

Abstract
Background
The Polima (pol) system of cytoplasmic male sterility (CMS) and its fertility restoration gene Rfp have been used in hybrid breeding in Brassica napus, which has greatly improved the yield of rapeseed. However, the mechanism of the male sterility transition in pol CMS remains to be determined.
Results
To investigate the transcriptome during the male sterility transition in pol CMS, a near-isogenic line (NIL) of pol CMS was constructed. The phenotypic features and sterility stage were confirmed by anatomical analysis. Subsequently, we compared the genomic expression profiles of fertile and sterile young flower buds by RNA-Seq. A total of 105,481,136 sequences were successfully obtained. These reads were assembled into 112,770 unigenes, which composed the transcriptome of the bud. Among these unigenes, 72,408 (64.21%) were annotated using public protein databases and classified into functional clusters. In addition, we investigated the changes in expression of the fertile and sterile buds; the RNA-seq data showed 1,148 unigenes had significantly different expression and they were mainly distributed in metabolic and protein synthesis pathways. Additionally, some unigenes controlling anther development were dramatically down-regulated in sterile buds.
Conclusions
These results suggested that an energy deficiency caused by orf224/atp6 may inhibit a series of genes that regulate pollen development through nuclear-mitochondrial interaction. This results in the sterility of pol CMS by leading to the failure of sporogenous cell differentiation. This study may provide assistance for detailed molecular analysis and a better understanding of pol CMS in B. napus.
Keywords: Brassica napus, pol CMS, Anther, Transcriptome
Background
A very important source of vegetable oil worldwide, Brassica napus has extremely high oil production efficiency [1], and pol cytoplasm has been widely used in most of cultivated breeds [2]. Cytoplasmic male sterility (CMS) prevents self-pollination through pollen abortion, enabling the use of heterosis in hybrid crops for genetic improvement [3,4]. The sterility mechanism of pol has been examined in many studies, in which the orf224/atp6 and Rfp genes have mainly been analyzed. Results have shown that the sterility is caused by chimeric mitochondrial genes regulated by nuclear genes [2,5-8].
In recent years, high-throughput sequencing methods such as Illumina SOLEXA, ABI SOLiD and Roche 454 have observably increased the efficiency and reduced the cost of sequencing, making the study of transcriptomes and even genome levels easier and more feasible [9]. Nowadays, the transcriptomes of many higher plants have been sequenced for different purposes, including Arabidopsis thaliana[10,11], Myrica rubra[12], Fagopyrum[13], Citrus sinensis[14], Carthamus tinctorius[15], and even Brassica napus for fertility studies [16]. By RNA-Seq, researchers can obtain almost all of the expressed genes, especially genes with very low abundance. Therefore, genes with abundant expression differences and interesting pathways can be analyzed exhaustively [17]. Additionally, RNA-Seq also has great advantages in the identification of new genes and SNPs, and even in genome-wide association studies (GWAS) [18-20]. In polyploid plants, it is used to study the fate of duplicated genes as well, such as in soybean and bread wheat [21,22].
In this work, fertile and sterile flower buds of pol CMS with a length of 0–1 mm were sequenced using the Illumina high-throughput sequencing platform, representing the first study of the pol CMS genome at the transcriptome level. The aim of this work was to identify the differences between the fertile and sterile buds at the transcriptional level, and find out the different bioprocesses involved and their related functions. These results will help the elucidation of the sterility molecular mechanism, and assist the breeding of B. napus.
Results
Phenotypic characterization of fertile and sterile floral buds
Sterile flowers were visually smaller than fertile flowers. In the developmental process, the anthers and filaments of the sterile flowers were always shorter than those of the fertile flowers. In addition, the sterile anthers produced little or no pollen, but the pistil was normal (Figure 1). To accurately determine the stage and tissue in which sterility occurs, fertile and sterile anthers were cytologically observed in different development stages. As shown in Figure 2, there was no clear difference between them until stage 4 of anther development. In stage 4, the sterile anthers did not differentiate sporogenous cells; additionally, the middle layer, endothecium and tapetum could not be distinguished (Figure 2C,I). After that, the sterile anthers were filled with numerous highly vacuolated cells. They also showed cavities in later stages (Figure 2K,L). In sterile anthers, locules with pollen sacs in stage 4 were very few and did not produce normal tetrads. In contrast, the fertile anthers developed normally in all stages.
Figure 1.

Phenotypic characterization of fertile and sterile floral buds. A: phenotype of sterile floral buds; B: phenotype of fertile floral buds. Bar for front-view is 5 mm, and for vertical-view is 15 mm.
Figure 2.
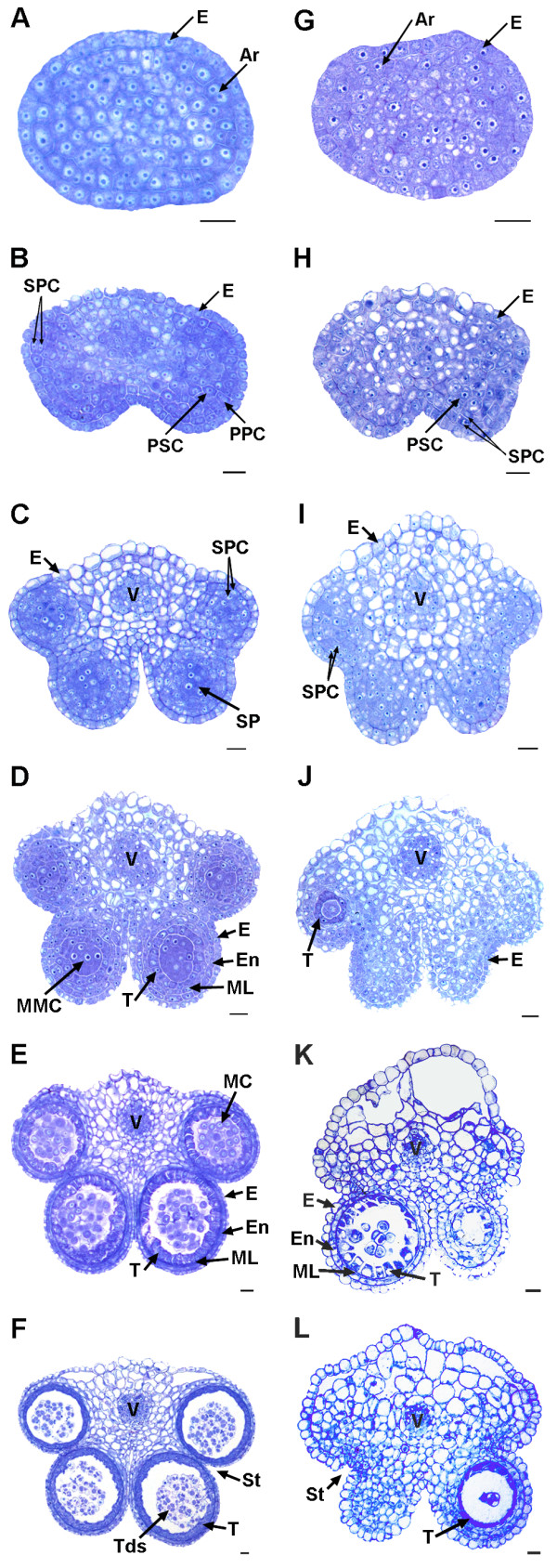
Development of fertile (A-F) and sterile (G-L) anthers in pol CMS. Bar = 10 μm for all the stages. Ar, archesporial cell; E, epidermis; En, endothecium; MC, meiotic cell; ML, middle layer; MMC, microspore mother cells; PPC, primary parietal cell; PSC, primary sporogenous cell; SP, sporogenous cell; SPC, secondary parietal cell; St, stomium; T, tapetum; Tds, tetrads; V, vascular region. A to F represent the anther development stage 2 to 7, respectively. So do G to L [25].
Transcriptome sequencing and assembly
After the raw data were trimmed, 62,417,937 clean reads for fertile samples and 43,063,199 for sterile samples were obtained (Additional file 1). All clean reads (105,481,136) were assembled by running Trinity, as no reference genome was available for B. napus[23]. As a result, 164,874 contigs (> 200 bp) were generated with an average length of 621 bp and a N50 of 927 bp. After clustering, 112,770 unigenes were obtained, including 22,306 clusters and 90,464 singletons. The average length of these unigenes was 638 bp and the N50 was 982 bp. There were 70,956 unigenes (62.92%) with a length range from 200 to 500 bp, 20,178 unigenes (17.89%) longer than 1,000 bp and no unigenes shorter than 200 bp (Table 1). The singletons mentioned above refer to contigs that matched no other contig, and the clusters and singletons generated from the contigs covered all of the obtained unigenes [24].
Table 1.
Summary of de novo transcriptome
| Length (bp) | Contigs | Percentage (%) | Unigenes | Percentage (%) |
|---|---|---|---|---|
|
200-499 |
105,020 |
63.70 |
70,956 |
62.92 |
|
500-999 |
32,032 |
19.43 |
21,636 |
19.19 |
|
1000-1499 |
13,678 |
8.30 |
9,677 |
8.58 |
|
1500-1999 |
7,304 |
4.43 |
5,404 |
4.79 |
| > = 2000 | 5,840 | 3.54 | 5,097 | 4.52 |
Functional annotation
For annotation, the 112,770 unigenes were subjected to BLASTX searches against the sequences in the NR (non-redundant protein sequences in NCBI), Swiss-Prot and COG databases (E-value ≤ 1e-5). As a result, 72,217 (64.04%), 47,919 (42.49%) and 23,295 (20.66%) unigenes were aligned against the three protein databases, respectively. Among these unigenes, 22,551 (20.00%) were annotated by all three databases and 72,408 (64.21%) were annotated by at least one database (Figure 3).
Figure 3.
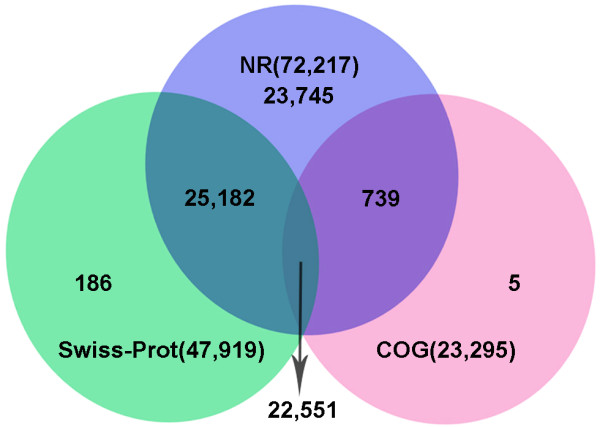
Unigenes annotated with public databases. The numbers of annotated unigenes were signified in the different regions.
There were 23,290 unigenes annotated by both the NR and COG databases. To examine the distribution of the assembly results, these unigenes were categorized into at least 24 COG (Clusters of Orthologous Groups) function clusters. Among the 24 clusters, the “general function prediction only” cluster comprised the highest number of unigenes (5,113, 21.95%), and the “replication, recombination and repair” cluster had the second largest number of unigenes. In contrast, only 23 unigenes were classified into “nuclear structure” (Figure 4). GO (Gene ontology) classifications were also obtained to investigate the functions of the unigenes; 47,733 (42.33%) unigenes that were annotated by both the NR and Swiss-Prot databases were classified into 47 groups. Among the 47,733 unigenes, 34,252 were assigned 158,341 GO annotations. All 47 groups can be categorized into three main classifications: “cellular component”, “molecular function” and “biological process”. There were 24,928 (72.78%) unigenes in the “cell part”, 18,352 (53.58%) in the “binding” and 19,118 (55.82%) in the “cellular process” categories, which were the major categories in each of the three main classifications motioned above, respectively. In addition, “organelle”, “catalytic activity” and “metabolic process” also had large proportions (> 45%) of unigenes. Conversely, some groups such as “metallochaperone activity”, “cell killing”, “proteasome regulator activity” and “auxiliary transport protein activity” had small numbers of unigenes (Figure 5).
Figure 4.
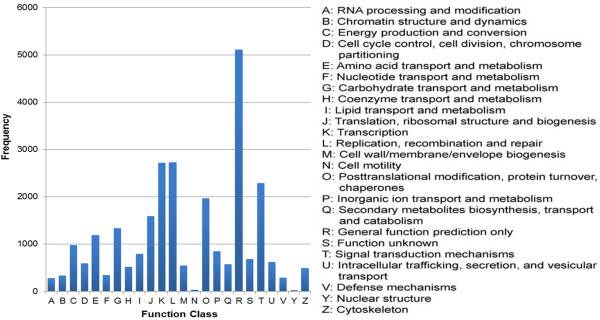
COG function classification. All the unigenes aligned in COG database were assorted in 24 clusters.
Figure 5.
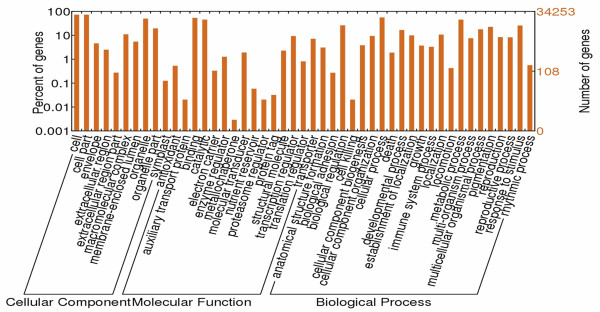
Classification of GO annotations. The x-axis indicates the sub-categories; the left y-axis indicates the percentage of a sub-category of genes in that category and the right y-axis indicates the number of unigenes in a sub-category.
Putative genes related with pollen development
The development of pollen is a fundamental and complex process in flowering plants. It is essential for propagation and evolution. As a model plant, Arabidopsis thaliana has been well studied for this bioprocess. Therefore, all of the unigenes identified here were annotated to the TAIR database (http://www.arabidopsis.org/). Subsequently, unigenes annotated to 19 genes from AG to MS1, which are considered to regulate pollen development and act before meiosis, were extracted for analysis [25-29]. The results showed that among the 19 genes, some were significantly down-regulated in sterile buds, such as BnNZZ/SPL, BnSPL8, BnEXS/EMS1 and BnER, but no gene was dramatically up-regulated (Additional file 2). Additionally, most of these genes did not show significant differences in expression between the fertile and sterile buds.
Transcripts differentially expressed in fertile and sterile buds
The FPKM (Fragments per Kilobase of transcript per Million mapped reads) value was calculated to test the expression levels of the unigenes. We noticed that in some basic bioprocesses these unigenes showed high expression levels in both the fertile and sterile buds. For example, unigene78635 and unigene78636, which were involved in RNA transport, showed very high expression in both fertile buds (FPKM = 14,059.7 and FPKM = 9,451.37) and sterile buds (FPKM = 12,954.3 and FPKM = 8,286.57). Additionally, some unigenes related to metabolism, such as unigene10390 (FPKM > 4,300), showed high abundance as well.
With the restrictive conditions of log2 ratio > 1.0 and P-value < 0.001, a total of 1,148 (1.02% of all unigenes) unigenes that were significantly differentially expressed were obtained, including 629 up-regulated and 519 down-regulated unigenes in sterile buds (Additional file 3). The up- and down-regulated unigenes were submitted to KAAS (KEGG Automatics Annotation Server) and classified with the SBH (single directional best hit) method [30]. Most of the up-regulated unigenes were included in metabolic pathways (Figure 6A). In contrast, a relatively large number of the down-regulated unigenes functioned in transcription and translation besides metabolic pathways (Figure 6A), such as the unigenes related with spliceosomes, RNA transport and ribosome biogenesis. Specifically, we found that there were seven callose synthase (K11000) genes in the down-regulated unigenes. This result was consistent with the observation of anther semi-thin sections.
Figure 6.

Comparison of annotations between up- and down-regulated unigenes in sterile buds. A: a pie chart of KEGG annotation in up-(left) and down-(right) regulated unigenes (n > 4), respectively; B: a statistical column diagram of GO annotation; C: an enriched graph of molecular function in GO annotation. They were filtered using default value.
In addition, GO analysis was performed using Blast2GO [31]. After enrichment analysis, only 9 GO terms showed significant differences. Among these terms, four (“oxidoreductase activity”, “cellular aromatic compound metabolic process”, “secondary metabolic process” and “cell wall”) were associated with the up-regulated unigenes and five (mainly hydrolase, pyrophosphatase and nucleoside-triphosphatase activity, and “nuclear lumen”) were associated with down-regulated unigenes (Figure 6B). In the molecular function category, only oxidoreductase and hydrolase activities were different between the up- and down-regulated unigenes (Figure 6C).
In particular, orf224, which is believed to be a key ORF of pol CMS in the mitochondrial genome [5], showed a 4-fold transcript increase in sterile buds compared with fertile buds. Atp6, a chimeric gene of orf224, also showed minor up-regulation.
qRT-PCR verification
The differentially expressed unigenes were selected to verify the accuracy of the RNA-Seq analysis by qRT-PCR, using the same RNA that was used for RNA-Seq. Even though most qRT-PCR results indicated smaller differences compared with the RNA-Seq analysis, there was a consistent expression tendency (Figure 7). However, unigene3022 was an exception. In qRT-PCR, unigene3022 showed no difference in the fertile and sterile buds, while RNA-Seq analysis indicated a significant difference. qRT-PCR verified that among the five unigenes annotated to pollen development, four (unigene28529, unigene13440, unigene88831, unigene21180) showed low abundance in sterile buds and one (unigene19392, which was the earliest expressed) showed no difference between fertile and sterile buds (Figure 7). Unigene19577 and unigene2959, which were predicted to encode callose synthases, showed down-regulation in sterile buds. Unigene87043 and unigene2751, mitochondrial genes that influence fertility directly, were confirmed to be up-regulated in sterile buds. In addition, unigene44866, unigene24663 and unigene42471, which participate in the glycolysis/gluconeogenesis pathway, amino acid metabolism and oxidative phosphorylation, respectively, were all up-regulated in sterile buds.
Figure 7.

qRT-PCR verification of differentially expressed unigenes. S means sterile sample and F means fertile sample.
Discussion
Here, the transcriptome reads of fertile and sterile buds were acquired using the Illumina sequencing platform. Then, because of the unavailability of the genome and transcriptome of B. napus at present, we assembled the transcriptome of the buds for further research. Altogether, 112,770 unigenes were obtained, but only 1,148 unigenes (1.02% of all unigenes) showed notable differences in expression, indicating that though the development of buds is a complicated and polygenic process, changes in a relatively small number of genes can transform the trait observably.
In the present study, unigenes that were annotated as direct regulatory genes of pollen development showed higher abundance in fertile buds. Interestingly, unigene28529 (BnNZZ/SPL) was the first unigene that showed a difference in the regulatory network of anther development. Unigene19392 (BnAG), which acts as upstream gene of unigene28529, showed no difference in expression [32]. Therefore, it can be presumed that the lack of Rfp emancipates the male-sterile gene in mitochondria. Through a series of bioprocesses, this male-sterile gene leads to serial inhibition of the downstream genes by regulating BnNZZ/SPL, a pivotal regulator of sporogenesis [33,34]. Consequently, the fertile buds become sterile before the formation of sporogenous cells. It has been reported that nuclear-mitochondrial interaction results in CMS in previous studies [35,36]. These genes were inhibited rather than silenced, which consequently led to the formation of only a few pollen sacs in the sterile buds.
Although orf224 and atp6 were both up-regulated in sterile buds, the change in orf224 expression was twice that of atp6, which might have been caused by the truncation of most of the orf224 transcripts in the presence of Rfp[8]. Nevertheless, what has been discussed above indicates that energy supply was much more plentiful in the fertile samples. We speculate that even though the orf224/atp6 gene was normally transcribed, the translation of it was defective, or the translated protein could not form a functional advanced structure. After all, the existence of integral orf224 led to the insufficiency of ATPase protein 6, which caused the energy deficiency in the sterile buds. In other studies, the candidate cytoplasmic male sterile genes have also had a close relationship with ATPase genes, such as atp2, atp6[37,38].
Comparative profiling was performed between the two transcriptomes. It was found that metabolism was still the principal activity in buds. Unigenes that involved in pyrophosphatase and nucleoside-triphosphatase activity were down-regulated, suggesting an energy deficiency in sterile buds. The depauperate bud activity was elucidated by the weakening of transcription and translation, which is similar to the results of an earlier study of Arabidopsis[39,40]. Different distributions of energy and materials may strongly influence fertility, and the regulation of sterility is very complicated. At the transcriptional level, mRNA plays an important role; besides that, small RNA also participates in the process and makes the regulation network more complete [41,42].
We hypothesized that orf224 was integrally transcribed because of the lack of the Rfp gene in pol CMS and that this led to malformation of the advanced structure of atp6, which subsequently caused an energy deficiency. Therefore, the exchange of materials between the nucleus and cytoplasm was blocked. The expression of some genes that were located in the nucleus and function in pollen development was inhibited, and BnNZZ/SPL was the first inhibited gene in temporal order. Finally, anthers could not differentiate sporogenous cells and became sterile in pol CMS (Figure 8).
Figure 8.

Proposed model for sterile and restored mechanism of pol CMS.
Conclusions
In this study, the different bioprocesses and genes that regulate pollen development were analyzed through a high-throughput sequencing approach. Combined with the investigation of the orf224/atp6 gene region, we propose a speculative model for the sterility and restoration mechanism of pol CMS. These results will promote the study of sterility mechanisms in more detail and the cloning of restorer genes. Additionally, the assembled B. napus unigenes obtained in this study will contribute to other transcriptomic and genomic studies.
Methods
Plant materials and RNA preparation
Fertile and sterile flower buds of pol CMS in B. napus were used in this study. They were near-isogenic lines (NIL) and were both cultivated in the same experimental plot in Huazhong Agriculture University (Wuhan, Hubei Province, China). Buds with a length of 0–1 mm were stripped from three different plants for transcriptomic profiling. All fertile buds were gathered together (as were sterile buds), snap-frozen in liquid nitrogen and kept at -80°C for further use. Total RNA was isolated according to the instructions of the TRIzol kit (Invitrogen, USA) and purified using a mRNA purification kit (Promega, China) following the manufacturer’s protocol.
cDNA library construction and RNA-Seq
The mRNA was reverse transcribed by PowderscriptTM II (Takara, China), and then double-stranded cDNA was amplified according to the user manual of the SMARTTM cDNA construction kit. Finally, double-stranded cDNA was purified using a DNA purification kit (QIAGEN, Germany) to generate high quality cDNA. Approximately 10 μg of sheared cDNA was then prepared for Illumina sequencing according to the manufacturer’s protocols. Libraries were prepared from a 300–500 bp size-selected fraction following adapter ligation and agarose gel separation. The libraries were sequenced using a paired-end read protocol with 100 bp of data collected per run on the Illumina Hiseq 2000.
Analysis of Illumina sequencing results
Clean reads were obtained from raw data by filtering the adaptor sequences and the low-quality sequences using SolexaQA [43]. Then, the pair-end clean reads were de novo assembled into contigs by Trinity. After that, TGI Clustering tools were used to obtain the non-redundant unigenes [44].
Functional annotation of unigenes was performed using BLASTX searches against the NR, Swiss-Prot and COG databases (E-value < 1e-5). The GO annotations for the unigenes were determined using Blast2GO [31]. Then, the results were submitted to WEGO to obtain the GO classification graph. The raw reads were deposited in the NCBI SRA (Short Read Archive) with the accession number SRA069852. The assembled unigenes are shown in Additional file 4.
Mapping and expression level analysis
The reads of the fertile and sterile samples were mapped back to our de novo assembling results separately using Tophat [45]. Then, the differentially expressed unigenes were obtained using Cufflinks [46]. The results were submitted to KAAS and Blast2GO for further interpretation.
Real-time quantitative PCR (qRT-PCR) verification
First-strand cDNA was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo, USA), from 1,200 ng total RNA. Gene-specific primers were designed based on the selected unigene sequences (Additional file 5). Reactions were performed with the SYBR Green Realtime PCR Master Mix (TOYOBO, Japan) in a Bio-Rad CFX96 instrument. Three biological replicates for each sample and three technical replicates were performed and the relative expression level was calculated using the 2-ΔΔCt method. The actin gene was used to normalize the gene expression.
Semi-thin sections and light microscopy
Sterile and fertile flower buds were fixed and embedded in resin using Technovit Embedding Kits (Germany). Then, semi-thin (2.5 μm) sections were obtained using an automatic microtome (Microm HM 360, Thermo). The sections were stained with 0.1% toluidine blue O for 30–60 sec at room temperature and observed with a Nikon Eclipse 80i microscope (Nikon, Japan). Images of the anthers in different stages were captured with a Nikon DS-Ri1 camera (Nikon, Japan).
Availability of supporting data
The project was submitted to NCBI BioProject with BioProject ID: PRJNA193477 (http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA193477). The raw reads were deposited in NCBI SRA (Short Read Archive) with the accession number SRA069852 (http://www.ncbi.nlm.nih.gov/sra/?term=SRA069852). The assembled unigenes were deposited in the Additional file 4: De novo assembled transcripts sequences. The DGEs and their annotations were deposited in the Additional file 3: Expressions and annotations of the differentially expressed unigenes.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BY conceived of the study. HA carried out the data analysis and molecular woks. ZY obtained the pol near-isogenic line (NIL) and prepared the RNA. HA and ZY executed anatomical work. HA and BY drafted the manuscript. JW, JS, JT, CM and TF provided suggestions, funds and experimental conditions. All authors have read and approved the final manuscript.
Supplementary Material
Summary of sequencing results.
19 genes related to pollen development. *: detected or confirmed by qRT-PCR.
Expressions and annotations of the differentially expressed unigenes. *: detected or confirmed by qRT-PCR.
De novo assembled transcripts sequences.
Primer sequences for qRT-PCR.
Contributor Information
Hong An, Email: anhong@webmail.hzau.edu.cn.
Zonghui Yang, Email: ksprings@163.com.
Bin Yi, Email: yibin@mail.hzau.edu.cn.
Jing Wen, Email: wenjing@mail.hzau.edu.cn.
Jinxiong Shen, Email: jxshen@mail.hzau.edu.cn.
Jinxing Tu, Email: tujx@mail.hzau.edu.cn.
Chaozhi Ma, Email: yuanbeauty@mail.hzau.edu.cn.
Tingdong Fu, Email: futing@mail.hzau.edu.cn.
Acknowledgements
This project was supported by the Natural Science Foundation of China (Grant No. 31371488) and the National High Technology Research and Development Program of China (Grant No. 2012AA101107).
References
- Han-zhong W. Review and future development of rapeseed industry in china. Chin J Oil Crop Sci. 2010;15(2):3. [Google Scholar]
- Chen J, Guan R, Chang S, Du T, Zhang H, Xing H. Substoichiometrically different mitotypes coexist in mitochondrial genomes of Brassica napus L. PLoS One. 2011;15(3):e17662. doi: 10.1371/journal.pone.0017662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Xu H, Liu Z, Guo J, Li H, Chen L, Fang C, Zhang Q, Bai M, Yao N, Wu H, Ji C, Zheng H, Chen Y, Ye S, Li X, Zhao X, Li R, Liu Y. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat Genet. 2013;15:573–577. doi: 10.1038/ng.2570. [DOI] [PubMed] [Google Scholar]
- Woodson JD, Chory J. Coordination of gene expression between organellar and nuclear genomes. Nat Rev Genet. 2008;15(5):383–395. doi: 10.1038/nrg2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Homme Y, Stahl RJ, Li XQ, Hameed A, Brown GG. Brassica nap cytoplasmic male sterility is associated with expression of a mtDNA region containing a chimeric gene similar to the pol CMS-associated orf224 gene. Curr Gen. 1997;15(4):325–335. doi: 10.1007/s002940050212. [DOI] [PubMed] [Google Scholar]
- Yuan M, Yang GS, Fu TD, Li Y. Transcriptional control of orf224/atp6 by the pol CMS restorer Rfp gene in Brassica napus L. Yi Chuan Xue Bao. 2003;15(5):469–473. [PubMed] [Google Scholar]
- Singh M, Hamel N, Menassa R, Li XQ, Young B, Jean M, Landry BS, Brown GG. Nuclear genes associated with a single Brassica CMS restorer locus influence transcripts of three different mitochondrial gene regions. Genetics. 1996;15(1):505–516. doi: 10.1093/genetics/143.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Brown GG. Suppression of cytoplasmic male sterility by nuclear genes alters expression of a novel mitochondrial gene region. Plant Cell. 1991;15(12):1349–1362. doi: 10.1105/tpc.3.12.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC. Next-generation sequencing transforms today’s biology. Nat Methods. 2008;15(1):16–18. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- Torti S, Fornara F, Vincent C, Andres F, Nordstrom K, Gobel U, Knoll D, Schoof H, Coupland G. Analysis of the Arabidopsis shoot meristem transcriptome during floral transition identifies distinct regulatory patterns and a leucine-rich repeat protein that promotes flowering. Plant Cell. 2012;15(2):444–462. doi: 10.1105/tpc.111.092791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Farmer AD, Langley RJ, Mudge J, Crow JA, May GD, Huntley J, Smith AG, Retzel EF. Meiosis-specific gene discovery in plants: RNA-Seq applied to isolated Arabidopsis male meiocytes. BMC Plant Biol. 2010;15:280. doi: 10.1186/1471-2229-10-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Chen M, Xu CJ, Bai L, Yin XR, Li X, Allan AC, Ferguson IB, Chen KS. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genomics. 2012;15:19. doi: 10.1186/1471-2164-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logacheva MD, Kasianov AS, Vinogradov DV, Samigullin TH, Gelfand MS, Makeev VJ, Penin AA. De novo sequencing and characterization of floral transcriptome in two species of buckwheat (Fagopyrum) BMC Genomics. 2011;15:30. doi: 10.1186/1471-2164-12-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu Q, Da X, Guo F, Ding Y, Deng X. Transcriptome changes during fruit development and ripening of sweet orange (Citrus sinensis) BMC Genomics. 2012;15(1):10. doi: 10.1186/1471-2164-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulin H, Xiao Y, Pei S, Wen T, Shangqin H. The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PLoS One. 2012;15(6):e38653. doi: 10.1371/journal.pone.0038653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Dong C, Yu J, Liu W, Jiang C, Liu J, Hu Q, Fang X, Wei W. Transcriptome profile analysis of young floral buds of fertile and sterile plants from the self-pollinated offspring of the hybrid between novel restorer line NR1 and Nsa CMS line in Brassica napus. BMC Genomics. 2013;15:26. doi: 10.1186/1471-2164-14-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;15(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn DA, Ragland GJ, Shoemaker DD, Denlinger DL. Gene discovery using massively parallel pyrosequencing to develop ESTs for the flesh fly Sarcophaga crassipalpis. BMC Genomics. 2009;15:234. doi: 10.1186/1471-2164-10-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trick M, Long Y, Meng J, Bancroft I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol J. 2009;15(4):334–346. doi: 10.1111/j.1467-7652.2008.00396.x. [DOI] [PubMed] [Google Scholar]
- Harper AL, Trick M, Higgins J, Fraser F, Clissold L, Wells R, Hattori C, Werner P, Bancroft I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat Biotechnol. 2012;15:798–802. doi: 10.1038/nbt.2302. [DOI] [PubMed] [Google Scholar]
- Roulin A, Auer PL, Libault M, Schlueter J, Farmer A, May G, Stacey G, Doerge RW, Jackson SA. The fate of duplicated genes in a polyploid plant genome. Plant J. 2012;15(1):143–153. doi: 10.1111/tpj.12026. [DOI] [PubMed] [Google Scholar]
- Pont C, Murat F, Confolent C, Balzergue S, Salse J. RNA-seq in grain unveils fate of neo- and paleopolyploidization events in bread wheat (Triticum aestivum L.) Genome Biol. 2011;15(12):R119. doi: 10.1186/gb-2011-12-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;15(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, Luan JB, Li JM, Bao YY, Zhang CX, Liu SS. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genomics. 2010;15:400. doi: 10.1186/1471-2164-11-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders PM, Bui AQ, Weterings K, Mclntire KN, Hsu YC, Lee PY, Truong MT, Beals TP, Goldberg RB. Anther developmental defects in Arabidopsis thaliana male-sterile mutants. Sex Plant Reprod. 1999;15(6):297–322. doi: 10.1007/s004970050158. [DOI] [Google Scholar]
- Unte US, Sorensen AM, Pesaresi P, Gandikota M, Leister D, Saedler H, Huijser P. SPL8, An SBP-Box gene that affects pollen sac development in Arabidopsis. Plant Cell Online. 2003;15(4):1009–1019. doi: 10.1105/tpc.010678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RJ, Spielman M, Dickinson HG. Stamen structure and function. Plant Cell. 2004;15(Suppl):S46–60. doi: 10.1105/tpc.017012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ZA, Zhang DB. From Arabidopsis to rice: pathways in pollen development. J Exp Bot. 2009;15(5):1479–1492. doi: 10.1093/jxb/erp095. [DOI] [PubMed] [Google Scholar]
- Ma H. Molecular genetic analyses of microsporogenesis and microgametogenesis in flowering plants. Annu Rev Plant Biol. 2005;15:393–434. doi: 10.1146/annurev.arplant.55.031903.141717. [DOI] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;15:W182–185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;15(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Ito T, Wellmer F, Yu H, Das P, Ito N, Alves-Ferreira M, Riechmann JL, Meyerowitz EM. The homeotic protein AGAMOUS controls microsporogenesis by regulation of SPOROCYTELESS. Nature. 2004;15(6997):356–360. doi: 10.1038/nature02733. [DOI] [PubMed] [Google Scholar]
- Yang WC, Ye D, Xu J, Sundaresan V. The SPOROCYTELESS gene of Arabidopsis is required for initiation of sporogenesis and encodes a novel nuclear protein. Genes Dev. 1999;15(16):2108–2117. doi: 10.1101/gad.13.16.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Song M, Fan S, Yu S. Transcriptomic analysis of differentially expressed genes during anther development in genetic male sterile and wild type cotton by digital gene-expression profiling. BMC Genomics. 2013;15(1):97. doi: 10.1186/1471-2164-14-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase CD. Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet. 2007;15(2):81–90. doi: 10.1016/j.tig.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Jing B, Heng S, Tong D, Wan Z, Fu T, Tu J, Ma C, Yi B, Wen J, Shen J. A male sterility-associated cytotoxic protein ORF288 in Brassica juncea causes aborted pollen development. J Exp Bot. 2012;15(3):1285–1295. doi: 10.1093/jxb/err355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Yamada M, Fujita M, Itabashi E, Hamada K, Yano K, Kurata N, Toriyama K. Cytoplasmic-nuclear genomic barriers in rice pollen development revealed by comparison of global gene expression profiles among five independent cytoplasmic male sterile lines. Plant Cell Physiol. 2010;15(4):610–620. doi: 10.1093/pcp/pcq026. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Yang X, Zhang M. Mitochondrially-targeted expression of a cytoplasmic male sterility-associated orf220 gene causes male sterility in Brassica juncea. BMC Plant Biol. 2010;15:231. doi: 10.1186/1471-2229-10-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yang C, Yuan Z, Zhang D, Gondwe MY, Ding Z, Liang W, Zhang D, Wilson ZA. The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell. 2010;15(1):91–107. doi: 10.1105/tpc.109.071803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Lu D, Ma X, Peng Y, Sun Y, Ning G, Ma H. Regulation of the Arabidopsis anther transcriptome by DYT1 for pollen development. Plant J. 2012;15(4):612–624. doi: 10.1111/j.1365-313X.2012.05104.x. [DOI] [PubMed] [Google Scholar]
- Shen Y, Zhang Z, Lin H, Liu H, Chen J, Peng H, Cao M, Rong T, Pan G. Cytoplasmic male sterility-regulated novel microRNAs from maize. Funct Integr Genom. 2011;15(1):179–191. doi: 10.1007/s10142-010-0202-3. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Xu B, Zhao N, Yang X, Zhang M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of brassica juncea. BMC Genomics. 2013;15:9. doi: 10.1186/1471-2164-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;15:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;15(5):651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;15(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;15(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of sequencing results.
19 genes related to pollen development. *: detected or confirmed by qRT-PCR.
Expressions and annotations of the differentially expressed unigenes. *: detected or confirmed by qRT-PCR.
De novo assembled transcripts sequences.
Primer sequences for qRT-PCR.