

Abstract
Physiological evidence indicates that the supraoptic nucleus (SON) is an important region for integrating information related to homeostasis of body fluids. Located bilaterally to the optic chiasm, this nucleus is composed of magnocellular neurosecretory cells (MNCs) responsible for the synthesis and release of vasopressin and oxytocin to the neurohypophysis. At the cellular level, the control of vasopressin and oxytocin release is directly linked to the firing frequency of MNCs. In general, we can say that the excitability of these cells can be controlled via two distinct mechanisms: 1) the intrinsic membrane properties of the MNCs themselves and 2) synaptic input from circumventricular organs that contain osmosensitive neurons. It has also been demonstrated that MNCs are sensitive to osmotic stimuli in the physiological range. Therefore, the study of their intrinsic membrane properties became imperative to explain the osmosensitivity of MNCs. In addition to this, the discovery that several neurotransmitters and neuropeptides can modulate their electrical activity greatly increased our knowledge about the role played by the MNCs in fluid homeostasis. In particular, nitric oxide (NO) may be an important player in fluid balance homeostasis, because it has been demonstrated that the enzyme responsible for its production has an increased activity following a hypertonic stimulation of the system. At the cellular level, NO has been shown to change the electrical excitability of MNCs. Therefore, in this review, we focus on some important points concerning nitrergic modulation of the neuroendocrine system, particularly the effects of NO on the SON.
Keywords: Magnocellular neurons, Supraoptic nucleus, Vasopressin, Oxytocin, Nitric oxide, Homeostasis
Nitric oxide as a messenger molecule
Studied in 1772 by Joseph Priestly (1), nitric oxide (NO) was taken, at first, to be a toxic gas. However, this view was changed when it was shown that NO was endogenously produced by living organisms. Initially characterized as an endothelium-derived relaxation factor (2), NO was later postulated to be a possible neurotransmitter in the central nervous system by Garthwaite et al. (3). In 1992, NO was named “Molecule of the Year”, and its physiological and pharmacological importance became clear through the studies of Louis J. Ignarro, Robert F. Furchgott, and Ferid Murad, who were awarded the Nobel Prize in Physiology or Medicine in 1998 (4). These scientists showed that NO could act as a signaling molecule in the central nervous system, suggesting its role as a neurotransmitter/neuromodulator.
This view changed the classical concepts used to explain communication between neurons, because NO cannot be stored, released, or inactivated by conventional regulatory mechanisms. NO signaling in excitable tissues requires rapid and controlled release to specific cellular targets, since its average lifetime is on the order of a few seconds (5). In principle, NO could spread out from its site of production to influence different types of tissues (neuronal, glial, and vascular) that are not necessarily in anatomical juxtaposition, acting as an autocrine or paracrine signaling molecule. At the present time, there is vast literature focused on the study of the biological effects of NO. This messenger is involved in the function of a great diversity of tissues and is shown to be involved in many different physiological processes. For a review on other aspects of NO function, not addressed in this review, see Calabrese et al. (6).
Synthesis of NO
In the organism, NO originates from L-arginine in a process catalyzed by three distinct NO synthase (NOS) enzymes: neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). eNOS and nNOS are constitutively expressed enzymes, which are stimulated by increases in intracellular calcium concentration (7). The immunological functions of NO are mediated by a calcium-independent iNOS that is activated by products released in inflammatory processes such as cytokines, interferon gamma, and interleukins 1 and 2 (8). However, all NOS enzymes use NADPH as an electron donor and require five cofactors as well as the presence of calmodulin to catalyze the oxidation of L-arginine to NO, with a stoichiometric formation of citrulline. The result of this reaction is one molecule of NO plus L-citrulline (9). L-citrulline is recycled back to regenerate L-arginine for a new NO synthesis, closing the cycle of NO production (Figure 1) (10). Since the amount of NADPH and citrulline correspond to the amount of NO produced, they were extensively used as markers of NO production in the brain including the supraoptic nucleus (SON) (11). Nowadays, other approaches can be used to detect NO production with the same precision: for example, fluorescent dyes like DAF and DAF-FM (12), or electrochemical sensors (13).
Figure 1. Nitric oxide synthesis. In the presence of NADPH, oxygen and co-factors, such as tetrahydrobiopterin (BH4), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), nitric oxide (NO) synthase (NOS) plus calcium (Ca2+), catalyze the breakdown of L-arginine to citrulline and NO. Citrulline will be recycled back to regenerated L-arginine and NO will produce autocrine and/or paracrine effects as well as negative feedback on NOS.
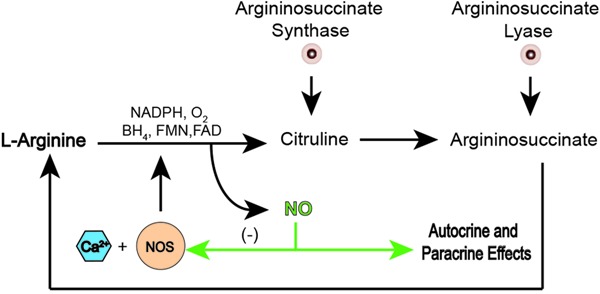
mRNA transcripts of all NOS isoforms are present in the hypothalamus, with the order of expression being nNOS>eNOS>iNOS (14). In the SON, the main isoform is nNOS (15), and its expression is likely to be controlled in an activity-dependent manner, i.e., increases in activity of the neuron induce an equivalent increase of nNOS in magnocellular neurons (16). However, the effects seen when nNOS is elevated are not completely understood. In the literature, there is a consensus that vasopressinergic neurons constitute the major neuronal phenotypes expressing nNOS, suggesting a role for this new neurotransmitter in the mechanisms regulating fluid homeostasis (17).
Donors and inhibitors of NO
To help in the understanding of the biological functions of NO, exogenous sources and inhibitors of this neuromodulator have been developed as research tools. To increase NO levels, both donors and substrates are used. Donors are compounds that release NO, and several compounds have already been described, for example, sodium nitroprusside (SNP), nitroso-N-acetylpenicillamine (SNAP), 3-morpholinosydnonimine (SIN-1), and diazeniumdiolates (NONOates).
Chemically, all donors have nitrate functionality within the molecule, and a nitroso functional group is present in all of these compounds (18). Some donors have functional nitrosothiol groups, S-nitrosothiols, whose decomposition is catalyzed by copper ions (Cu+) to form NO and disulfide. Interestingly, S-nitrosothiols were found endogenously, supposedly acting as NO stores for release when required (19). Another functionally important group is nitrosyl, commonly found in sodium nitroprusside (Na2[Fe(CN)5NO]-SNP), which is a mixed nitrosyl-cyano complex (20). Additionally, other metal complexes have been developed including a nitrosyl-ruthenium complex, which has the advantage of low cytotoxicity and readily releasing NO upon illumination (21). Although the classification of all NO donors is a complicated task, we can say that, when they show similar chemical structures, they usually have similar NO-releasing mechanisms.
SNP was used by several authors in their studies of the SON (22-24). SNP is a complex of ferrous ion with five cyanide anions (CN-) and a nitrosonium ion (NO+). Interaction of SNP with a reducing agent, such as thiols, leads to the formation of NO. Its use in biological systems has the inconvenience that formation of NO is accompanied by the formation of ferricyanide, a biologically active and toxic compound (25). To avoid the effects of ferricyanide, other types of donors have been used, for example, SNAP, SIN-1, and NONOates. Regarding SNAP, it is a derivative of the nitrosothiol group that seems to provide higher concentrations of NO, but micromolar concentrations of Cu2+ are required for its action (26). SIN-1 also produces NO and originates a breakdown product, SIN-1C, which is biologically inactive (27). A side effect of SIN-1 is the production of superoxide anion, which can react with NO and H+ to form peroxinitrite (ONOO−) and hydrogen peroxide (H2O2), both with deleterious effects on membranes (28). NONOates release NO spontaneously in solution at physiological pH and temperature. The NO derived from NONOates is not accompanied by the cytotoxic effects of hydrogen, alkyl hydroperoxide, or hypoxanthine/xanthine. In addition, other NO donors have been developed, which promise advantages over the previous ones, such as spontaneous release of NO under controlled rates. In the literature, we can find descriptions of a variety of these, i.e., donors with higher levels of NO release without being photosensitive or releasing cyanide: for example, Rut-bpy (Cis-[Ru(bpy)2(SO3)(NO)]PF6) (29) and/or donors with protection against hydrogen peroxide-mediated cytotoxicity diethylamine (DEA/NO) and propylamine propylamine (PAPA/NO) (30). The amount and duration of NO release depend on the pharmacological properties of each donor. Thus, some compounds could have a fast action in small quantities or a slow action when NO is released for long periods.
Besides donors, production of endogenous NO can be controlled by using inhibitors of NOS. These inhibitors can be divided into two groups: 1) inhibitors that target cofactor-binding sites and 2) inactive L-arginine analog molecules. The first interfere with flavin, calmodulin, or dioxygen binding sites. These NOS inhibitors have no selectivity for a particular isoform and interfere with the activity of other enzymes that need the same cofactors to become active (31).
In an opposite way, substrates analogous to L-arginine have a higher specificity toward the NOS isoforms. These inhibitors act as false substrates by binding to the L-arginine binding site (32). Although over one-hundred NOS inhibitors have been described as possible pharmacological tools (31) reducing or preventing the biological effects of NO (2,5), the majority of them are nonselective, and just a few compounds, such as 7-nitro-indazole, amidines, and some amino acid derivatives, are able to selectively inhibit nNOS (33,34). Nω-nitro-L-arginine methyl ester (L-NAME), a nonselective NOS inhibitor, has been extensively used in studies of the SON (23,35,36).
Mechanisms of action
Guanylate cyclase
Although the number of newly discovered potential targets of NO increases continually, its major effect, under physiological conditions, appears to be mediated mainly through the activation of soluble guanylate cyclase (sGC), the intracellular NO receptor. sGC is a heterodimer composed of α- and β-subunits. Due to the electronic structure of NO, an uncharged molecule, it activates sGC by binding directly to the heme portion, leading to a conformational change that augments enzyme activity by forming a ferrous-nitrosyl-heme complex. At this point, there is conversion of GTP to cGMP followed by intracellular activation of several effectors (37). Some of the effectors were identified as 1) cGMP-dependent protein kinase, 2) cGMP-regulated phosphodiesterases, and 3) cGMP-gated ion channels (38). Although it seems that the effects of NO are cGMP dependent, in the SON the evidence is controversial. Yang and Hatton (23) have shown that cGMP enhances dye coupling and excitability of supraoptic neurons. On the other hand, studies have shown that nitrergic modulation of magnocellular neurons of the SON involves an increase in the frequency of γ-aminobutyrate (GABA)ergic events (24) and that the signal transduction is independent of cGMP (39). The same type of result was observed in the neurosecretion of vasopressin (VP) and oxytocin (OT) in experiments with awake rats (39). In brain slices of the SON, using the outside-out configuration of the patch-clamp technique, results show that NO acts directly on the N-methyl-D-aspartate receptor (NMDA) channel complex, without mobilization of cGMP (40). However, to make the subject more complicated, our group has demonstrated that nitrergic modulation can be independent of synaptic events, suggesting a possible direct action on ion channels (35). As can be seen, the pathway used by NO to change the excitability of magnocellular neurons is still a matter of debate.
An alternative pathway
The classical view of cGMP being the exclusive mediator of the effects of NO has been questioned by findings suggesting that NO can modify proteins through direct chemical reactions (41). One of those alternative pathways involves S-nitrosylation, in which the NO molecule interacts with cysteine thiol groups in a covalent bond, resulting in an S-nitrosothiol complex (Figure 2). This mechanism has been described as an important NO reaction, which preserves its biological actions, and can be expressed as a key posttranslational modification of ion channels increasing or decreasing protein activity (42,43). Thus, protein functions can be controlled by either interaction or deletion of NO from the cysteine thiol group. For more details about S-nitrosylation, see Hess et al. (42).
Figure 2. S-nitrosylation at cysteine residues. After synthesis, nitric oxide (NO) reacts with cysteine residues (red) of proteins forming a nitrosylated compound.
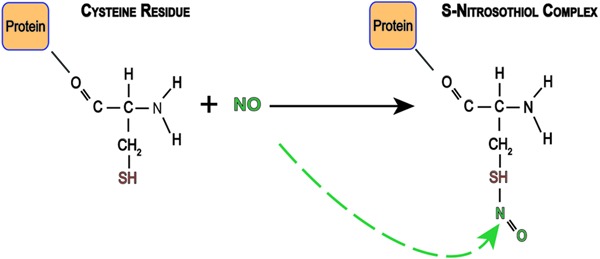
In the central nervous system, this process was first described in NMDA-type glutamate receptors, where the cysteine (residue 399) in the NR2A subunit was nitrosylated (44). However, emerging results have demonstrated that other proteins can also be nitrosylated, such as, calcium-activated potassium channels (45), cyclic nucleotide activated cation channels (46), and hyperpolarizing activated and cyclic nucleotide gated cation channels (47). In addition, Jaffrey et al. (48) verified that some proteins are endogenously nitrosylated, reinforcing the idea that this signaling mechanism for NO is of physiological significance.
NO effects on the mammalian SON
The observation that osmotic stimulation upregulated NOS as well as VP and OT mRNA expression in magnocellular neurosecretory cells (MNCs) of the SON was taken as evidence that NO could play a role as a neuromessenger involved in the control of neurohypophysial hormone secretion (16). Additional immunohistochemical evidence colocalizing NOS with VP or OT and detection of NOS mRNA, NOS protein, and its product L-citrulline in MNCs lends further support to this idea. Moreover, in vivo and in vitro studies also produced a wealth of results that revealed a significant participation of NO in the control of VP and OT secretions (49,50).
Through a dynamic approach using confocal microscopy and NO-sensitive indicators, a basal production of NO at the cellular level was detected within the SON, but not the surrounding nuclei (51). Moreover, hyperosmotic stimulation induced NO production in MNCs in slices of the SON (52). This finding adds support to the hypothesis that osmotic stimulation induces an increase in NO production as a consequence of NOS overexpression (16,53).
Controversial effects of NO on VP and OT secretion
Results concerning the central effects of NO on VP and OT plasma levels are not always coincident and are sometimes contradictory. Table 1 shows a collection of the main findings from experiments using intracerebroventricular (icv) injections of NO donors, L-arginine, or NOS blockers (L-NAME). Under physiological conditions, icv injection of L-NAME elicits an increase in both VP and OT plasma levels (54). This result indicates that NO tonically inhibits neurohormonal secretion. On the other hand, increased or unmodified effects on plasma VP levels were shown after icv injections of NO donors and L-arginine treatment (54-56).
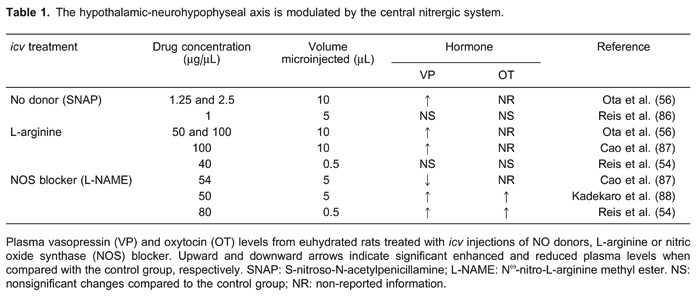
Since increased plasma levels of VP and OT were observed after blockade of endogenous NO production, it would be expected that increased NO availability, after treatment with NO donors or L-arginine, would induce opposite effects. However, similar to the blocking of endogenous NO production, a larger NO availability also increased VP and OT plasma levels. On the contrary, in vitro studies reveal different effects of NO on neurohypophysial hormone secretion. In rodent hypothalamic explants, NO suppressed VP secretion, an effect seen with NO donors SIN-1 and SNP (49,57). L-arginine also reduced VP release in this preparation, an effect reversed and reduced, respectively, by the NOS blocker L-NMMA and the addition of human hemoglobin, an NO scavenger (49). In microinjection experiments, interpretation of the results needs to take into consideration the microenvironments of the nuclei. Different brain nuclei have different sizes and can be damaged by microinjections with relatively large volumes. In situations like this, the effects observed are subjected to severe criticism because of the possibility of mechanical lesions and tissue edema. Furthermore, nuclei in the surroundings of the injection site can also be affected by the injected drug, and the final measured response may be misleading (58). A third and very important point is the concentration of drug used. As can be seen in Table 1, icv microinjections of donor and substrate of NO resulted, at the higher doses, in an increase in the release of VP. Such an effect is opposite to that observed in in vitro studies, where the release of VP was inhibited. However, in experiments where the NOS enzyme was blocked, the results obtained with microinjections are more similar to those obtained from in vitro experiments. Thus, although results from in vivo studies are controversial, findings from in vitro studies are more consistent, indicating a general inhibitory effect of NO on neurohypophysial hormone secretion. On the other hand, in dehydrated rats icv microinjections of L-NAME, an NOS blocker, induced an acute increase in OT, but not VP plasma levels, suggesting that the postulated tonic nitrergic inhibition of VP secretion is removed during dehydration (59). Such an effect was also reported after icv injection of angiotensin II (AngII), hypertonic solution treatment (60), and in hypovolemic rats (36). Besides this, NO seems to induce an increase in VP, but not in OT plasma levels induced by hypertonic blood volume expansion (61). Taken together, these findings indicate that, similar to what happens during hypovolemia, total and intracellular dehydration removes tonic inhibitory nitrergic modulation on VP neurons, but not on OT neurons. Therefore, it seems that nitrergic modulation on the hypothalamic-neurohypophysis axis can be strongly controlled by reflex responses activated by osmotic imbalance and depletion of body fluid compartments.
From the above discussions, the question that remains is: How could osmotic and volume challenges induce such diverse nitrergic effects on VP and OT secretions? It is known that dehydration and salt load induce overexpression of neuronal NOS mRNA in MNCs (53,62), a response controlled by the anteroventral third ventricular (AV3V) region (63). Thus, it is expected that 24-h dehydration would increase the levels of NO into the SON, with a consequent inhibition of VP and OT secretion. In order to address this problem, we should recall that hypovolemia, hypotension, and total dehydration, but not intracellular dehydration, significantly increase in AngII plasma levels. Circulating AngII may induce VP (64) and OT (65) secretion by acting on circumventricular organ neurons, where the blood-brain barrier is absent (66). Thus, circulating AngII may activate neurons at the subfornical organ (67), which sends axonal projections to the SON, increasing MNC activity via AngII release and activation of postsynaptic AngII receptors type-1 (AT1). This hypothesis is supported by experiments showing that administration of AT1 receptor antagonist suppresses the AngII response (68). Similarly, cellular dehydration induced by hypertonic solution activates subfornical organ neurons enhancing AngII transmission to MNCs (64). How can a blood-borne signal like AngII modulate the nitrergic system present in the SON? Experimental evidence shows that AngII could modulate nNOS mRNA expression in MNCs (69). Such modulation is dependent on nNOS activity itself, since its blockade prevented AngII-induced nNOS mRNA overexpression in the SON. Therefore, we can assume that AngII receptor (AT) activation modulates the nitrergic system in MNCs. In neurons, increased NO production as a consequence of the AT1 receptor antagonist, losartan, is a response that may be blocked by an AT2 receptor antagonist, such as PC123319 (70). This indicates that, while AngII binding to AT1 receptors inhibits tonically produced NO, binding to AT2 receptors stimulates it. In this case, NO production was exclusively dependent on nNOS (70). How could the modulation of activity of AngII receptors explain selective nitrergic inhibition on OT secretion? According to Wang et al. (70), since AngII receptor activation stimulates or inhibits NO production, the balance between AT1 and AT2 activities may be decisive for VP and OT release. However, during dehydration, icv microinjection of losartan did not induce any changes in VP and OT plasma levels. On the other hand, icv losartan plus icv L-NAME elicited increased OT secretion. It is likely that, in MNCs, enhanced NO production is the consequence of both neuronal NOS stimulation via AT2 receptor activation and reduction of NOS inhibition via AT1 inactivation by losartan. Therefore, it seems that, when AT1 receptors are inactivated, AngII activates AT2 receptors in MNCs, eliciting overactivity of NOS, increased NO production, and inhibition of VP and OT secretion. Such a hypothesis would explain the increased OT plasma levels after icv L-NAME treatment, but would not explain nonsignificant changes on VP secretion. We could speculate that AT2 receptors are absent in VP neurons, but they have been detected in rodent VP neurons (71). Thus, to this point, we may assume that AngII may induce NO production through AT2 receptors, via nNOS activation, with consequent inhibition of both VP and OT secretion. However, another question remains: Why is OT, and not VP secretion, affected by L-NAME treatment? A possible explanation emerges from studies involving stimulation of AT2 receptors and consequent production of phospholipase A2 and arachidonic acid (72). Arachidonic acid is the substrate for cyclooxygenase enzymes (COX-1 and COX-2) to produce prostaglandins (PGs), also involved in the control of neurohypophysial hormone secretion. Indeed, central microinjections of different types of PGs elicit VP and OT secretion (73,74), an effect also observed in in vitro preparations (75). Interestingly, Yamaguchi et al. (73,74) observed that, in rats, brain PGs were synthesized during hyperosmolarity and volume depletion, but not under physiological conditions, suggesting that COX enzymes are activated by osmotic stress and hypovolemia. This is a plausible assumption, since COX-2 is also a constitutively expressed enzyme in the brain (76), lending support to the hypothesis that PGs participate in selective nitrergic inhibition on OT secretion during dehydration. Since NO can directly activate COXs (77,78), we speculate that dehydration-induced NOS overexpression (79) would lead to an enhanced activation of the COX enzymes and synthesis of PGs, thereby inducing neurohypophysial hormone secretion. Central microinjection of meclofenamate, a COX inhibitor, increased VP plasma levels during osmotic stimulation and hypovolemia, an effect not observed in euvolemic and euhydrated rats. Such results indicate that osmotic and volume imbalances stimulate central synthesis of PGs, which tonically induce the secretion of VP (73,74). Interestingly, it was reported that icv microinjections of PGE2 or PGF2α elicit OT, but not VP secretion (77). The activation of SON neurons by PGE2 occurs via binding to prostanoid receptors. In the SON, electrophysiological investigations showed that PGE2 increases neuronal activity through postsynaptic PGE2 and PGF2α receptors. Indirectly, PGE2 induces MNC activation by reducing the inhibitory GABA inputs via presynaptic EP3 receptors (80). Thus, we may speculate that, during dehydration, NO and PGs are two immediately synthesized factors with opposing effects to control MNC activity and neurohormonal secretion. As NO may induce additional synthesis of PGs through direct effects on COX, PG levels may increase during dehydration in parallel with NO production. The counterbalance between both factors maintains OT and VP secretions at optimal levels. Even when icv L-NAME treatment interrupts NO production, COX enzymes S-nitrosylated by NO may maintain the PG levels. In addition, the high sensitivity of OT neurons to PGE2 and PGF2α may explain the exclusive increase in OT plasma levels after L-NAME treatment (77), since icv indomethacin microinjection suppressed L-NAME-induced OT secretion and did not change VP levels (81).
In summary, although in vivo experiments brought relevant contributions to understanding the control of neuroendocrine function by the brain nitrergic system, in vitro studies were also needed to unveil the nitrergic mechanisms controlling VP and OT secretion. In this regard it is worth noting that, although in vivo studies suggest that the activity of MNCs is modulated by synaptic neurotransmission, it is also well known that NO plays an important modulator effect by controlling their activity through both indirect (synaptic) and direct (intrinsic) mechanisms.
Indirect effects of NO on MNCs: synaptic mechanism
In vitro electrophysiological experiments were necessary to understand how NO induces changes in VP and OT plasma levels. Since release of VP and OT is correlated with the electrical activity of MNCs, several papers have investigated the effects of NO on the pattern and frequency of firing neurons of action potentials. To investigate how the effect of NO inhibits electrical activity of SON neurons, spontaneous excitatory (EPSCs) and inhibitory (IPSCs) postsynaptic currents were recorded using the whole cell patch-clamp technique in unidentified SON neurons (82). NO reversibly increased only the frequency of IPSCs and did not change the amplitudes of either IPSCs or EPSCs. Since both IPSCs and EPSCs were recorded in the presence of tetrodotoxin, the spontaneous synaptic currents are, in fact, miniature IPSCs and EPSCs representing local release of GABA and glutamate, respectively (83). Contrary to the above results, it was reported that SNP, a donor of NO, and L-arginine, precursor of NO synthesis, increase both the frequency and amplitude of GABAA miniature IPSCs in VP and OT neurons (24). This finding was taken to indicate that NO modulates GABA neurotransmission at both pre- and postsynaptic sites. Such evidence lends support to the idea that NO increases the presynaptic quantal release of GABA and the open probability (P 0) of GABAA channels, since IPSC currents were abolished by the GABAA channel blocker, picrotoxin (82). Although controversy still remains about a possible postsynaptic effect, the increase in amplitude elicited by L-arginine and the reversal of this effect by the nNOS inhibitor, 7-nitroindazole, suggest that postsynaptic GABA channel activity may be influenced by endogenous NO (24). Also, the NO donor, SNP, is known to negatively modulate glutamatergic neurotransmission in SON neurons (22). However, such evidence must be considered with caution, because it is well known that ferrocyanide ions, a byproduct of SNP photolysis, mimicked the effects of SNP on NMDA currents in neurons (25). Although Ozaki et al. (82) did not observe any effect of SNAP (NO donor) on the spontaneous EPSCs in SON neurons, there are reports that NMDA currents in striatal neurons are negatively modulated by other NO donors (27). In summary, endogenous NO acts indirectly on MNCs by enhancing fast inhibitory synaptic transmission through the induction of presynaptic GABA release, as well as by modulating the conductance and/or P 0 of postsynaptic GABAA channels.
Direct effects of NO on MNCs: intrinsic mechanisms
NO may also inhibit the electrical activity of MNCs by acting independently of inhibitory synaptic input. Such a hypothesis has arisen from experiments where the effects of NO were observed in the presence of blockers of both GABAergic and glutamatergic neurotransmissions. Under these conditions, treatment of MNCs with L-arginine reduced their firing rate while treatment with L-NAME, an NOS blocker, produced the opposite effect. These observations are clear indications that endogenous NO directly inhibits the electrical activity of MNCs, independently of an effect via GABAergic neurotransmission. A more detailed analysis of the phenomenon showed that NO promotes a slower depolarization by increasing the hyperpolarizing after potential amplitude (35), a component involved with spike frequency adaptation. Depolarization after potentials are determined by inward cation currents flowing through, but not only, hyperpolarization-activated and cyclic nucleotide-gated channels (HCN channels). Current flowing through the HCN channels (Ih) is involved in the control of neuronal rhythmic activity, also regulating the spontaneous activity of MNCs. It has been observed in our lab that NO decreases the amplitude of Ih currents. This effect on the HCN channels seems to be independent of the soluble guanylyl cyclase-cGMP pathway (da Silva MP and Varanda WA, unpublished results), since the soluble guanylyl cyclase inhibitor, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), did not prevent the effects of L-arginine. Alternative routes by which NO may modulate the kinetics of HCN channels may involve other intracellular messenger pathways (84) and/or the S-nitrosylation mechanism proposed to occur in other cell types.
Hyperosmolality and NO signaling in MNCs
Nitrergic signaling in MNCs seems also to be influenced by osmosensitive mechanisms. Blockade of NOS depolarizes the resting membrane potential both in hyperosmotic and isotonic conditions. Indeed, L-arginine significantly decreased the action potential firing rate elicited by hypertonicity, and blocking NOS induced a further increase in the frequency of action potential firing induced by the hypertonic solution. This suggests that, during the osmotic challenge, endogenous NO is synthesized, and modulates the electrical activity of MNCs (52).
Since these results were obtained without major excitatory and inhibitory synaptic input, they suggest that MNCs exhibit intrinsic osmosensitivity, which may induce the synthesis of NO. How do intrinsic osmosensitive mechanisms affect nitrergic signaling in MNCs? A cationic conductance is augmented in MNCs during hyperosmotic stress, suggesting that intrinsic osmosensitivity may directly modulate the synthesis of NO. Recently, it was reported that an N-terminal variant of the transient receptor potential cation channel subfamily V member 1 (TRPV1) is essential for osmosensory transduction in supraoptic MNCs. A splice variant of the TRPV1 channel mediated hyperosmotic stimulus-induced depolarizing potential and action potential discharge (85). As the activation of TRPV channels is known to occur during hypertonicity and may lead to NO synthesis, it is suggested that an osmotic stimulus may also indirectly elicit NO synthesis in MNCs through this pathway.
Concluding remarks
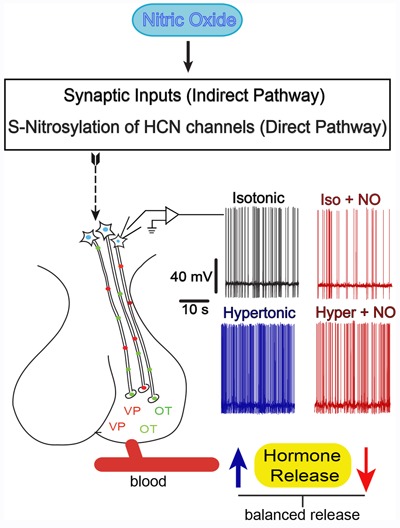
Although NO is a gas, its diffusion radius limits the extension of its actions. Thus, it is thought that endogenous NO produced within the SON represents an important neuromessenger to quickly control the activity of MNCs acting at synaptic elements and/or the MNCs themselves. NO may be seen as a fine tuner modulating MNC activity by increasing GABA neurotransmission and reducing Ih currents. In this way, NO is part of a feedback compensatory mechanism that is set to avoid overactivity of MNCs and, hence, oversecretion of VP and OT. Osmotic stress increases NO levels in MNCs, indirectly through glutamatergic neurotransmission and, directly, possibly through TRPV activation. Increased NO levels would avoid excessive MNC activation, and hence VP and OT depletion. Figure 3 and Table 2 depict the relevant points involved in the control of VP and OT secretion as described earlier.
Figure 3. Direct and indirect mechanisms involved in the control of magnocellular neurosecretory cells (MNCs) firing frequency by nitric oxide (NO). NO controls the firing rate of the MNCs in order to prevent over-secretion of the neurohypophysial hormones through an indirect (increase in synaptic inputs) and/or a direct pathway by S-nitrosylation of hyperpolarization-activated and cyclic nucleotide-gated (HCN) channels (see text). Traces shown on each side of the neurohypophysis are action potentials recorded directly from the MNCs under the conditions indicated. Increased firing rate (blue trace), induced by a hypertonic solution, is associated with an increased VP/OT secretion (blue arrow). NO decreases firing rates (red traces) in both basal and osmotic stress conditions. Voltage and time scales are the same for all records.

Acknowledgments
Research supported by FAPESP (#2012/19750-7) to W.A. Varanda. M.P. da Silva is the recipient of a fellowship from CAPES (Proex #23038.006588/2011-44) and P.L. Cedraz-Mercez from FAPESP (#2012/01859-2). W.A. Varanda is a research fellow from CNPq.
Footnotes
First published online January 17, 2014.
References
- 1.Ignarro LJ. Biosynthesis and metabolism of endothelium-derived nitric oxide. Annu Rev Pharmacol Toxicol. 1990;30:535–560. doi: 10.1146/annurev.pa.30.040190.002535. [DOI] [PubMed] [Google Scholar]
- 2.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 3.Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- 4.Derentowicz P, Markiewicz K, Wawrzyniak M, Czerwinska-Kartowicz I, Bulawa E, Siwinska-Golebiowska H. Nitric oxide (NO) - Nobel prize in medicine and physiology for 1998. Med Wieku Rozwoj. 2000;4:209–217. [PubMed] [Google Scholar]
- 5.Santos RM, Lourenco CF, Ledo A, Barbosa RM, Laranjinha J. Nitric oxide inactivation mechanisms in the brain: role in bioenergetics and neurodegeneration. Int J Cell Biol. 2012;2012: doi: 10.1155/2012/391914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 7.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 8.Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 9.Groves JT, Wang CC. Nitric oxide synthase: models and mechanisms. Curr Opin Chem Biol. 2000;4:687–695. doi: 10.1016/S1367-5931(00)00146-0. [DOI] [PubMed] [Google Scholar]
- 10.Bush PA, Gonzalez NE, Griscavage JM, Ignarro LJ. Nitric oxide synthase from cerebellum catalyzes the formation of equimolar quantities of nitric oxide and citrulline from L-arginine. Biochem Biophys Res Commun. 1992;185:960–966. doi: 10.1016/0006-291X(92)91720-B. [DOI] [PubMed] [Google Scholar]
- 11.Miyagawa A, Okamura H, Ibata Y. Coexistence of oxytocin and NADPH-diaphorase in magnocellular neurons of the paraventricular and the supraoptic nuclei of the rat hypothalamus. Neurosci Lett. 1994;171:13–16. doi: 10.1016/0304-3940(94)90592-4. [DOI] [PubMed] [Google Scholar]
- 12.Kojima H, Urano Y, Kikuchi K, Higuchi T, Hirata Y, Nagano T. Fluorescent Indicators for Imaging Nitric Oxide Production. Angew Chem Int Ed Engl. 1999;38:3209–3212. doi: 10.1002/(SICI)1521-3773(19991102)38:21<3209::AID-ANIE3209>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Malinski T, Mesaros S, Tomboulian P. Nitric oxide measurement using electrochemical methods. Methods Enzymol. 1996;268:58–69. doi: 10.1016/S0076-6879(96)68009-4. [DOI] [PubMed] [Google Scholar]
- 14.Bhat G, Mahesh VB, Aguan K, Brann DW. Evidence that brain nitric oxide synthase is the major nitric oxide synthase isoform in the hypothalamus of the adult female rat and that nitric oxide potently regulates hypothalamic cGMP levels. Neuroendocrinology. 1996;64:93–102. doi: 10.1159/000127104. [DOI] [PubMed] [Google Scholar]
- 15.Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience. 1992;46:755–784. doi: 10.1016/0306-4522(92)90184-4. [DOI] [PubMed] [Google Scholar]
- 16.Kadowaki K, Kishimoto J, Leng G, Emson PC. Up-regulation of nitric oxide synthase (NOS) gene expression together with NOS activity in the rat hypothalamo-hypophysial system after chronic salt loading: evidence of a neuromodulatory role of nitric oxide in arginine vasopressin and oxytocin secretion. Endocrinology. 1994;134:1011–1017. doi: 10.1210/en.134.3.1011. [DOI] [PubMed] [Google Scholar]
- 17.Calka J, Block CH. Relationship of vasopressin with NADPH-diaphorase in the hypothalamo-neurohypophysial system. Brain Res Bull. 1993;32:207–210. doi: 10.1016/0361-9230(93)90177-D. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto T, Bing RJ. Nitric oxide donors. Proc Soc Exp Biol Med. 2000;225:200–206. doi: 10.1046/j.1525-1373.2000.22525.x. [DOI] [PubMed] [Google Scholar]
- 19.Al-Sa'doni H, Ferro A. S-Nitrosothiols: a class of nitric oxide-donor drugs. Clin Sci. 2000;98:507–520. doi: 10.1042/CS19990267. [DOI] [PubMed] [Google Scholar]
- 20.Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, et al. Nitric oxide donors: chemical activities and biological applications. Chem Rev. 2002;102:1091–1134. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 21.Lunardi CN, da Silva RS, Bendhack LM. New nitric oxide donors based on ruthenium complexes. Braz J Med Biol Res. 2009;42:87–93. doi: 10.1590/S0100-879X2009000100013. [DOI] [PubMed] [Google Scholar]
- 22.Cui LN, Inenaga K, Nagatomo T, Yamashita H. Sodium nitroprusside modulates NMDA response in the rat supraoptic neurons in vitro . Brain Res Bull. 1994;35:253–260. doi: 10.1016/0361-9230(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 23.Yang QZ, Hatton GI. Nitric oxide via cGMP-dependent mechanisms increases dye coupling and excitability of rat supraoptic nucleus neurons. J Neurosci. 1999;19:4270–4279. doi: 10.1523/JNEUROSCI.19-11-04270.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stern JE, Ludwig M. NO inhibits supraoptic oxytocin and vasopressin neurons via activation of GABAergic synaptic inputs. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1815–R1822. doi: 10.1152/ajpregu.2001.280.6.R1815. [DOI] [PubMed] [Google Scholar]
- 25.Manzoni O, Prezeau L, Desagher S, Sahuquet A, Sladeczek F, Bockaert J, et al. Sodium nitroprusside blocks NMDA receptors via formation of ferrocyanide ions. Neuroreport. 1992;3:77–80. doi: 10.1097/00001756-199201000-00020. [DOI] [PubMed] [Google Scholar]
- 26.Dicks AP, Williams DL. Generation of nitric oxide from S-nitrosothiols using protein-bound Cu2+ sources. Chem Biol. 1996;3:655–659. doi: 10.1016/S1074-5521(96)90133-7. [DOI] [PubMed] [Google Scholar]
- 27.Manzoni O, Prezeau L, Marin P, Deshager S, Bockaert J, Fagni L. Nitric oxide-induced blockade of NMDA receptors. Neuron. 1992;8:653–662. doi: 10.1016/0896-6273(92)90087-T. [DOI] [PubMed] [Google Scholar]
- 28.Feelisch M. The use of nitric oxide donors in pharmacological studies. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:113–122. doi: 10.1007/PL00005231. [DOI] [PubMed] [Google Scholar]
- 29.Campelo MW, Campelo AP, Lopes LG, Santos AA, Guimaraes SB, Vasconcelos PR. Effects of Rut-bpy (Cis-[Ru(bpy)2(SO3)(NO)]PF 6), a novel nitric oxide donor, in L-NAME-induced hypertension in rats. Acta Cir Bras. 2011;26(Suppl 1):57–59. doi: 10.1590/S0102-86502011000700012. [DOI] [PubMed] [Google Scholar]
- 30.Wink DA, Cook JA, Pacelli R, Liebmann J, Krishna MC, Mitchell JB. Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicol Lett. 1995;8283:221–226. doi: 10.1016/0378-4274(95)03557-5. [DOI] [PubMed] [Google Scholar]
- 31.Moore PK, Handy RL. Selective inhibitors of neuronal nitric oxide synthase - is no NOS really good NOS for the nervous system? Trends Pharmacol Sci. 1997;18:204–211. doi: 10.1016/s0165-6147(97)01064-x. [DOI] [PubMed] [Google Scholar]
- 32.Bryk R, Wolff DJ. Pharmacological modulation of nitric oxide synthesis by mechanism-based inactivators and related inhibitors. Pharmacol Ther. 1999;84:157–178. doi: 10.1016/S0163-7258(99)00030-3. [DOI] [PubMed] [Google Scholar]
- 33.Moore PK, Babbedge RC, Wallace P, Gaffen ZA, Hart SL. 7-Nitro indazole, an inhibitor of nitric oxide synthase, exhibits anti-nociceptive activity in the mouse without increasing blood pressure. Br J Pharmacol. 1993;108:296–297. doi: 10.1111/j.1476-5381.1993.tb12798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H, Martasek P, Roman LJ, Masters BS, Silverman RB. N(omega)-Nitroarginine-containing dipeptide amides. Potent and highly selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- 35.Ventura RR, Aguiar JF, Antunes-Rodrigues J, Varanda WA. Nitric oxide modulates the firing rate of the rat supraoptic magnocellular neurons. Neuroscience. 2008;155:359–365. doi: 10.1016/j.neuroscience.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Kadekaro M, Terrell ML, Liu H, Gestl S, Bui V, Summy-Long JY. Effects of L-NAME on cerebral metabolic, vasopressin, oxytocin, and blood pressure responses in hemorrhaged rats. Am J Physiol. 1998;274:R1070–R1077. doi: 10.1152/ajpregu.1998.274.4.R1070. [DOI] [PubMed] [Google Scholar]
- 37.Waldman SA, Murad F. Cyclic GMP synthesis and function. Pharmacol Rev. 1987;39:163–196. [PubMed] [Google Scholar]
- 38.Vaandrager AB, de Jonge HR. Signalling by cGMP-dependent protein kinases. Mol Cell Biochem. 1996;157:23–30. doi: 10.1007/BF00227877. [DOI] [PubMed] [Google Scholar]
- 39.Terrell ML, Salas N, Bui V, Summy-Long JY, Kadekaro M. NO inhibition of the magnocellular neuroendocrine system in rats is independent of cGMP signaling pathway. Exp Neurol. 2003;184:846–856. doi: 10.1016/S0014-4886(03)00305-4. [DOI] [PubMed] [Google Scholar]
- 40.Fagni L, Olivier M, Lafon-Cazal M, Bockaert J. Involvement of divalent ions in the nitric oxide-induced blockade of N-methyl-D-aspartate receptors in cerebellar granule cells. Mol Pharmacol. 1995;47:1239–1247. [PubMed] [Google Scholar]
- 41.Ahern GP, Klyachko VA, Jackson MB. cGMP and S-nitrosylation: two routes for modulation of neuronal excitability by NO. Trends Neurosci. 2002;25:510–517. doi: 10.1016/S0166-2236(02)02254-3. [DOI] [PubMed] [Google Scholar]
- 42.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 43.Gaston BM, Carver J, Doctor A, Palmer LA. S-nitrosylation signaling in cell biology. Mol Interv. 2003;3:253–263. doi: 10.1124/mi.3.5.253. [DOI] [PubMed] [Google Scholar]
- 44.Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, et al. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- 45.Lang RJ, Harvey JR, McPhee GJ, Klemm MF. Nitric oxide and thiol reagent modulation of Ca2+-activated K+ (BKCa) channels in myocytes of the guinea-pig taenia caeci. J Physiol. 2000;525((Part 2)):363–376. doi: 10.1111/j.1469-7793.2000.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Broillet MC, Firestein S. Direct activation of the olfactory cyclic nucleotide-gated channel through modification of sulfhydryl groups by NO compounds. Neuron. 1996;16:377–385. doi: 10.1016/S0896-6273(00)80055-0. [DOI] [PubMed] [Google Scholar]
- 47.Wenker IC, Benoit JP, Chen X, Liu H, Horner RL, Mulkey DK. Nitric oxide activates hypoglossal motoneurons by cGMP-dependent inhibition of TASK channels and cGMP-independent activation of HCN channels. J Neurophysiol. 2012;107:1489–1499. doi: 10.1152/jn.00827.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 49.Yasin S, Costa A, Trainer P, Windle R, Forsling ML, Grossman A. Nitric oxide modulates the release of vasopressin from rat hypothalamic explants. Endocrinology. 1993;133:1466–1469. doi: 10.1210/en.133.3.1466. [DOI] [PubMed] [Google Scholar]
- 50.Summy-Long JY, Bui V, Mantz S, Koehler E, Weisz J, Kadekaro M. Central inhibition of nitric oxide synthase preferentially augments release of oxytocin during dehydration. Neurosci Lett. 1993;152:190–193. doi: 10.1016/0304-3940(93)90515-M. [DOI] [PubMed] [Google Scholar]
- 51.Stern JE, Zhang W. Cellular sources, targets and actions of constitutive nitric oxide in the magnocellular neurosecretory system of the rat. J Physiol. 2005;562:725–744. doi: 10.1113/jphysiol.2004.077735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.da Silva MP, Ventura RR, Varanda WA. Hypertonicity increases NO production to modulate the firing rate of magnocellular neurons of the supraoptic nucleus of rats. Neuroscience 2013; 250:70–79. doi: 10.1016/j.neuroscience.2013.06.067. [DOI] [PubMed] [Google Scholar]
- 53.Ueta Y, Levy A, Chowdrey HS, Lightman SL. Water deprivation in the rat induces nitric oxide synthase (NOS) gene expression in the hypothalamic paraventricular and supraoptic nuclei. Neurosci Res. 1995;23:317–319. doi: 10.1016/0168-0102(95)00956-6. [DOI] [PubMed] [Google Scholar]
- 54.Reis WL, Saad WA, Camargo LA, Elias LL, Antunes-Rodrigues J. Central nitrergic system regulation of neuroendocrine secretion, fluid intake and blood pressure induced by angiotensin-II. Behav Brain Funct. 2010;6:64. doi: 10.1186/1744-9081-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calka J, Block CH. Angiotensin-(1-7) and nitric oxide synthase in the hypothalamo-neurohypophysial system. Brain Res Bull. 1993;30:677–685. doi: 10.1016/0361-9230(93)90099-W. [DOI] [PubMed] [Google Scholar]
- 56.Ota M, Crofton JT, Festavan GT, Share L. Evidence that nitric oxide can act centrally to stimulate vasopressin release. Neuroendocrinology. 1993;57:955–959. doi: 10.1159/000126459. [DOI] [PubMed] [Google Scholar]
- 57.Rossi NF, Beierwaltes WH. Nitric oxide modulation of ET(B) receptor-induced vasopressin release by rat and mouse hypothalamo-neurohypophyseal explants. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1208–R1215. doi: 10.1152/ajpregu.00701.2005. [DOI] [PubMed] [Google Scholar]
- 58.Nicholson C. Diffusion from an injected volume of a substance in brain tissue with arbitrary volume fraction and tortuosity. Brain Res. 1985;333:325–329. doi: 10.1016/0006-8993(85)91586-0. [DOI] [PubMed] [Google Scholar]
- 59.Liu H, Terrell ML, Bui V, Summy-Long JY, Kadekaro M. Nitric oxide control of drinking, vasopressin and oxytocin release and blood pressure in dehydrated rats. Physiol Behav. 1998;63:763–769. doi: 10.1016/S0031-9384(97)00528-3. [DOI] [PubMed] [Google Scholar]
- 60.Kadekaro M, Summy-Long JY. Centrally produced nitric oxide and the regulation of body fluid and blood pressure homeostases. Clin Exp Pharmacol Physiol. 2000;27:450–459. doi: 10.1046/j.1440-1681.2000.03264.x. [DOI] [PubMed] [Google Scholar]
- 61.Ventura RR, Gomes DA, Reis WL, Elias LL, Castro M, Valenca MM, et al. Nitrergic modulation of vasopressin, oxytocin and atrial natriuretic peptide secretion in response to sodium intake and hypertonic blood volume expansion. Braz J Med Biol Res. 2002;35:1101–1109. doi: 10.1590/S0100-879X2002000900011. [DOI] [PubMed] [Google Scholar]
- 62.Serino R, Ueta Y, Hanamiya M, Nomura M, Yamamoto Y, Yamaguchi KI, et al. Increased levels of hypothalamic neuronal nitric oxide synthase and vasopressin in salt-loaded Dahl rat. Auton Neurosci. 2001;87:225–235. doi: 10.1016/S1566-0702(00)00279-4. [DOI] [PubMed] [Google Scholar]
- 63.Aguila FA, Oliveira-Pelegrin GR, Yao ST, Murphy D, Rocha MJ. Anteroventral third ventricle (AV3V) lesion affects hypothalamic neuronal nitric oxide synthase (nNOS) expression following water deprivation. Brain Res Bull. 2011;86:239–245. doi: 10.1016/j.brainresbull.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 64.McKinley MJ, McAllen RM, Davern P, Giles ME, Penschow J, Sunn N, et al. The sensory circumventricular organs of the mammalian brain. Adv Anat Embryol Cell Biol. 2003;172:III–122. doi: 10.1007/978-3-642-55532-9. back. [DOI] [PubMed] [Google Scholar]
- 65.Ferguson AV, Kasting NW. Angiotensin acts at the subfornical organ to increase plasma oxytocin concentrations in the rat. Regul Pept. 1988;23:343–352. doi: 10.1016/0167-0115(88)90235-2. [DOI] [PubMed] [Google Scholar]
- 66.Tanaka J, Saito H, Kaba H, Seto K. Subfornical organ neurons act to enhance the activity of paraventricular vasopressin neurons in response to intravenous angiotensin II. Neurosci Res. 1987;4:424–427. doi: 10.1016/0168-0102(87)90008-3. [DOI] [PubMed] [Google Scholar]
- 67.Okuya S, Inenaga K, Kaneko T, Yamashita H. Angiotensin II sensitive neurons in the supraoptic nucleus, subfornical organ and anteroventral third ventricle of rats in vitro. Brain Res. 1987;402:58–67. doi: 10.1016/0006-8993(87)91047-X. [DOI] [PubMed] [Google Scholar]
- 68.Jhamandas JH, Lind RW, Renaud LP. Angiotensin II may mediate excitatory neurotransmission from the subfornical organ to the hypothalamic supraoptic nucleus: an anatomical and electrophysiological study in the rat. Brain Res. 1989;487:52–61. doi: 10.1016/0006-8993(89)90939-6. [DOI] [PubMed] [Google Scholar]
- 69.Zhang L, Tong M, Xiao M, Li L, Ding J. Nitric oxide mediates feedback inhibition in angiotensin II-induced upregulation of vasopressin mRNA. Peptides. 2009;30:913–917. doi: 10.1016/j.peptides.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 70.Wang G, Coleman CG, Glass MJ, Zhou P, Yu Q, Park L, et al. Angiotensin II type 2 receptor-coupled nitric oxide production modulates free radical availability and voltage-gated Ca2+ currents in NTS neurons. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1076–R1083. doi: 10.1152/ajpregu.00571.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Coleman CG, Anrather J, Iadecola C, Pickel VM. Angiotensin II type 2 receptors have a major somatodendritic distribution in vasopressin-containing neurons in the mouse hypothalamic paraventricular nucleus. Neuroscience. 2009;163:129–142. doi: 10.1016/j.neuroscience.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu M, Gelband CH, Moore JM, Posner P, Sumners C. Angiotensin II type 2 receptor stimulation of neuronal delayed-rectifier potassium current involves phospholipase A2 and arachidonic acid. J Neurosci. 1998;18:679–686. doi: 10.1523/JNEUROSCI.18-02-00679.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamaguchi K, Hama H, Watanabe K. Possible roles of prostaglandins in the anteroventral third ventricular region in the hyperosmolality-evoked vasopressin secretion of conscious rats. Exp Brain Res. 1997;113:265–272. doi: 10.1007/BF02450324. [DOI] [PubMed] [Google Scholar]
- 74.Yamaguchi K, Hama H, Watanabe K. Possible participation of prostaglandins generated in the anteroventral third ventricular region in the hypovolemia-induced vasopressin secretion of conscious rats. Eur J Endocrinol. 1998;138:206–215. doi: 10.1530/eje.0.1380206. [DOI] [PubMed] [Google Scholar]
- 75.Negro-Vilar A, Snyder GD, Falck JR, Manna S, Chacos N, Capdevila J. Involvement of eicosanoids in release of oxytocin and vasopressin from the neural lobe of the rat pituitary. Endocrinology. 1985;116:2663–2668. doi: 10.1210/endo-116-6-2663. [DOI] [PubMed] [Google Scholar]
- 76.Hetu PO, Riendeau D. Cyclo-oxygenase-2 contributes to constitutive prostanoid production in rat kidney and brain. Biochem J. 2005;391:561–566. doi: 10.1042/BJ20050451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knigge U, Kjaer A, Kristoffersen U, Madsen K, Toftegaard C, Jorgensen H, et al. Histamine and prostaglandin interaction in regulation of oxytocin and vasopressin secretion. J Neuroendocrinol. 2003;15:940–945. doi: 10.1046/j.1365-2826.2003.01079.x. [DOI] [PubMed] [Google Scholar]
- 78.Salvemini D, Kim SF, Mollace V. Reciprocal regulation of the nitric oxide and cyclooxygenase pathway in pathophysiology: relevance and clinical implications. Am J Physiol Regul Integr Comp Physiol. 2013;304:R473–R487. doi: 10.1152/ajpregu.00355.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ueta Y, Levy A, Lightman SL. Gene expression in the supraoptic nucleus. Microsc Res Tech. 2002;56:158–163. doi: 10.1002/jemt.10020. [DOI] [PubMed] [Google Scholar]
- 80.Shibuya I, Setiadji SV, Ibrahim N, Harayama N, Maruyama T, Ueta Y, et al. Involvement of postsynaptic EP4 and presynaptic EP3 receptors in actions of prostaglandin E2 in rat supraoptic neurones. J Neuroendocrinol. 2002;14:64–72. doi: 10.1046/j.1365-2826.2002.00741.x. [DOI] [PubMed] [Google Scholar]
- 81.Kadekaro M, Terrell ML, Liu H, Bui V, Summy-Long JY. Indomethacin prevents the L-NAME-induced increase in plasma levels of oxytocin in dehydrated rats. Brain Res. 2000;877:371–373. doi: 10.1016/S0006-8993(00)02699-8. [DOI] [PubMed] [Google Scholar]
- 82.Ozaki M, Shibuya I, Kabashima N, Isse T, Noguchi J, Ueta Y, et al. Preferential potentiation by nitric oxide of spontaneous inhibitory postsynaptic currents in rat supraoptic neurones. J Neuroendocrinol. 2000;12:273–281. doi: 10.1046/j.1365-2826.2000.00448.x. [DOI] [PubMed] [Google Scholar]
- 83.Kabashima N, Shibuya I, Ibrahim N, Ueta Y, Yamashita H. Inhibition of spontaneous EPSCs and IPSCs by presynaptic GABAB receptors on rat supraoptic magnocellular neurons. Part 1J Physiol. 1997;504:113–126. doi: 10.1111/j.1469-7793.1997.113bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martinez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic Biol Med. 2011;51:17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 85.Sharif Naeini R, Witty MF, Seguela P, Bourque CW. An N-terminal variant of Trpv1 channel is required for osmosensory transduction. Nat Neurosci. 2006;9:93–98. doi: 10.1038/nn1614. [DOI] [PubMed] [Google Scholar]
- 86.Reis WL, Giusti-Paiva A, Ventura RR, Margatho LO, Gomes DA, Elias LL, et al. Central nitric oxide blocks vasopressin, oxytocin and atrial natriuretic peptide release and antidiuretic and natriuretic responses induced by central angiotensin II in conscious rats. Exp Physiol. 2007;92:903–911. doi: 10.1113/expphysiol.2007.037911. [DOI] [PubMed] [Google Scholar]
- 87.Cao L, Sun X, Shen E. Nitric oxide stimulates both the basal and reflex release of vasopressin in anesthetized rats. Neurosci Lett. 1996;221:49–52. doi: 10.1016/S0304-3940(96)13284-5. [DOI] [PubMed] [Google Scholar]
- 88.Kadekaro M, Liu H, Terrell ML, Gestl S, Bui V, Summy-Long JY. Role of NO on vasopressin and oxytocin release and blood pressure responses during osmotic stimulation in rats. Am J Physiol. 1997;273:R1024–R1030. doi: 10.1152/ajpregu.1997.273.3.R1024. [DOI] [PubMed] [Google Scholar]