

Abstract
Suid Herpesvirus 1 (SHV-1) is the etiological agent of Aujeszky’s disease (AD), which affects swine herds worldwide and causes substantial economic losses due to animal mortality and lost productivity. In order to eradicate SHV-1, vaccination programs using viruses lacking the gene encoding glycoprotein E (gE) are ongoing in several countries. These eradication programs have generated a currently unmet demand for affordable and sensitive tests that can detect SHV-1 infection, yet distinguish between infected and vaccinated pigs. To meet this demand, we used the baculovirus-insect cell system to produce immunologically authentic full-length recombinant gE protein for use in a serum ELISA assay. As previous efforts to clone the gE gene had failed due to its extremely high GC-content (75% average), we used betaine as a PCR enhancer to facilitate amplification of the entire gE gene from the Argentinian CL15 strain of SHV-1. The cloned gE gene was expressed at high levels in recombinant baculovirus-infected insect cells and reacted strongly with sera from SHV-1 infected pigs. We used the recombinant gE protein to develop a local indirect ELISA test with sensitivity and specificity comparable to currently available commercial tests. Thus, recombinant gE produced in baculovirus-infected insect cells is a viable source of antigen for the detection of SHV-1 in ELISA tests. We also provide evidence supporting a potential application of this recombinant form of gE as a SHV-1 subunit vaccine.
Keywords: Suid Herpesvirus 1, Baculovirus, Insect cell, Glycoprotein, ELISA, GC content
Introduction
Suid Herpesvirus 1 (SHV-1), also known as Pseudorabies virus (PRV), is a double-stranded DNA virus that is the etiologic agent of Aujeszky’s disease (AD) in swine [1]. AD is an economically significant swine disease characterized by a range of clinical signs, including central nervous system disorders, respiratory disease, reproductive failure and death, depending on the age, reproductive status, and immune status of the host [2]. AD eradication programs are either under way or have already been successful in several countries such as the UK, Sweden and the USA [3-5]. A major development in the control of this disease has been the use of marker vaccines consisting of viral particles lacking specific structural proteins in combination with serological tests that detect serum antibodies against these structural proteins, which enable differentiation between infected and vaccinated pigs [6]. Although vaccination is effective in reducing the circulation of wild SHV-1 and preventing the illness, it generally does not prevent infection and establishment of latency [7]. In Argentina, a voluntary vaccination program was implemented in 1998, using a gE-deleted imported vaccine. However, the vaccine and differential test were discontinued between 2001 and 2002 due to an economic crisis and the use of this vaccine is currently prohibited by Argentinian authorities (Resolution 474/2009 SENASA (National Service of Animal Health) (www.senasa.gov.ar).
Glycoprotein E (gE), also known as US8, is one of the six structural glycoproteins found in the SHV-1 viral envelope [1]. Because gE was shown to be non-essential for virus replication, recombinant vaccines can be formulated with SHV-1 particles lacking gE. One advantage of this vaccine formulation is that it enables serological discrimination between vaccinated or infected pigs in the field because infection with the wild-type virus always gives rise to detectable anti-gE antibodies [8, 9]. Moreover, although gE is not required for virus growth, it is involved in virulence, playing a role in virus spread to the central nervous system [1, 10] and in virus egression from infected cells [10].
Recombinant SHV-1 gE could be used to develop a test that can detect SHV-1 infection and distinguish between infected and vaccinated pigs. However the gE DNA coding sequence has a high guanine-cytosine content of around 75%, which makes PCR amplification extremely difficult.
In this study, we used a modified PCR method that included enhancers designed to reduce the melting temperature of GC-rich sequences [11, 12] to successfully amplify the full-length gE coding sequence. We subsequently produced the recombinant gene product in the baculovirus insect cell system (BICS), which is commonly used to produce large quantities of fully processed, antigenically active foreign glycoproteins [13]. We then used the recombinant gE produced in the BICS to develop a local indirect ELISA assay with sensitivity and specificity comparable to commercially available tests. Finally, we evaluated the immunogenicity of the recombinant gE in mice to determine its potential utility as a SHV-1 subunit vaccine.
Materials and Methods
DNA isolation
DNA was isolated from the Continuous Porcine Kidney cell line [14] (CPK) infected with the CL-15 SHV-1 strain [15] at a multiplicity of infection between 1 and 5, as follows. CPK cells were grown in 75 cm2 plastic flasks, inoculated with 1 ml of supernatant from positive cell cultures and, after extensive cytopathic effect was observed, cell pellets were washed and suspended in TEN buffer (100 mM Tris-HCl; pH7.5, 12.5 mM EDTA; pH 8.0, 150 mM NaCl, and 1% SDS). Proteinase K (Promega, Madison, WI) was added to a final concentration of 0.2 mg/ml and samples were incubated at 50° C for 4 h. The lysate was then extracted once with TE (10 mM Tris-HCl pH 8.0 and 1 mM EDTA pH 8.0) buffer-saturated phenol and once with a mixture of phenol-chloroform-isoamyl alcohol (25:24:1). Finally, total DNA was precipitated with two volumes of 99% ethanol, rinsed twice with 70% ethanol, dried and dissolved in 40 μl of sterilized distilled water. DNA was quantified and its purity was assayed by measuring absorbance at an OD260/OD280 ratio in a nanoVue Plus™ spectrophotometer (GE Healthcare, Piscataway, NJ).
Amplification of the CL-15 SHV-1 gE gene
The 1736 bp CL-15 SHV-1 gE gene was amplified in a polymerase chain reaction (PCR) with the following primers: gE-SP 5′- C ACC ATG CGG CCC TTT CTG CTG C -3′ and gE-ASP 5′- AGC GGG GCG GGC ATT CAA CAG GC -3′ (IDT, Coralville, IO). The underlined CACC sequence was introduced into the forward primer to facilitate directional cloning into the pENTR™ entry vector using the Directional TOPO® Cloning Kit, according to the manufacturer’s instructions (Life Technologies Corporation, Carlsbad, CA). The PCRs included 0.2 μl of Phusion DNA polymerase (New England Biolabs, Beverly, MA), 10 μl of Phusion GC Buffer, all four dNTPs at final concentrations of 0.2 μM each, primers at a final concentration of 1 μM each, and 10 μl of 5M betaine freebase (Sigma-Aldrich, St Louis, MO) in a total volume of 50 μl. The cycling conditions included an initial denaturation step at 98° C for 30 s, and then 30 cycles consisting of 20 s denaturation at 98° C, 15 s annealing at 58° C, and 80 s extension at 72° C, followed by a final extension for 10 min at 72° C. The PCRs were performed in a Biometra TProfessional thermal cycler (Biometra, Göttingen, Germany) and the amplification product was purified on a 1% (w/v) agarose gel in TAE buffer, stained with ethidium bromide, and extracted using a commercial kit (QIAquick Gel Extraction kit, Qiagen), according to the manufacturer’s instructions.
CL-15 SHV-1 gE sequence analysis
The purified CL-15 SHV-1 PCR product was cloned into pENTR™/D-TOPO® (Life Technologies) according to the manufacturer’s instructions. Three independent clones were sequenced with M13 forward and reverse primers and the sequences were analyzed using Vector NTI® 10.3 software (Life Technologies). The consensus sequence was used as the query for a BLASTP search with standard parameters at the NCBI interface. Hits representing full-length or near full-length SHV-1 gE sequences were downloaded (tabulated in Table 1), used to generate a multiple sequence alignment with ClustalX 2 [16], and exported to the PHYLIP format. Protdist (PHYLIP Version 3.69 [17]) was used to generate a distance matrix from the multiple sequence alignment with the Jones-Taylor-Thornton model. An unrooted tree was then produced from the distance matrix using the neighbor-joining method (Neighbor; PHYLIP package) and drawn using the PHYLIP drawtree postscript generator.
Table 1.
Identities of full-length or near full-length gE protein sequences deposited in GenBank™ compared to the CL-15 strain gE protein, and the countries in which they were isolated
| Strain | Identity (residues/total) |
Genbank™ Accession # |
Ref. | Origin |
|---|---|---|---|---|
| NiA1 | 575/577 | ACU43472.1 | - | Spain |
| NiA3 | 574/577 | ACA97995.1 | - | Spain |
| Consensus* | 575/577 | YP_068389.1 | [40] | - |
| Rice | 574/577 | P08354.1 | [41] | USA |
| 75V19 | 572/577 | ACU43469.1 | - | Belgium |
| Kaplan | 571/577 | AEM64041.1 | [42] | Hungary |
| 89V87 | 568/577 | ACU43470.1 | - | Belgium |
| GZ-Z1 | 566/577 | AEH93984.1 | [43] | China |
| 00V72 | 567/577 | ACU43468.1 | - | Belgium |
| NS374 | 566/577 | ACU43471.1 | - | Belgium |
| Min-A | 546/579 | AAO11838.1 | - | China |
| Guangdong | 531/556 | AAK95640.1 | [32] | China |
| Yangsan | 555/577 | AAP04400.1 | - | S. Korea |
| Guizhou-DY | 550/577 | AFS64557.1 | - | China |
| LXB6 | 556/579 | ADN78282.1 | - | China |
| P-PrV | 554/578 | ACI24005.1 | - | Malaysia |
| HB/LF | 549/579 | AGF86404.1 | - | China |
| HB/HD | 550/579 | AGF86402.1 | - | China |
| Ea | 554/578 | AAD51327.1 | - | China |
| HNJZ | 548/579 | ACB37376.1 | - | China |
| HB/HS | 548/579 | AGF86403.1 | - | China |
| HB/BD | 548/579 | AGF86401.1 | - | China |
| LXB88 | 552/578 | ADN78283.1 | - | China |
| PRV-SH | 549/577 | AAF19200.1 | - | China |
| LA | 545/579 | AAN65185.1 | - | China |
| Fa | 525/558 | AAK95639.1 | [44] | China |
strains Kaplan, Becker, Rice, Indiana-Funkhauser, NIA-33 and TNL
Recombinant baculovirus production and analysis
A recombinant baculovirus encoding CL-15 SHV-1 gE was isolated in Sf9 cells using a consensus pENTR™/D-TOPO® clone and the BaculoDirect™ C-Term Baculovirus Expression System (Life Technologies) according to the manufacturer’s instructions. Recombinant baculovirus clones were plaque-purified once in Sf9 cells, tested for gE expression by immunoblotting, and a clone expressing the full-length, membrane bound version of the gE protein with C-terminal 6xHis and V5 epitope tags was identified and designated AcgE. Immunoblotting analysis was performed using AcgE-infected High Five™ cell [18] lysates prepared in Laemmli sample buffer [19] at three days post-infection. Duplicate samples were resolved on discontinuous 10% SDS-PAGE gels, one was stained with Coomassie Brilliant Blue G-250 and the other was used for immunoblotting with a mouse anti-gE SHV-1 monoclonal antibody (courtesy of Dr Barbara Klupp, Federal Research Center for Virus Disease of Animals, Germany) or swine polyclonal SHV-1 as the primary and horseradish peroxidase (HRP)-conjugated rabbit anti-mouse antibody or rabbit anti-swine antibody (Sigma Aldrich, St. Louis, MO) as the secondary antibodies. After three washes with PBS-T 3,3 diaminobenzidine tetrahydrochloride (DAB, Sigma Chemical, was used to visualize the recombinant protein.
Recombinant protein (gEr) purification
gEr was extracted from 2 × 106 AcgE-infected High Five™ cells with 5 ml of lysis buffer (PBS containing 0.3% SDS) lysed and homogenised by repeated drawing through a narrow 18-gauge needle with a syringe and purified by immobilized metal affinity chromatography using the Ni-NTA Purification System (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Eluted fractions were analyzed for gE by immunoblotting, as described above. Protein concentration was determined by the Bradford total protein content assay using the Bio-Rad Protein Assay kit (Bio-Rad) with bovine serum albumin (BSA) as the standard.
Standardization of the indirect ELISA-gEr
Optimal dilutions of gEr and sera were determined by a checkerboard titration test with SHV-1 positive and negative sera previously analyzed by virus neutralization. The antigen was coated in 96-well ELISA plates (PolySorb, Nunc, Roskilde, Denmark) ranging in concentration from 8 to 0.0156 μg/ml diluted in 50 mM carbonate/bicarbonate buffer (pH 9.6). Reference positive and negative sera were both diluted serially from 1:4 to 1:32 and tested to determine the optimal serum dilution. The dilutions that gave the maximum difference in absorbance at 405 nm between positive and negative sera were selected to test the sera panel. The working dilution of rabbit anti-swine HRP-IgG (Sigma Aldrich, St. Louis, MO), the reaction temperature, time and other conditions also were optimized.
ELISA-gEr procedure
A panel of 94 swine serum samples previously analyzed by virus neutralization was obtained from the serum bank of the Virology Laboratory of the Veterinary Faculty of La Plata City (Buenos Aires, Argentina) and used for ELISA tests with the gEr protein as the antigen (ELISA-gEr). Microtiter plates were coated with 100 μl of 0.25 μg/ml gE protein in 50 mM carbonate/bicarbonate buffer pH 9.6, and incubated overnight at 4° C. After removing excess unbound antigen, 100 μl of blocking solution (PBS - 0.1% bovine serum albumin) were added to each well and incubated for 1 h at 37° C. Plates were washed five times with PBS - 0.5% Tween-20 (PBS-T). After washing, 50 μl of serum panel samples diluted 1/8 in PBS-T containing 0.5% skim milk powder (PBS-T–SMP) were added and incubated for 1 h at 37°C. After three washes, 50 μl of rabbit-anti-swine IgG HRP conjugated (Sigma Aldrich, St. Louis, MO) diluted 1:2000 in PBST-SMP, were added. Finally, after four washes, 100 μl of 1 mM 2,2-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) ABTS (Sigma Aldrich, St. Louis, MO) substrate solution was added to each well and incubated at room temperature for 40 min. The optical density (OD) at 405 nm was determined using an automatic ELISA reader. Concordance testing was performed between the ELISA-gE and VN test. In order to determine the cut-off value of the ELISA-gE, the OD values were analyzed with Stata® SE 9.2 software (Stata Corporation). In the first step, the values obtained were analyzed by ROC (Receiver Operating Characteristic) curve where the true positive rate (Sensitivity) is plotted as a function of the false positive rate (100–Specificity) for different cut-off points. Each point on the ROC plot represents a sensitivity/specificity pair corresponding to a particular decision threshold. With the purpose of selecting the optimal cut-off value, sensitivity (Se) and specificity (Sp) were analyzed and compared with the gold standard neutralization values using Win Episcope 2.0 [20].
Determination of immunogenicity by mouse inoculation
Five male Balb/c mice were intraperitoneally injected with 2.5 × 104 AcgE-infected High Five™ cells that had been suspended in PBS and emulsified with an equal volume of Freund’s adjuvant [21]. Two mice were injected with PBS alone as negative controls. All animals were bled at 28 days post-immunization and the sera were tested in an immunoblotting assay with semi-purified SHV-1 obtained from a CPK cell line culture as the target [22]. Briefly, local SHV-1 CL-15 strain was propagated in CPK cell cultures. The virus culture was harvested when 90% of the cells exhibited cytopathic effect, and the culture was clarified by centrifugation. Nonidet P-40 at a concentration of 1% in Tris ethylenediaminetetraacetic acid (EDTA)-saline buffer was used to lyse and solubilize the pellet of SHV-1-infected cells. These lysates were used as the source of antigen for western blot.
Results
PCR amplification and sequencing of the SHV-1 gE gene
The SHV-1 gE open reading frame (ORF) is extremely GC-rich, with an average of 74.8%, a low of approximately 56%, and a high of approximately 94% GC when analyzed by a sliding window approach (50 base pair window, Fig. 1a). Previous attempts to amplify the gE ORF by PCR consistently failed, presumably due to its high GC content (data not shown). Thus, we adopted a PCR method specifically designed to amplify high GC sequences [12]. Analysis of the reaction products by agarose gel electrophoresis and ethidium bromide staining revealed a specific amplimer of the expected size (approximately 1700 bp; Fig. 1b). This result indicated that adding 1M betaine to the PCR enabled amplification of the GC-rich SHV-1 gE ORF. The PCR amplimer was subsequently gel purified, cloned into the pENTR™/D-TOPO® Gateway entry vector and three independent clones were sequenced. The CL-15 SHV-1 gE nucleotide and protein sequences were deposited in GenBank (Accession Numbers AET36924.1 and JF460026.1, respectively). BLASTp analysis revealed that the CL-15 gE protein is similar to gE proteins encoded by several other SHV-1 strains isolated across the world (Table 1). Surprisingly, further sequence analysis using the clustering algorithms of the PHYLIP package showed that gE proteins from the various strains listed in Table 1 cluster according to the geographical site from which they were isolated (Fig. 2). This analysis revealed that all SHV-1 isolates and strains from Asia are distinct from SHV-1 isolates and strains isolated in the Western hemisphere, with the only exception being the GZ-Z1 strain. The CL-15 strain gE sequence reported here was found to be most closely related to the Rice and NiA strains isolated in the USA and Spain, respectively.
Figure 1.
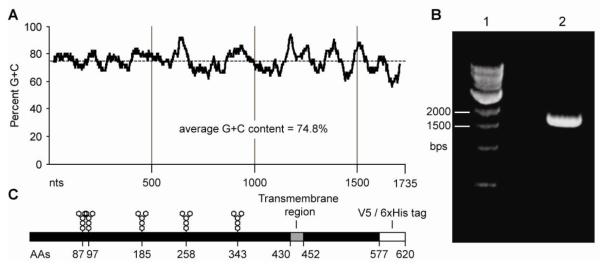
gE structural features and PCR amplification (A) GC content of the gE gene. 50 bp sliding window scan of the gE gene open reading frame. The overall GC content is 75%, ranging from 56% to a high of 94% (B) PCR amplification of the gE gene using betaine as a PCR enhancer. Lane 1: 1000 bp marker (NEB), Lane 2: PCR reaction. (C) Diagram of the recombinant gE gene product. N-linked glycosylation sites, the predicted transmembrane domain, as well as the position of tags picked up from the baculoviral vector are indicated.
Figure 2.

Unrooted tree showing relationships between gE protein sequences from various SHV-1 isolates. The sequences fall into two distinct clusters: a cluster comprising isolates from the Western world (on the left) and a cluster with isolates from Asia (on the right). The CL-15 isolate from the present study (bold) clusters with the Western world group.
Expression and purification of gEr
A consensus Gateway entry clone was used to produce a baculovirus encoding a C-terminally 6X His-tagged form of the full-length gE protein under the transcriptional control of the polyhedrin promoter (AcgE). AcgE-infected High Five cell extracts were then prepared and analyzed for the presence of gE by SDS-PAGE and immunoblotting analyses (Fig.3). The AcgE-infected cell extracts included a protein of slightly more than 72 kDa, which was absent in mock-infected cell extracts. The identity of this protein as gE was confirmed by immunoblotting with monoclonal and polyclonal anti-SHV-1 gE antibodies (Fig. 3a). After purification using Ni-NTA, immunoblotting with an anti-SHV-1 gE monoclonal antibody revealed a specific band of the same size (Fig. 3b and 3c). The positive immunoblotting results suggested that we could use the recombinant SHV gE expressed and purified in baculovirus-infected insect cells to develop an ELISA test. A total of 80 μg/ml of purified recombinant protein was obtained.
Figure 3.

SDS-PAGE and Western Blot analysis of recombinant gE protein expressed in High Five™ cells. M: prestained molecular weight marker. (A) Cell lysate obtained from cells infected with Ac-gE: 1: anti-SHV-1 polyclonal serum; 2 and 4: anti-SHV-1 monoclonal serum; 3: mock infected cells with polyclonal anti-SHV-1 serum. (B) Recombinant gE protein purified by Ni-NTA immobilized metal affinity chromatography probed with a monoclonal anti-SHV-1 antibody. (C) SDS-PAGE gel of gEr protein after purification (5)
ELISA-gEr
Optimization experiments were performed to examine the utility of the gEr protein as a diagnostic antigen for an indirect ELISA assay (ELISA-gEr). ROC analysis of the ELISA-gEr results showed that the area under the curve was 0.9236. Further detailed analysis of the sensitivity and specificity of our ELISA-gEr using the STATA program yielded a recommended cut-off value of 0.55, at which the optimum number of serum samples could be classified correctly (88.54%). This cut-off value corresponded to a high Sensitivity (91.94 %) combined with a good Specificity (82.35 %) (Fig. 4, Fig. 5). We then compared the results obtained using our newly developed ELISA-gEr to those obtained with a virus neutralization test using a two-sided contingency table. This analysis showed that 56 out of 94 pig serum samples were positive in both assays, five serum samples that scored positive by neutralization scored negative with the ELISA-gEr, and six samples that scored negative by neutralization scored positive with the ELISA-gEr. Using the selected cut-off value, we obtained a Kappa value of 0.74, (CI 95% 0.608-0.888), which indicated substantial agreement between the two methods [23] (Table 2).
Figure 4.
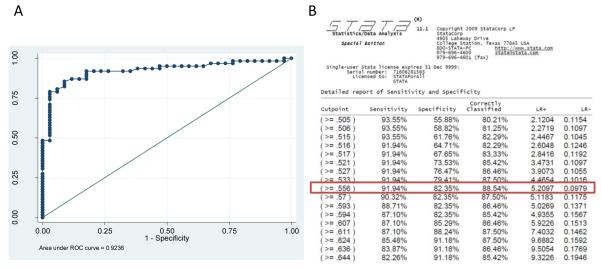
B>: Statistical analysis of ELISA-gEr results (A) ROC analysis using STATA SE 9.2 statistical analysis software (CI 95% 0.855-0.970). (B) report of sensitivity and specificity by STATA program. Cut-off value of 0.55 correctly classify serum samples in the maximum value (88.54%) with the highest Se (91.94 %) combined with a good Sp (82.35 %)
Figure 5.

Characterization of pig field serum samples using ELISA-gEr. Cut-off values of the assay are indicated as dotted lines (0.55)
Table 2.
Results of antibody detection in field serum samples using virus neutralization test and ELISA-gEr (cut-off 0.55)
| VN positive | VN negative | Total | |
|---|---|---|---|
| gE-ELISA positive | 56 | 6 | 62 |
| gE-ELISA negative | 5 | 27 | 32 |
|
| |||
| Total | 61 | 33 | 94 |
gEr Immunogenicity
Finally, in order to determine the potential utility of baculovirus-expressed gE protein as a SHV-1 subunit vaccine, we assayed the ability of gEr to elicit an immune response and trigger the production of anti-gE antibodies. We immunized five mice with AcgE infected High-Five™ cells and used immunoblotting to compare their serum reactivity against a semi-purified SHV-1 virus extract to that of sera from two mock-immunized mice. The results showed that sera from all five immunized, but not mock-immunized mice reacted strongly with a protein whose relative electrophoretic mobility corresponded to the size of the native SHV-1 gE gene product (Fig. 6).
Figure 6.

Immunogenicity of the BICS-produced recombinant gE protein in mice detected by Western blot.. M: prestained molecular weight marker; 1 – 5: mice immunized with Ac-gE infected insect cells (High Five™). C: mouse inoculated with mock infected cells as negative control
Discussion
A sensitive diagnostic immunoassay that can discriminate between SHV-infected pigs and those immunized with an attenuated, gene-deleted SHV-1 vaccine has substantial value in the swine industry. Ideally, such an assay relies not on antibody binding to an antigen containing only one or a few epitopes, but on binding to the whole, correctly processed antigen. Previously published diagnostic SHV-1 immunoassays were based on SHV-1 gE protein fragments expressed in a number of systems including the BICS [6, 24], E. coli [25], and yeast [9]. In the present study, we extended these results by expressing the full-length SHV-1 Argentinian strain CL-15 gE protein in the BICS and using the product to develop a local indirect ELISA.
A major reason for our success in this project was the use of a modified PCR protocol specifically designed to amplify GC-rich templates, which enabled us to amplify the full-length CL-15 SHV-1 gE ORF (1735 bps, average GC content 75%). Previous attempts to PCR amplify the gE gene ORF had failed, presumably due to its extremely high GC content. Our modified PCRs included 1 M betaine, which reduces DNA melting temperatures and reduces the formation of secondary structures [11, 12]. In addition, we used Phusion™ DNA polymerase, which can withstand a higher denaturation temperature (98°C) than Taq DNA polymerase (95°C). Thus, the use of betaine and higher temperatures permitted by Phusion™ DNA polymerase apparently provided more effective denaturation, which facilitated amplification of the GC-rich template and enabled us to produce adequate amounts of a specific PCR product.
The ability to obtain the SHV-1 CL-15 strain gE amplification product allowed us to clone it into pENTR™/D-TOPO® for subsequent sequence analysis. BLAST searches [26] revealed the CL-15 strain gE sequence is very similar to other gE sequences deposited in GenBank. We also found that CL-15 gE is more similar to the gE proteins from Western isolates than to Asian isolates of SHV-1. This suggests that gE proteins from Asian SHV-1 isolates are genetically distinct from gE proteins encoded by SHV-1 strains found in the Western world, which has not been noted in previous studies [27, 28]. SDS-PAGE analysis of AcgE-infected High Five cell lysates revealed the presence of a protein around 72 kDa, which was not present in lysates from control baculovirus-infected cells and was immunoreactive with monoclonal and polyclonal SHV-1 gE-specific antibodies.
We subsequently expressed and purified the full-length recombinant gE protein using a standard immobilized metal affinity approach and used it in conjunction with a large panel of sera from infected and vaccinated pigs to develop a local diagnostic ELISA assay. We showed that this new assay could be used to detect positive and negative sera with similar sensitivity and specificity compared to an established viral neutralization testing method. At the selected 0.55 cutoff level, our assay was in agreement in 88.54% of cases. It also identified 5/56 serum samples that scored positive by virus neutralization test as false negatives. One possible explanation is that those animals were never infected but, instead, were vaccinated before 2002 with vaccine consisting of gE-deleted SHV-1. Our local indirect ELISA also identified six negative serum samples by virus neutralization as positive, which probably reflects the higher sensitivity of the ELISA assay relative to the virus neutralization assay. Nonetheless, the Kappa value obtained after concordance analysis (0.748) revealed a high level of agreement between the results obtained using both tests [23].
The ability to distinguish between swine infected with naturally occurring SHV-1 strains and swine vaccinated with inactivated, gE-gene deleted SHV-1 continues to be of great importance for Aujeszky’s disease control worldwide. Although AD has been eradicated in some countries including the USA [29] and Germany [30], outbreaks continue to occur with some frequency in Asian and South American countries, including Argentina. Apart from the threat that SHV-1 poses to the swine industry in countries in which it has not yet been eradicated, the continued presence of SHV-1 in commercial swine herds also presents a virus reservoir that could allow spread to other countries. This unacceptable risk lends urgency to continue and complete eradication campaigns. An integral part of such campaigns is the need to develop ELISAs that can detect antibodies against the SHV-1 gE protein, as a complement to vaccines formulated using gE-defective viruses. However, current expression platform limitations impair inexpensive production of gE-derived antigens. Moreover, available antigens that differ from native full-length proteins can result in immunoassays with reduced sensitivity and specificity.
The success of a program focused on eradicating Aujeszky’s disease using gene-deleted vaccines and serological tests depends on the ability of those tests to discriminate between infected and vaccinated pigs. Furthermore, the tests will need to be specific and sensitive. The sensitivity of diagnostic tests can be evaluated in different ways, including their ability to detect the early humoral response to infection, latency in infected pigs, or low levels of antibodies at any stage of the infection [31]. Both full length gE and its immunodominant epitopes have been suggested for use as antigens in serological tests designed to discriminate between naturally infected and vaccinated animals [6, 25, 32]. Reported difficulties in the detection of antibodies to gE include false-negative results, false-positive results, nonspecific reactions, and high rates of doubtful test results [32] [33]. A successful eradication campaign involving vaccination would result in very low seroprevalence, which would be expected to give rise to a high rate of false-positive test results [24].
In addition, the high rate of gE antigenic drift [34, 35] and variation in the epitope-specific immune response to gE [36-38] may produce loss of sensitivity of diagnostic assays due to difficulty in detecting antibodies raised against native field versions of the protein. In our previous report, we developed an ELISA test using the immunodominant epitopes of gE SHV-1 expressed in the BICS as an antigen, however, that test was compromised by low sensitivity [39]. Based on this observation, we decided to use the entire gE protein, and speculated that the presence of the native environment surrounding might allow better folding of the recombinant product. The preliminary results obtained with recombinant full-length gE as the antigen for a new ELISA assay were very promising and demonstrate its potential as a standard tool for diagnosis. We expect to further improve the performance of our ELISA-gEr with continued development and validation of the technique, including a larger number of serum samples. These features make this recombinant antigen an attractive option for SHV-1 diagnostics opening the possibility of developing unexpensive diagnostic tests that can be used as molecular tools for large epidemiological studies in Argentina.
Finally, we performed a preliminary experiment to examine the potential utility of AcgE for the production of a SHV-1 subunit vaccine, which would be easier and cheaper to produce than attenuated or inactivated virus. As anti-gE antibodies have been shown to have the capacity to neutralize SHV-1 virus, immunization with recombinant gE could provide effective SHV-1 protection. Our results indicated that vaccinated mice mounted a robust immune response against SHV-1 gE, suggesting that insect cells infected with AcgE could be useful as a SHV-1 subunit vaccine.
Acknowledgements
This work was supported by the Faculty of Veterinary Sciences of La Plata University, Argentina, and by Award no. R01GM49734 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences, or the National Institutes of Health. The authors thank Dr. Barbara Klupp at the Federal Research Center for Virus Disease of Animals, Germany, for providing the mouse anti-gE SHV-1 monoclonal antibody.
References
- [1].Mettenleiter TC. Aujeszky’s disease (pseudorabies) virus: the virus and molecular pathogenesis - state of the art, June 1999. Vet Res. 2000;31:99–115. doi: 10.1051/vetres:2000110. [DOI] [PubMed] [Google Scholar]
- [2].Kluge J, Beran G, Hill H, Platt K. Pseudorabies. In: Leman A, Straw B, Mengeling W, D’Allaire S, Taylor D, editors. Diseases of Swine. Iowa State University Press; Ames, Iowa: 1999. pp. 312–323. [Google Scholar]
- [3].Pensaert M, Labarque G, Favoreel H, Nauwynck H. Aujeszky’s disease vaccination and differentiation of vaccinated from infected pigs. Dev Biol (Basel) 2004;119:243–254. [PubMed] [Google Scholar]
- [4].Pensaert M, Morrison R, B. Challenges of the final stages of the ADV eradication program. Vet Res. 2000;31:141–145. doi: 10.1051/vetres:2000112. [DOI] [PubMed] [Google Scholar]
- [5].I.O.o.E.B.S. Commission, I.O.o.E.I. Committee . Aujeszky’s Disease, Manual of diagnostic tests and vaccines for terrestrial animals: mammals, birds and bees. Office international des épizooties; 2012. [Google Scholar]
- [6].Gomez-Sebastian S, Perez-Filgueira DM, Gomez-Casado E, Nunez MC, Sanchez-Ramos I, Tabares E, Escribano JM. DIVA diagnostic of Aujeszky’s disease using an insect-derived virus glycoprotein E. J Virol Methods. 2008;153:29–35. doi: 10.1016/j.jviromet.2008.06.017. [DOI] [PubMed] [Google Scholar]
- [7].Wittmann G. Spread and control of Aujeszky’s disease (AD) Comp Immunol Microbiol Infect Dis. 1991;14:165–173. doi: 10.1016/0147-9571(91)90129-2. [DOI] [PubMed] [Google Scholar]
- [8].van Oirschot JT, Gielkens AL, Moormann RJ, Berns AJ. Marker vaccines, virus protein-specific antibody assays and the control of Aujeszky’s disease. Vet Microbiol. 1990;23:85–101. doi: 10.1016/0378-1135(90)90139-m. [DOI] [PubMed] [Google Scholar]
- [9].Ao JQ, Wang JW, Chen XH, Wang XZ, Long QX. Expression of pseudorabies virus gE epitopes in Pichia pastoris and its utilization in an indirect PRV gE-ELISA. J Virol Methods. 2003;114:145–150. doi: 10.1016/j.jviromet.2003.09.012. [DOI] [PubMed] [Google Scholar]
- [10].Fuchs W, Rziha HJ, Lukacs N, Braunschweiger I, Visser N, Lutticken D, Schreurs CS, Thiel HJ, Mettenleiter TC. Pseudorabies virus glycoprotein gI: in vitro and in vivo analysis of immunorelevant epitopes. J Gen Virol. 1990;71(Pt 5):1141–1151. doi: 10.1099/0022-1317-71-5-1141. [DOI] [PubMed] [Google Scholar]
- [11].Henke W, Herdel K, Jung K, Schnorr D, Loening SA. Betaine Improves the PCR Amplification of GC-Rich DNA Sequences. Nucleic Acids Research. 1997;25:3957–3958. doi: 10.1093/nar/25.19.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shi X, Jarvis DL. A new rapid amplification of cDNA ends method for extremely guanine plus cytosine-rich genes. Analytical Biochemistry. 2006;356:222–228. doi: 10.1016/j.ab.2006.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Luckow VA. Trends in the Development of Baculovirus Expression Vectors. Nature Biotechnology. 1988;6:47. [Google Scholar]
- [14].Komaniwa H, Fukusho A, Shimizu Y. Micro method for performing titration and neutralization test of hog cholera virus using established porcine kidney cell strain. Natl Inst Anim Health Q (Tokyo) 1981;21:153–158. [PubMed] [Google Scholar]
- [15].Echeverria MG, Norimine J, Galosi CM, Oliva GA, Etcheverrigaray ME, Nosetto EO, Tohya Y, Mikami T. The genotype of Aujeszky’s disease viruses isolated in Argentina. J Vet Med Sci. 1994;56:985–987. doi: 10.1292/jvms.56.985. [DOI] [PubMed] [Google Scholar]
- [16].Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- [17].Felsenstein J. PHYLIP. ver. 3.69 Univ of Washington; 2009. [Google Scholar]
- [18].Wickham TJ, Davis T, Granados RR, Shuler ML, Wood HA. Screening of insect cell lines for the production of recombinant proteins and infectious virus in the baculovirus expression system. Biotechnol Prog. 1992;8:391–396. doi: 10.1021/bp00017a003. [DOI] [PubMed] [Google Scholar]
- [19].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- [20].Thrusfield M, Ortega C, De Blas I, Noordhuizen J, Frankena K. WIN EPISCOPE 2.0: improved epidemiological software for veterinary medicine. Veterinary Record. 2001;148:567–572. doi: 10.1136/vr.148.18.567. [DOI] [PubMed] [Google Scholar]
- [21].Tami C, Peralta A, Barbieri R, Berinstein A, Carrillo E, Taboga O. Immunological properties of FMDV-gP64 fusion proteins expressed on SF9 cell and baculovirus surfaces. Vaccine. 2004;23:840–845. doi: 10.1016/j.vaccine.2004.03.070. [DOI] [PubMed] [Google Scholar]
- [22].Echeverria MG, Nosetto EO, Etcheverrigaray ME. Evaluation of a blocking ELISA using a urease conjugate for the detection of antibodies to pseudorabies virus. J Vet Diagn Invest. 2000;12:266–268. doi: 10.1177/104063870001200313. [DOI] [PubMed] [Google Scholar]
- [23].Dohoo IR, Martin W, Stryhn H. Screening and Diagnostic tests, Veterinary epidemiologic research. AVC Incorporated; 2010. [Google Scholar]
- [24].Kimman TG, de Leeuw O, Kochan G, Szewczyk B, van Rooij E, Jacobs L, Kramps JA, Peeters B. An indirect double-antibody sandwich enzyme-linked immunosorbent assay (ELISA) using baculovirus-expressed antigen for the detection of antibodies to glycoprotein E of pseudorabies virus and comparison of the method with blocking ELISAs. Clin Diagn Lab Immunol. 1996;3:167–174. doi: 10.1128/cdli.3.2.167-174.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yong T, Huan-chun C, Shao-bo X, Ya-li Q, Qi-gai H, Yu-qi R. Development of a latex agglutination test using the major epitope domain of glycoprotein E of pseudorabies virus expressed in E. coli to differentiate between immune responses in pigs naturally infected or vaccinated with pseudorabies virus. Vet Res Commun. 2005;29:487–497. doi: 10.1007/s11259-005-1865-4. [DOI] [PubMed] [Google Scholar]
- [26].Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Serena M, Metz G, OCAMPOS GM, Gambaro S, Mórtola E, Echeverría M. Characterization of Suid herpesvirus 1 field isolates from Argentina. Revista Argentina de Microbiología. 2010;42:179–183. [PubMed] [Google Scholar]
- [28].Serena MS, Metz GE, Mórtola EC, Echeverría MG. Phylogenetic analysis of Suid Herpesvirus 1 isolates from Argentina. Veterinary Microbiology. 2011;154:78–85. doi: 10.1016/j.vetmic.2011.06.028. [DOI] [PubMed] [Google Scholar]
- [29].Anderson LA, Animal US, Service PHI. Pseudorabies (Aujeszky’s Disease) and Its Eradication: A Review of the U.S. Experience. U.S. Department of Agriculture, Animal and Plant Health Inspection Service; 2008. [Google Scholar]
- [30].Müller T, Bätza HJ, Schlüter H, Conraths F, Mettenleiter T. Eradication of Aujeszky’s disease in Germany. Journal of Veterinary Medicine, Series B. 2003;50:207–213. doi: 10.1046/j.1439-0450.2003.00666.x. [DOI] [PubMed] [Google Scholar]
- [31].Kinker DR, Swenson SL, Wu L-L, Zimmerman JJ. Evaluation of serological tests for the detection of pseudorabies gE antibodies during early infection. Veterinary Microbiology. 1997;55:99–106. doi: 10.1016/s0378-1135(96)01308-9. [DOI] [PubMed] [Google Scholar]
- [32].Ao J, Lou G, Yang L, Long Q, Wang X. Cloning and sequencing of gE gene fragment exclude signal peptide of pseudorabies virus Guangdong strain. Acta scientiarum naturalium Universitatis Sunyatseni. 2001;40:115–117. [Google Scholar]
- [33].Morenkov FN. OS, Fodor I, Indirect ELISAs based on recombinant and affinity-purified glycoprotein E of Aujeszky’s Disease virus to differentiate between vaccinated and infected animals. Acta Vet Hung. 1999;47:137–150. doi: 10.1556/AVet.47.1999.1.15. [DOI] [PubMed] [Google Scholar]
- [34].Ben-Porat T, DeMarchi JM, Lomniczi B, Kaplan AS. Role of glycoproteins of pseudorabies virus in eliciting neutralizing antibodies. Virology. 1986;154:325–334. doi: 10.1016/0042-6822(86)90458-7. [DOI] [PubMed] [Google Scholar]
- [35].Mettenleiter TC, Schreurs C, Thiel HJ, Rziha HJ. Variability of pseudorabies virus glycoprotein I expression. Virology. 1987;158:141–146. doi: 10.1016/0042-6822(87)90247-9. [DOI] [PubMed] [Google Scholar]
- [36].Jacobs L, Kimman TG. Epitope-specific antibody response against glycoprotein E of pseudorabies virus. Clinical and Diagnostic Laboratory Immunology. 1994;1:500–505. doi: 10.1128/cdli.1.5.500-505.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jacobs L, Moonen-Leusen BM, Bianchi AT, Kimman TG. Glycoprotein gI of pseudorabies virus: epitope-specific antibody response in mice and pigs. Acta Vet Hung. 1994;42:347–351. [PubMed] [Google Scholar]
- [38].Katz JB, Pederson JC. Analysis of glycoprotein I (gI) negative and aberrant pseudorabies viral diagnostic isolates. Am J Vet Res. 1992;53:2259–2263. [PubMed] [Google Scholar]
- [39].Serena MS, Metz GE, Corva SG, Mortola EC, Echeverria MG. A differential ELISA based on recombinant immunodominant epitopes of the gE gene of SHV-1 in a baculovirus-insect cell system to discriminate between pigs infected naturally with pseudorabies and vaccinated pigs. J Virol Methods. 2011;171:388–393. doi: 10.1016/j.jviromet.2010.12.005. [DOI] [PubMed] [Google Scholar]
- [40].Klupp BG, Hengartner CJ, Mettenleiter TC, Enquist LW. Complete, annotated sequence of the pseudorabies virus genome. J Virol. 2004;78:424–440. doi: 10.1128/JVI.78.1.424-440.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Petrovskis EA, Timmins JG, Post LE. Use of lambda gt11 to isolate genes for two pseudorabies virus glycoproteins with homology to herpes simplex virus and varicella-zoster virus glycoproteins. J Virol. 1986;60:185–193. doi: 10.1128/jvi.60.1.185-193.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 2011;7:e1002282. doi: 10.1371/journal.ppat.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zeng Z-Y, Zhou L, Tang D-Y, Liu Z, Liang H-Y, Li Q, Xiao C-N, Wang B, Wang F, Gan Z-L. Construction of Prokaryotic Expression Plasmid of gE Gene of PRV GZ-Z1 Strain. Guizhou Agricultural Sciences. 2011;39:149–152. [Google Scholar]
- [44].Lou G, Ao J, Yang L, Du W, Wang X. Cloning and sequencing of gE gene encoding area of pseudorabies virus Fa strain excluding signal peptide. Chinese journal of veterinary science. 2001;21:568–570. [Google Scholar]