y41 leads to release of transcriptional repressor HP1α from the chromatin and concomitant expression of genes repressed by HP1α. The mechanism of JAK2 activation in the nucleus still needs to be determined [37]. Surprisingly, only JAK2 protein of MPN patients that carry the V617F mutation accumulates in the nucleus of CD34+ progenitor cells and not JAK2 from patients that carry the wildtype protein [38]. The functional significance of this difference is unclear. JAK2 has additional downstream targets such as the erythropoietin receptor [39], VAV [40], p27KIP [41], PAK1 [42], SH2-Bβ [43], PRMT5 [44] and other proteins may significantly contribute to JAK2 dependent malignancies and are potential candidates for targeted approaches (Figure 2).
Figure 2. Cytoplasmic and nuclear targets of JAK2.
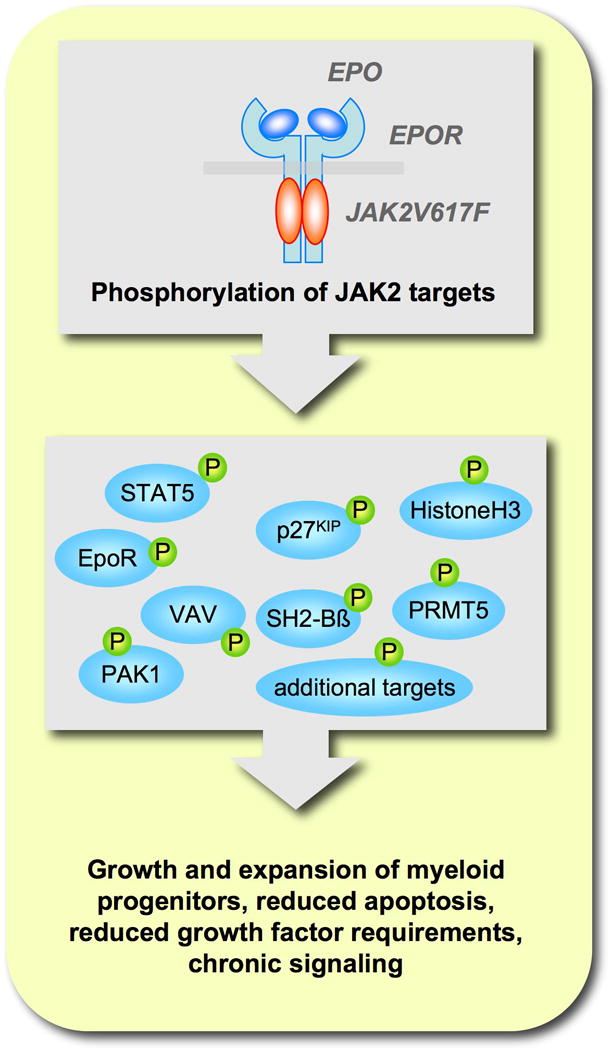
Activation of the JAK2/receptor complex leads to direct phosphorylation and functional regulation of cytoplamsic and nuclear proteins.
Signaling through the JAK2V617F gain-of-function mutation is not limited to a set of specific JAK2 target proteins. The expression of JAK2V617F is thought to confer hypersensitivity to growth factors as well as factor independent growth, thus aberrantly activating growth and survival pathways that can normally be stimulated by signaling through cytokine and growth factor receptors. In addition to the STAT pathway, the PI3K (phosphatidylinositol -3′-kinase) and MAPK (mitogen activated protein kinase) pathways are also prominent targets of activation in JAK2V617F expressing cells [45,46]. The importance of the PI3K downstream target mTOR (mammalian target of rapamycin) has recently been demonstrated in phase1/2 clinical trials of patients with MF [47]. mTOR activity within the multi-functional mTORC1 or mTORC2 complex was inhibited by everolimus. mTOR is thought to function as a nutrient and energy sensor as well as a regulator of cell growth, in part through phosphorylation of the translation initiation factor 4E-BP1 and the ribosomal p70S6 kinase [48]. Everolimus was overall well tolerated and in about 50% of the patients a reduction of splenomegaly was observed with the majority of the patients experiencing complete resolution of their clinical symptoms [47]. Interestingly, the response was independent of the presence of JAK2V617F. Larger cohorts will be needed to further evaluate the efficacy of this approach but these initial results would indicate that targeting mTOR with everolimus may be a viable option for the treatment of MF and related diseases. At present, it is not known whether concomitant inhibition of JAK2 and mTOR is beneficial for this patient group. The importance of the MAPK pathway in JAK2V617F signaling has not been worked out in great detail. However, in vitro experiments with CD34+ cells from PV patients suggest that inhibition of p38MAPK with SB203580 does not suppress growth of JAK2V617F-positive hematopoietic progenitor cells [49]. Interestingly, in these experiments interferon-α targeted this cell population and the growth inhibition could be overwritten by SB203580 treatment.
4. Targeting JAK2 in myeloproliferative neoplasms
Activating JAK2 mutations in MPNs have been suggested to be an ideal target for inhibition due to their prevalence and apparent role in the disease phenotype. Initial clinical trials have been focusing specifically on MF due to a limited number of efficient targeted approaches and immediate therapeutic need. Clinical trials are in the process of being expanded to all MPNs and include PV and ET as well (see also http://cliniclatrials.gov). The current treatment options for MF patients depend on individual circumstances and can include drug treatment (such as hydroxyurea, danazol, thalidomide, corticosteroids or others), splenectomy, radiotherapy (locally for extramedulary hematopoiesis) or stem cell transplant. Aggressive treatment options are frequently a last resort and may be accompanied by significant side effects or even death [50]. Even though MPNs can often be managed for years with little complications, the onset of pain, splenomegaly and anemia are some of the main intermediate concerns that require additional palliative care. There are several small molecule drugs currently in clinical trials that attempt to target JAK2 function at different levels (Figure 3).
Figure 3. Schematic diagram of strategies targeting the JAK2 pathway in MPNs.
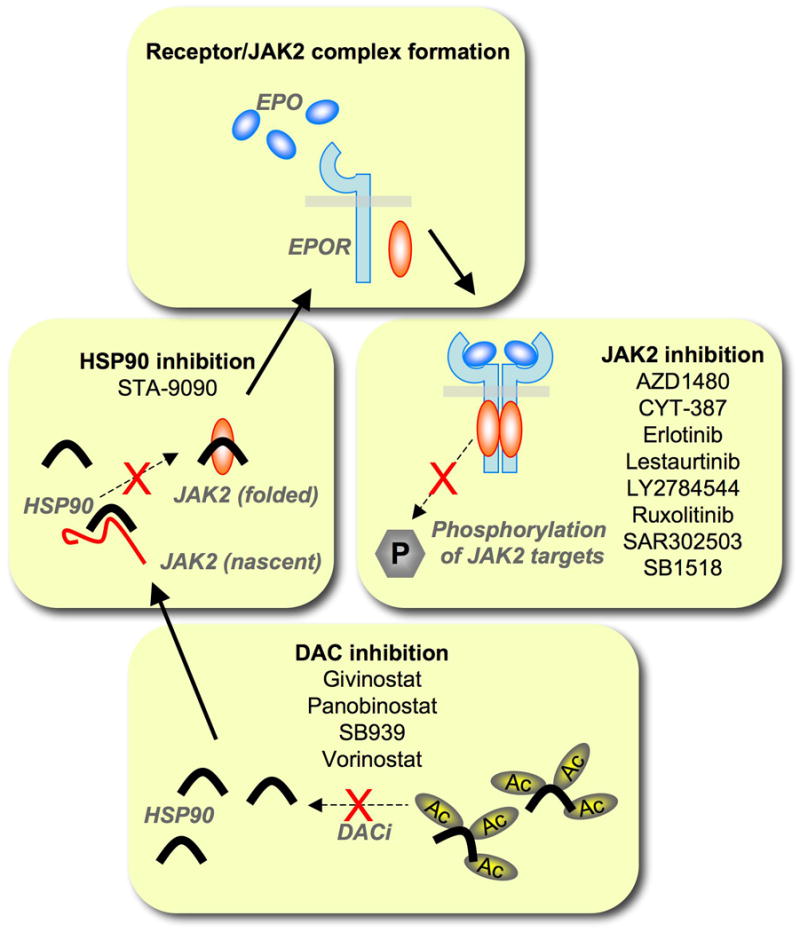
Functional expression of JAK2 can be blocked at several levels. Deacetylase inhibitors (DACi) increase the acetylation and thus inhibit the chaperone function of HSP90 (heat shock protein 90). Inhibition of HSP90 leads to a reduction in levels of matured JAK2. Specific tyrosine kinase inhibitors prevent activation of downstream signaling cascades. Indicated are drugs that are currently in clinical trials for the treatment of MPNs. (see also http://cliniclatrials.gov)
4.1. JAK2 small molecule kinase inhibitors
The tremendous success of small molecule tyrosine kinase inhibitors targeting BCR-ABL in chronic myeloid leukemia (CML) has led to the development of related drugs that specifically inhibit kinases required for transformation in various malignancies. In general, these drugs prevent interaction of ATP with its binding pocket in the catalytic site of the kinase. High affinity interactions with amino acids in this hydrophobic groove determine the specificity for each drug. Currently, there are seven JAK2 kinase inhibitors in clinical trials (Table 1, Figure 4). The JAK2 inhibitor ruxolitinib was recently approved by the FDA (U.S. Food and Drug Administration) for the treatment of intermediate or high-risk myelofibrosis. Even though these inhibitors were developed to target JAK2, they have been shown to have activity against some other kinases as well. Notably ruxolitinib (INCB018424), one of the first drugs tested for its efficacy in the treatment of MF, also inhibits the related JAK1 [51,52]. Similarly, the JAK2 inhibitors SAR302503 (TG101348), AZD1480 and CYT-387 display activity against JAK1 as well [53-56]. Surprisingly, the effect on JAK2V617F allele burden with ruxolitinib did not match the clinical outcome and it had been speculated that its clinical efficacy might be in part due to inhibition of JAK1-dependent cytokine signaling. Ruxolitinib demonstrated significant and dose-dependent efficacy in MF with dose-limiting thrombocytopenia [51]. There may be additional specific toxic effects that are caused by inhibiting JAK2 or secondary targets, such as dose-limiting hyperamylasaemia with SAR302503 or other effects with related drugs [57,58]. The JAK2 inhibitor lestaurtinib (CEP-701) belongs to a different group of compounds and is structurally related to the pan protein kinase inhibitor staurosporine with additional activity against FLT3, RET and Trk family members [59-62]. The JAK2 inhibitor SB1518 is a pyrimidine-based macrocycle with additional activity against FLT3 [63], whereas the EGFR inhibitor erlotinib is also effective against JAK2 [64], but little is known about the JAK2 inhibitor LY2784544. Despite these additional activities, all inhibitors tested for MF, including ruxolitinib, lestaurtinib, CYT-387, SB1518 and SAR302503, lead to reduction in splenomegaly in some patients, further validating JAK2 as an important therapeutic target in MF (see for detailed review of clinical trials [58]). The toxicity profile of each individual JAK2 inhibitor may be an important determinant for its success and it will be interesting to see whether this correlates with the known off-target effects.
Table 1. Inhibitors of JAK2 under clinical evaluation. (see also http://clinicaltrials.gov).
| Compound | Company | Additional Targets | Phase of study | |||
|---|---|---|---|---|---|---|
| I | II | III | IV | |||
| AZD1480 | Astra-Zeneca | JAK1, JAK3 | ||||
| CYT387 | YM Biosciences | JAK1, TYK2, JNK1, CDK2 | ||||
| Erlotinib | OsiPharma | EGFR | ||||
| Lestaurtinib (CEP-701) | Cephalon | FLT3, TrkA/B/C | currently suspended | |||
| LY2784544 | Elli-Lilly | |||||
| Ruxolitinib (INCB018424) | Novartis (Incyte) | JAK1 | ** | |||
| SAR302503 (TG101348) | Sanofi-Aventis (TargeGen) | FLT3, RET | ||||
| SB1518 | S*Bio | FLT3 | ||||
| XL019 | Exelixis | early termination | ||||
Ruxolitinib was approved by the FDA (U.S. Food and Drug Administration) for the treatment of intermediate or high-risk myelofibrosis in November 2011.
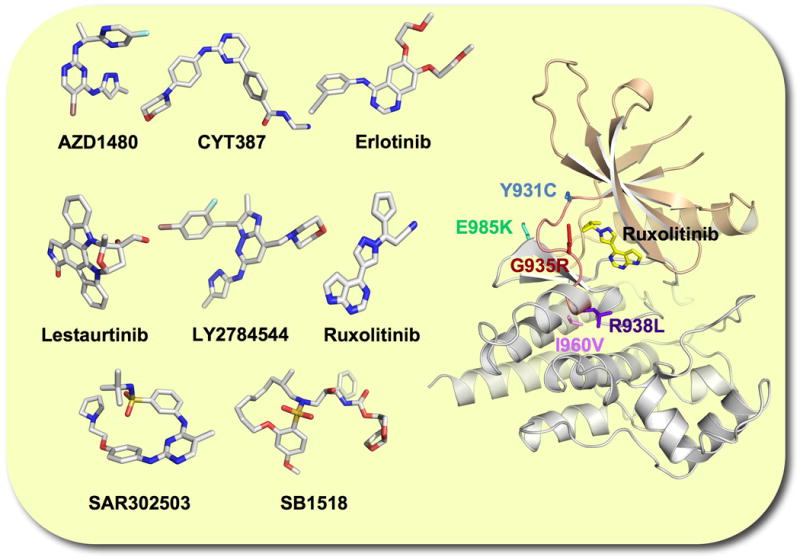
Figure 4. JAK2 tyrosine kinase inhibitors currently in clinical trials for the treatment of MPNs and tertiary structure of the JAK2 kinase domain containing drug resistant mutations.
4.2. HSP90 (heat shock protein 90) inhibitors
Heat shock proteins (HSPs) are family of related proteins that are involved in the maintenance of proper folding and stability of its client proteins. Their expression can be significantly increased not only upon heat exposure but also in response to a variety of other stress conditions. HSP90 is a member of this family with ATP dependent chaperone activity, towards several cellular proteins [65]. In myeloid malignancies, BCR-ABL and active forms of FLT3 or JAK2 are among prominent targets that are dependent on HSP90 for their functional conformation [66-68]. Thus, HSP90 plays a crucial role in the proper folding and activation of these oncoproteins and their downstream signaling pathways. HSP90 may not only act on the native oncoprotein but also mutated forms that are resistant to targeted approaches, such as BCR-ABL mutations in CML. In MPNs, JAK2 kinase inhibitors have only recently been introduced into clinical trials and drug resistant point mutations are not expected to emerge at this stage but may be of significant relevance in the near future. Nevertheless, recent approaches suggest that HSP90 inhibitors may show efficacy against active JAK2. Marubayashi et al. have shown that the novel HSP90 inhibitor PU-H71, is effective in vitro as well as in vivo using preclinical models of PV and ET [69]. Direct interaction between HSP90/PU-H71 and JAK2 protein in isogenic and leukemic cell lines that led to reduced growth was demonstrated. Mouse bone marrow transplant models and MPN primary samples confirmed the HSP90 inhibition mediated JAK2 degradation and further downregulation of its downstream signaling pathway. Targeting HSP90 may also overcome drug resistance. Fiskus et al. found that cells resistant to the JAK2 inhibitor TG101209 were sensitive towards the HSP90 inhibitor AUY922. Further, AUY922 in combination with TG101209 exerted synergistic in vitro activity in human CD34+ MF-MPN hematopoietic progenitors [68]. AUY922 depleted JAK2V617F protein expression in a dose dependent manner, thus effectively further inhibiting its downstream signaling and reducing the phosphorylation of STAT5, AKT and ERK1/2 in vitro. As would be predicted, this effect was enhanced by co-treatment with TG101209. Combination of both drugs in TG101209 resistant cells expressing JAK2V617F led to higher apoptosis as compared to each drug alone. Thus, combination treatment may enhance the efficacy of JAK2 targeted approaches in MPN patients.
4.3. Deacetylase inhibitors (DACi)
Histone deacetylase (HDACs) inhibitors have shown efficacy in preclinical models of MPNs [70,71]. Increased HDAC function was found in MF, even though no correlation was observed between JAK2 mutational status and its activity [72]. It is not known whether inhibition of histone deacetylation contributes to the efficacy of these drugs or whether inhibition of deacetylation of non-histone proteins by HDACs is more prominent, in particular HSP90 [73]. Currently, there are four deacetylase inhibitors (vorinostat, panobinostat, givinostat and SB939) (Figure 3) in clinical trials for the treatment of MPNs. In a limited study of patients with JAK2V617F positive MF, vorinostat (suberoylanilide hydroxamic acid, SAHA, MK-0683; Merck) showed clinical efficacy [74]. This drug binds to the Zinc ion in the catalytic domain to inhibit HDAC activity [75]. It recently gained FDA approval for the treatment of T cell lymphomas [76,77] and is currently in phase II clinical trials for the treatment of MPNs. Givinostat (ITF 2357; Italfarmaco, Milan, Italy), is currently in phase II clinical studies for the treatment of PV and permitted in the European Union for its treatment as an orphan drug. A phase IIA study in MPN patients with JAK2V617F showed hematological response in most PV patients [78]. It has also been shown to selectively target the cells from patients expressing JAK2V617F, but not wildtype JAK2. The phosphorylation levels of JAK2V617F, STAT5 and STAT3 were reduced in response to givinostat treatment [70]. Panobinostat (Novartis) was developed as a pan-deacetylase inhibitor and is currently in phase II trial for the treatment of MPNs either as a single agent or in combination with ruxolitinib in phase I study. Panobinostat treatment resulted in disruption of signaling downstream of JAK2V617F as well as HSP90 mediated folding of JAK2V617F in vitro. The efficacy of panobinostat was synergistically increased in CD34+ MPN cells when treated in combination with TG101209 [79]. Another deacetylase inhibitor, SB939 (S*BIO, Singapore) is currently in phase II clinical trials for the treatment of MPNs. This inhibitor has been shown to specifically accumulate to tumor tissue compared to healthy tissues in a colorectal cancer mouse model [80].
5. JAK2 inhibitor resistance
With the introduction of imatinib in CML and related tyrosine kinase inhibitors in other cancers, drug resistant mutations that interfere with the binding of the inhibitors rapidly emerged. For example, the T315I mutation in ABL shows imatinib resistance by interfering with drug binding [81,82]. Therefore, it would be of great clinical value to predict potential JAK2 inhibitor resistant mutations in MPNs. Recently, in a screen of randomly mutagenized JAK2V617F libraries, five non-synonymous point mutations in its JH1 domain were identified that led to ruxolitinib resistance in vitro [83] (Figure 4). These Y931C, I960V, G935R, E985K and R938L point mutations were either predicted to interact with ruxolitinib or were in proximity to the binding pocket. In particular mutation at tyrosine 931 would offer drug resistance as a result of loss of π–π interaction between the tyrosine ring and the inhibitor structure. JAK2V617F containing mutations also showed cross-resistance to the structurally unrelated JAK2 inhibitors CYT-387, SAR302503, AZD1480 and lestaurtinib [83], indicating an overlap in the interaction of these drugs with the ATP binding pocket. Similarly, ruxolitinib resistant mutations were identified in vitro in the JAK1 kinase domain. Homologous to the JAK1 F958C mutation identified in this screen, a Y931C mutation was introduced into JAK2 and not only led to cytokine independent growth of cells expressing JAK2 but also caused ruxolitinib resistance independent of the JAK2 mutation status [84]. Surprisingly, this screen did not identify a homologous ‘gate-keeper’ mutation that has been linked to secondary drug resistance in active forms of ABL, EGFR and KIT [82,85,86]. Introduction of a M929I mutation at the gatekeeper site in JAK2V617F showed only four-fold increase in EC50 for ruxolitinib compared to native JAK2V617F but not to CYT-387, SAR302503, AZD1480 and lestaurtinib [83]. It will be important to carefully look for potential JAK2 inhibitor resistance in MPN patients that have undergone prolonged treatment.
6. Metabolic reprogramming as an opportunity for targeted approaches
One of the hallmarks of malignant cells is dysregulated glucose metabolism, compared to their healthy counterparts. Transformed cells tend to utilize glycolysis and fermentation as a major energy generating process, even in the presence of oxygen instead of more efficient oxidative phosphorylation. This so-called ‘Warburg effect’ [87], named after its discoverer, is a result of increased expression, localization and/or activity of the genes involved in glucose transport and metabolism. Cancer cells exploit this altered metabolism to provide precursors for biomolecules needed for their uncontrolled proliferation and to evade apoptosis [88]. Even though there may be significant overlap between mechanisms that induce the Warburg effect in different types of cancers, little has been done to specifically investigate this effect in malignancies driven by active JAK2. For instance, it has been shown that active JAK2V617F was sufficient for increased cell surface expression of glucose transporter Glut1, concomitant increase in glucose uptake and increased lactate production [89]. In addition, JAK2 expressing cells also showed elevated expression of phosphofructokinase/fructose-bisphosphatase 3 (PFKFB3), a rate-limiting enzyme that affects phosphofructokinase-1 (PFK-1) activity in the glycolytic pathway. The metabolic end product of PFKFB3, fructose-2,6-bisphosphate is an allosteric activator of PFK1, that allows continuous activity, even in the presence of ATP, which normally suppresses PFK1 activity. The role of PFKFB3 in JAK2V617F mediated transformation was further confirmed in vivo using a murine xenograft model with knockdown of PFKFB3 [89]. PFKFB3 could also be targeted with the small molecule inhibitor 3-PO (3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one) [90], resulting in reduced cell growth and reversal of the Warburg effect [89]. Another important aspect of hyperactive metabolism associated with JAK2V617F is an excess production of mitochondrial reactive oxygen species (ROS), such as superoxide, hydrogen peroxide and hydroxyl radicals. ROS are a by-product of the electron transport chain and transiently elevated levels are required for some normal cellular functions such as cell growth, migration and signaling and may contribute to genomic instability [91]. In JAK2V617F expressing cells ROS are constitutively elevated [92] (Figure 5). In leukemia stem cells, the naturally occurring compound parthenolide further increased ROS levels and triggered apoptosis [93]. Additional strategies to target ROS are currently under investigation [94]. A minor source for superoxide production in metabolically active myeloid cells transformed by JAK2V617F and other oncogenic tyrosine kinases are NADPH oxidases [95]. Interestingly, knockdown of the NADPH oxidases resulted in decreased cell growth and migration [95], consistent with a localized production of superoxide. Targeting NADPH production or development of specific NADPH oxidase inhibitors may provide synergy to JAK2 inhibition.
Figure 5. The JAK2V617F/STAT5 pathway is a key regulator of growth and metabolism.
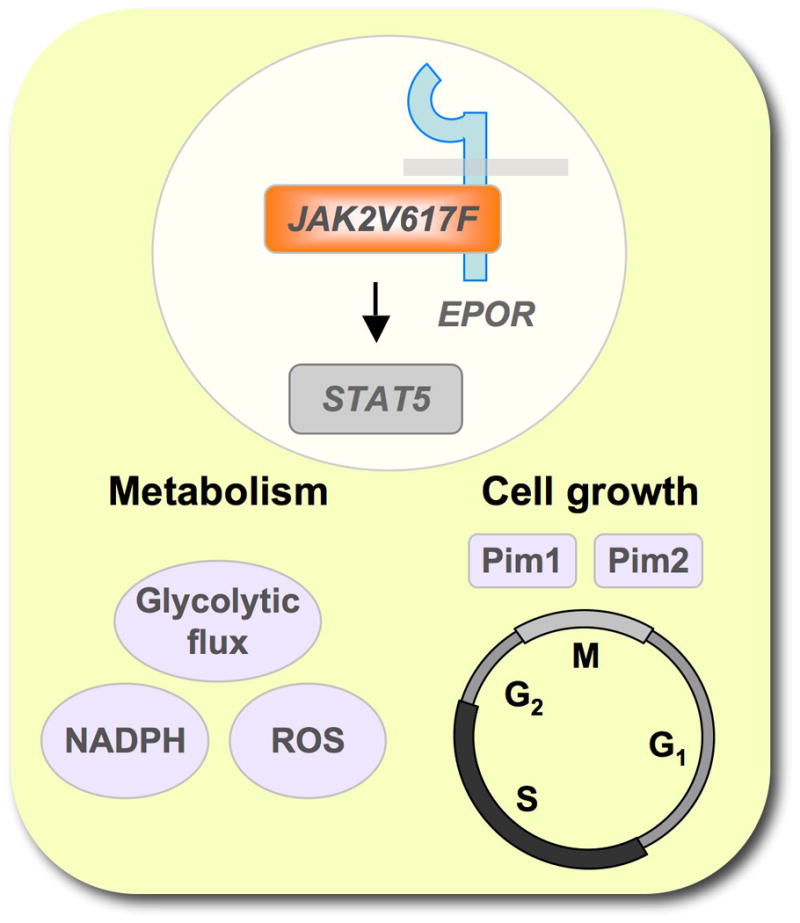
Signaling through JAK2V617F requires a functional interaction of the kinase with a cytokine receptor, such as the erythropoietin receptor (EPOR). Activation of the JAK2 target STAT5 is sufficient for increased cell growth (in part through Pim1 and Pim2) as well as increased metabolism (indicated by increased glycolytic flux or elevated ROS and NADPH levels).
7. Conclusion and expert opinion
The discovery of activating JAK2 mutations in MPNs has underlined the importance of the associated signaling pathway for the disease process. However, the lack of activating JAK2 or receptor mutations in a proportion of MPNs suggests that there may be additional activities that are independent of this pathway. Nevertheless, early results from clinical trials demonstrate efficacy of JAK2 inhibitors, even in MPNs without apparent JAK2 pathway mutations. These findings do not indicate whether the JAK2 signaling pathway is constitutively active in MPN patients, even though this would be an intriguing possibility. It is likely that in these MPNs, JAK activation through cytokine receptor stimulation plays a greater role than previously thought. Initial clinical trials with JAK2 inhibitors demonstrate dose-dependent efficacy that is limited by toxicity. The importance for JAK2 in the hematopoietic compartment may leave a small therapeutic window for treatment, thus causing adverse effects at higher inhibitor concentrations. It can also not be entirely excluded that JAK2 with activating mutation is insufficient to cause addiction to this oncogene. In vivo mouse models suggest that JAK2V617F by itself can initiate a myeloproliferative disease. However, this picture is somewhat more complicated in patients, where JAK2V617F is frequently associated with other mutations that have epigenetic effects. A two-hit model adapted for MPNs transformation (see for review [96]) suggests that in MPNs the activation of signaling molecules (such as JAK2V617F) may not be sufficient for the full transforming potential and that changes in the activity of epigenetic regulators (such as TET2) may be required for the disease process. Also, JAKV617F more efficiently supports late hematopoiesis and not expansion of stem cells, whereas TET2 mutations benefit the progenitor pool. Thus, additional therapeutics for co-treatment may well be required to efficiently inhibit MPNs associated with JAK2V617F and could have differential effects on the disease phenotype. Further, oncogene-targeted therapies in combination with additional signaling-targeted therapies have the potential to increase the efficacy of current approaches and further identification and characterization of signaling pathways that drive MPNs may lead to novel therapeutics. Lesson learned from imatinib and other ABL kinase inhibitors would suggest that even under optimal circumstances, JAK2 inhibition by itself is insufficient to lead to a curative therapy but may be sufficient to stabilize clinical symptoms in a large proportion of the patient pool for a long period of time. Additional characterization of the genetic alterations associated with these diseases and a better understanding of the affected genes may be required to achieve this goal. Whole genome sequencing has brought a new understanding about these changes. Nevertheless, analysis of future results needs to be further refined to obtain useful insights into the evolution of different malignant clones and the mutations that truly drive their expansion. These data will likely identify genes that provide growth advantages and help to prioritize target development. In this context it is also important to note that similar to other kinase inhibitors, it may be necessary to monitor for secondary drug resistance. Preliminary data hint at potential JAK2 inhibitor resistant mutations in the JAK2 kinase domain and this knowledge will help to develop next generation JAK2 inhibitors or drugs that target proteins such as HSP90 and DAC to overcome JAK2 inhibitor resistance. Similar to other malignancies with a complex phenotype, the field will likely move towards more individualized therapy. In particular knowledge about the aforementioned disease driving genes will allow to device more effective combination therapies that inhibit the expansion of malignant cells at multiple levels.
Article highlights.
Myeloproliferative neoplasms (MPNs) are frequently associated with the activating V617F mutation in JAK2. Dysregulation of the JAK2 pathway can also be found in other myeloid as well as non-myeloid malignancies.
Small molecule JAK2 kinase inhibitors are currently in clinical trials for the treatment of MPNs.
Secondary resistance to JAK2 inhibitors may be caused by mutations in the kinase domain. Mutational analysis of this domain should therefore be considered for patients that fail to respond to these drugs.
Inhibition of HSP90 (heat shock protein 90) deacetylation and HSP90 chaperone function may cooperate with JAK2 kinase inhibition.
Activation of the JAK2 pathway leads to increased carbon metabolism. JAK2V617F dependent and independent metabolic reprogramming hints at novel targets for drug development.
Acknowledgments
Declaration of Interest: This work is supported in part by National Institutes of Health grant CA134660-04, M.S.
Bibliography
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1*.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. First description of the JAK2V617F mutation. [DOI] [PubMed] [Google Scholar]
- 2*.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. First description of the JAK2V617F mutation. [DOI] [PubMed] [Google Scholar]
- 3*.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. First description of the JAK2V617F mutation. [DOI] [PubMed] [Google Scholar]
- 4*.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. First description of the JAK2V617F mutation. [DOI] [PubMed] [Google Scholar]
- 5*.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–22792. doi: 10.1074/jbc.C500138200. First description of the JAK2V617F mutation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112:2190–2198. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones AV, Chase A, Silver RT, et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009;41:446–449. doi: 10.1038/ng.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kilpivaara O, Mukherjee S, Schram AM, et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009;41:455–459. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olcaydu D, Harutyunyan A, Jager R, et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009;41:450–454. doi: 10.1038/ng.341. [DOI] [PubMed] [Google Scholar]
- 10.Jatiani SS, Baker SJ, Silverman LR, Reddy EP. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: approaches for targeted therapies. Genes Cancer. 2010;1:979–993. doi: 10.1177/1947601910397187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peeters P, Raynaud SD, Cools J, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90:2535–2540. [PubMed] [Google Scholar]
- 12.Lacronique V, Boureux A, Valle VD, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 13.Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 14.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roll JD, Reuther GW. CRLF2 and JAK2 in B-progenitor acute lymphoblastic leukemia: a novel association in oncogenesis. Cancer Res. 2010;70:7347–7352. doi: 10.1158/0008-5472.CAN-10-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaoka K, Saharinen P, Pesu M, et al. The Janus kinases (Jaks) Genome Biol. 2004;5:253. doi: 10.1186/gb-2004-5-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindauer K, Loerting T, Liedl KR, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 2001;14:27–37. doi: 10.1093/protein/14.1.27. [DOI] [PubMed] [Google Scholar]
- 18.Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277:47954–47963. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- 19.Saharinen P, Vihinen M, Silvennoinen O. Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol Biol Cell. 2003;14:1448–1459. doi: 10.1091/mbc.E02-06-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang LJ, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001;8:1327–1338. doi: 10.1016/s1097-2765(01)00401-4. [DOI] [PubMed] [Google Scholar]
- 21.Tanner JW, Chen W, Young RL, et al. The conserved box 1 motif of cytokine receptors is required for association with JAK kinases. J Biol Chem. 1995;270:6523–6530. doi: 10.1074/jbc.270.12.6523. [DOI] [PubMed] [Google Scholar]
- 22.Royer Y, Staerk J, Costuleanu M, et al. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem. 2005;280:27251–27261. doi: 10.1074/jbc.M501376200. [DOI] [PubMed] [Google Scholar]
- 23.Wernig G, Gonneville JR, Crowley BJ, et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood. 2008;111:3751–3759. doi: 10.1182/blood-2007-07-102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zanjani ED, Lutton JD, Hoffman R, Wasserman LR. Erythroid colony formation by polycythemia vera bone marrow in vitro. Dependence on erythropoietin. J Clin Invest. 1977;59:841–848. doi: 10.1172/JCI108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prchal JF, Axelrad AA. Letter: Bone-marrow responses in polycythemia vera. N Engl J Med. 1974;290:1382. doi: 10.1056/nejm197406132902419. [DOI] [PubMed] [Google Scholar]
- 26.Neubauer H, Cumano A, Muller M, et al. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 27.Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 28.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onishi M, Nosaka T, Misawa K, et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998;18:3871–3879. doi: 10.1128/mcb.18.7.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teglund S, McKay C, Schuetz E, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- 31.Socolovsky M, Fallon AE, Wang S, et al. Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98:181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Levine R, Tong W, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005;102:18962–18967. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Funakoshi-Tago M, Tago K, Abe M, et al. STAT5 activation is critical for the transformation mediated by myeloproliferative disorder-associated JAK2 V617F mutant. J Biol Chem. 2010;285:5296–5307. doi: 10.1074/jbc.M109.040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson EA, Walker SR, Weisberg E, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117:3421–3429. doi: 10.1182/blood-2009-11-255232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen LS, Redkar S, Bearss D, et al. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood. 2009;114:4150–4157. doi: 10.1182/blood-2009-03-212852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson MA, Bannister AJ, Gottgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sattler M, Griffin JD. JAK2 gets histone H3 rolling. Cancer Cell. 2009;16:365–366. doi: 10.1016/j.ccr.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Rinaldi CR, Rinaldi P, Alagia A, et al. Preferential nuclear accumulation of JAK2V617F in CD34+ but not in granulocytic, megakaryocytic, or erythroid cells of patients with Philadelphia-negative myeloproliferative neoplasia. Blood. 2010;116:6023–6026. doi: 10.1182/blood-2010-08-302265. [DOI] [PubMed] [Google Scholar]
- 39.Miura O, Nakamura N, Quelle FW, et al. Erythropoietin induces association of the JAK2 protein tyrosine kinase with the erythropoietin receptor in vivo. Blood. 1994;84:1501–1507. [PubMed] [Google Scholar]
- 40.Shigematsu H, Iwasaki H, Otsuka T, et al. Role of the vav proto-oncogene product (Vav) in erythropoietin-mediated cell proliferation and phosphatidylinositol 3-kinase activity. J Biol Chem. 1997;272:14334–14340. doi: 10.1074/jbc.272.22.14334. [DOI] [PubMed] [Google Scholar]
- 41.Jakel H, Weinl C, Hengst L. Phosphorylation of p27Kip1 by JAK2 directly links cytokine receptor signaling to cell cycle control. Oncogene. 2011;30:3502–3512. doi: 10.1038/onc.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rider L, Shatrova A, Feener EP, et al. JAK2 tyrosine kinase phosphorylates PAK1 and regulates PAK1 activity and functions. J Biol Chem. 2007;282:30985–30996. doi: 10.1074/jbc.M701794200. [DOI] [PubMed] [Google Scholar]
- 43.Rui L, Mathews LS, Hotta K, et al. Identification of SH2-Bbeta as a substrate of the tyrosine kinase JAK2 involved in growth hormone signaling. Mol Cell Biol. 1997;17:6633–6644. doi: 10.1128/mcb.17.11.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu F, Zhao X, Perna F, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19:283–294. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laubach JP, Fu P, Jiang X, et al. Polycythemia vera erythroid precursors exhibit increased proliferation and apoptosis resistance associated with abnormal RAS and PI3K pathway activation. Exp Hematol. 2009;37:1411–1422. doi: 10.1016/j.exphem.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grimwade LF, Happerfield L, Tristram C, et al. Phospho-STAT5 and phospho-Akt expression in chronic myeloproliferative neoplasms. Br J Haematol. 2009;147:495–506. doi: 10.1111/j.1365-2141.2009.07870.x. [DOI] [PubMed] [Google Scholar]
- 47.Guglielmelli P, Barosi G, Rambaldi A, et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood. 2011;118:2069–2076. doi: 10.1182/blood-2011-01-330563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fasolo A, Sessa C. Current and future directions in mammalian target of rapamycin inhibitors development. Expert Opin Investig Drugs. 2011;20:381–394. doi: 10.1517/13543784.2011.541154. [DOI] [PubMed] [Google Scholar]
- 49.Lu M, Zhang W, Li Y, et al. Interferon-alpha targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp Hematol. 2010;38:472–480. doi: 10.1016/j.exphem.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50**.Tefferi A. How I treat myelofibrosis. Blood. 2011;117:3494–3504. doi: 10.1182/blood-2010-11-315614. A comprehensive review on the current treatment options for myelofibrosis. [DOI] [PubMed] [Google Scholar]
- 51.Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52**.Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. First report demonstrating efficacy of a JAK2 inhibitor in myeloproliferative neoplasms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pardanani A, Lasho T, Smith G, et al. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23:1441–1445. doi: 10.1038/leu.2009.50. [DOI] [PubMed] [Google Scholar]
- 55.Tyner JW, Bumm TG, Deininger J, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood. 2010;115:5232–5240. doi: 10.1182/blood-2009-05-223727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wernig G, Kharas MG, Okabe R, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–320. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 57.Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29:789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tefferi A, Pardanani A. JAK inhibitors in myeloproliferative neoplasms: rationale, current data and perspective. Blood Rev. 2011 doi: 10.1016/j.blre.2011.1006.1002. [DOI] [PubMed] [Google Scholar]
- 59.George DJ, Dionne CA, Jani J, et al. Sustained in vivo regression of Dunning H rat prostate cancers treated with combinations of androgen ablation and Trk tyrosine kinase inhibitors, CEP-751 (KT-6587) or CEP-701 (KT-5555) Cancer Res. 1999;59:2395–2401. [PubMed] [Google Scholar]
- 60.Hexner EO, Serdikoff C, Jan M, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111:5663–5671. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levis M, Allebach J, Tse KF, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–3891. doi: 10.1182/blood.v99.11.3885. [DOI] [PubMed] [Google Scholar]
- 62.Strock CJ, Park JI, Rosen M, et al. CEP-701 and CEP-751 inhibit constitutively activated RET tyrosine kinase activity and block medullary thyroid carcinoma cell growth. Cancer Res. 2003;63:5559–5563. [PubMed] [Google Scholar]
- 63.William AD, Lee AC, Blanchard S, et al. Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011;54:4638–4658. doi: 10.1021/jm200326p. [DOI] [PubMed] [Google Scholar]
- 64.Li Z, Xu M, Xing S, et al. Erlotinib effectively inhibits JAK2V617F activity and polycythemia vera cell growth. J Biol Chem. 2007;282:3428–3432. doi: 10.1074/jbc.C600277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richter K, Buchner J. Hsp90: chaperoning signal transduction. J Cell Physiol. 2001;188:281–290. doi: 10.1002/jcp.1131. [DOI] [PubMed] [Google Scholar]
- 66.Gorre ME, Ellwood-Yen K, Chiosis G, et al. BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood. 2002;100:3041–3044. doi: 10.1182/blood-2002-05-1361. [DOI] [PubMed] [Google Scholar]
- 67.George P, Bali P, Cohen P, et al. Cotreatment with 17-allylamino-demethoxygeldanamycin and FLT-3 kinase inhibitor PKC412 is highly effective against human acute myelogenous leukemia cells with mutant FLT-3. Cancer Res. 2004;64:3645–3652. doi: 10.1158/0008-5472.CAN-04-0006. [DOI] [PubMed] [Google Scholar]
- 68.Fiskus W, Verstovsek S, Manshouri T, et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-11-1541. Published Online First October 5, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marubayashi S, Koppikar P, Taldone T, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120:3578–3593. doi: 10.1172/JCI42442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guerini V, Barbui V, Spinelli O, et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F) Leukemia. 2008;22:740–747. doi: 10.1038/sj.leu.2405049. [DOI] [PubMed] [Google Scholar]
- 71.Wang X, Zhang W, Tripodi J, et al. Sequential treatment of CD34+ cells from patients with primary myelofibrosis with chromatin-modifying agents eliminate JAK2V617F-positive NOD/SCID marrow repopulating cells. Blood. 2010;116:5972–5982. doi: 10.1182/blood-2010-02-269696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang JC, Chen C, Dumlao T, et al. Enhanced histone deacetylase enzyme activity in primary myelofibrosis. Leuk Lymphoma. 2008;49:2321–2327. doi: 10.1080/10428190802527699. [DOI] [PubMed] [Google Scholar]
- 73.Kovacs JJ, Murphy PJ, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 74.Lee J. Clinical efficacy of vorinostat in a patient with essential thrombocytosis and subsequent myelofibrosis. Ann Hematol. 2009;88:699–700. doi: 10.1007/s00277-008-0640-3. [DOI] [PubMed] [Google Scholar]
- 75.Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 76.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 77.Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 78.Rambaldi A, Dellacasa CM, Finazzi G, et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150:446–455. doi: 10.1111/j.1365-2141.2010.08266.x. [DOI] [PubMed] [Google Scholar]
- 79.Wang Y, Fiskus W, Chong DG, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood. 2009;114:5024–5033. doi: 10.1182/blood-2009-05-222133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Novotny-Diermayr V, Sausgruber N, Loh YK, et al. Pharmacodynamic evaluation of the target efficacy of SB939, an oral HDAC inhibitor with selectivity for tumor tissue. Mol Cancer Ther. 2011;10:1207–1217. doi: 10.1158/1535-7163.MCT-11-0044. [DOI] [PubMed] [Google Scholar]
- 81.Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 82.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 83*.Deshpande A, Reddy MM, Schade GO, et al. Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia. 2011 doi: 10.1038/leu.2011.255. First comprehensive description of in vitro JAK2 inhibitor resistance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hornakova T, Springuel L, Devreux J, et al. Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica. 2011;96:845–853. doi: 10.3324/haematol.2010.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tamborini E, Bonadiman L, Greco A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology. 2004;127:294–299. doi: 10.1053/j.gastro.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 86.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 88.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 89.Reddy MM, Fernandes MS, Deshpande A, et al. The JAK2V617F oncogene requires expression of inducible phosphofructokinase/fructose-bisphosphatase 3 for cell growth and increased metabolic activity. Leukemia. 2011 doi: 10.1038/leu.2011.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clem B, Telang S, Clem A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008;7:110–120. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 91.Rodrigues MS, Reddy MM, Sattler M. Cell cycle regulation by oncogenic tyrosine kinases in myeloid neoplasias: from molecular redox mechanisms to health implications. Antioxid Redox Signal. 2008;10:1813–1848. doi: 10.1089/ars.2008.2071. [DOI] [PubMed] [Google Scholar]
- 92.Walz C, Crowley BJ, Hudon HE, et al. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J Biol Chem. 2006;281:18177–18183. doi: 10.1074/jbc.M600064200. [DOI] [PubMed] [Google Scholar]
- 93.Guzman ML, Rossi RM, Karnischky L, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–4169. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 95.Reddy MM, Fernandes MS, Salgia R, et al. NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia. 2011;25:281–289. doi: 10.1038/leu.2010.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011;118:1723–1735. doi: 10.1182/blood-2011-02-292102. [DOI] [PubMed] [Google Scholar]