

Abstract
Aging is the main risk factor for most chronic diseases, disabilities, and declining health. It has been proposed that senescent cells—damaged cells that have lost the ability to divide—drive the deterioration that underlies aging and age-related diseases. However, definitive evidence for this relationship has been lacking. The use of a progeroid mouse model (which expresses low amounts of the mitotic checkpoint protein BubR1) has been instrumental in demonstrating that p16Ink4a-positive senescent cells drive age-related pathologies and that selective elimination of these cells can prevent or delay age-related deterioration. These studies identify senescent cells as potential therapeutic targets in the treatment of aging and age-related diseases. Here, we describe how senescent cells develop, the experimental evidence that causally implicates senescent cells in age-related dysfunction, the chronic diseases and disorders that are characterized by the accumulation of senescent cells at sites of pathology, and the therapeutic approaches that could specifically target senescent cells.
SENESCENT CELLS AND THEIR SECRETOME
In 1961, Leonard Hayflick and Paul Moorhead discovered that normal (diploid) human fibroblasts have a finite proliferative capacity in vitro, a phenomenon termed replicative senescence (Figure 1).1 They proposed that this predetermined “passage potential,” later termed the “Hayflick limit,” was due to an asynchronous self-replicating system,1 and they interpreted these findings as being the basis of the cellular mechanism underlying organismal aging. Consistent with this theory, telomeres are observed to shorten as diploid cells reach their passage potential, acting as mitotic clocks in regulating replicative senescence.2 Ectopic expression of the human telomerase reverse transcriptase catalytic domain in nontransformed human fibroblasts and epithelial cells was shown to enable cells to bypass their Hayflick limits and evade senescence.2 Together with the observation that immortalized cell lines and the majority of human tumors express human telomerase reverse transcriptase,2 these studies led to the idea that replicative senescence is an intrinsic anticancer mechanism that limits a cell’s proliferative ability, thereby preventing neoplastic transformation (Figure 1).
Figure 1.

Three distinct origins of senescent cells. Cells that have reached their Hayflick limits have lost their ability to proliferate further; this is termed “replicative senescence.” In cellular senescence, a variety of stimuli also cause an irreversible cell cycle arrest before cells lose their proliferative capacity. Emerging evidence suggests that there is a third path to the formation of senescent cells, one in which terminally differentiated, nonproliferative cells acquire key features of senescent cells, including a SASP. We have termed these cells SAD (senescence after differentiation) cells. SASP, senescence-associated secretory phenotype.
As suggested by Hayflick’s initial observations, senescent cells have been shown to increase with increasing age in various organs and tissues.3,4 This was first demonstrated in vivo using a pH-dependent β-galactosidase (β-Gal) now known as senescence-associated-β-Gal (SA-β-Gal). This allowed researchers, for the first time, to distinguish senescent cells from quiescent and postmitotic cells.4 However, replicative senescence is not believed to be the sole or primary cause for the accumulation of senescent cells in mammalian tissues over time. Instead, it is now thought that another form of senescence, termed cellular senescence, contributes to this phenomenon (Figure 1). Cellular senescence, like replicative senescence, is a terminal fate of mitotic cells, characterized by permanent cell-cycle arrest. Unlike replicative senescence, however, cellular senescence does not require telomere deterioration and can be induced by a variety of stressors including ultraviolet light, reactive oxygen species, chemotherapeutics, ionizing radiation, distortion of chromatin structure, and excessive mitogenic signaling (Figure 1). Through various signaling pathways, these stimuli engage either p53 or retinoblastoma protein (RB), or both, depending on the stressor and the severity of the stress.5 In this context, primarily two cell-cycle inhibitors linked to the p53 and/or RB signaling pathways, p21Cip1/Waf1 and p16Ink4a (encoded by Cdkn1a and Cdkn2a, respectively),5 have been implicated as biomarkers and effectors of senescence; p16Ink4a is currently regarded as being a premier indicator of the presence of senescent cells.3
Many mechanisms have been proposed to contribute to the process of aging, including genome maintenance impairments and oxidative damage or free radical accumulation. Both replicative and cellular senescence have been hypothesized as contributing to age-associated tissue dysfunction, reduced regenerative capacity, and disease. However, it was unclear how “dormant” (i.e., mitotically inactive) senescent cells could have such profoundly detrimental effects, until it was discovered that senescent cells have a drastically altered pattern of gene expression as compared with nonsenescent cells of the same lineage.5 Senescent cells have a complex phenotype characterized by irreversible cell-cycle arrest mediated predominantly by p21 and/or p16Ink4a, increased cell size, altered morphology, resistance to apoptosis, altered gene expression including upregulation of SA-β-Gal, and acquisition of a unique secretory profile, which is referred to as the senescence-associated secretory phenotype (SASP).5,6 The SASP is largely composed of three classes of secreted proteins: (i) inflammatory chemokines and cytokines, (ii) matrix-remodeling proteases, and (iii) growth factors (Figure 1). SASP signaling is complex and can have opposing effects under different experimental conditions. For example, the SASP plays a role in tumor suppression by reinforcing senescence in a cell-autonomous fashion7 and by contributing to antitumor immune surveillance;8,9 on the other hand, the SASP has also been implicated in tumor promotion by stimulating neovascularization, cell growth, and invasion via paracrine signaling.6,10,11
In vitro data suggest that the SASP is highly, albeit not entirely, conserved between mice and humans when cultured in physiologic oxygen.10 Furthermore, recent data indicate that the SASPs of senescent human fibroblasts and epithelial cells have considerable overlap, with some notable exceptions.6 Although the SASP is senescence-associated, it does not appear to be the result of p53/p21 or p16Ink4a-dependent cell-cycle arrest.12 Rather, the SASP is believed to be regulated by the DNA damage response11 and/or p38MAPK signaling,13 depending on the nature and context of the senescence-inducing stressor. Consequently, whereas cell-cycle arrest may indirectly limit the SASP by decreasing proliferation-induced DNA damage, it is not a causative factor in generating or establishing the SASP.12
We, and others, hypothesize that senescent cells contribute to aging and age-related diseases by altering tissue microenvironments via their SASP.5 In the subsequent sections of this review, we discuss the possible mechanisms through which the SASP is thought to contribute to the development, maintenance, and progression of each senescence-associated disease.
SENESCENT CELLS AND AGE-RELATED PATHOLOGY
Evidence that senescent cells cause age-related dysfunction
In addition to p16Ink4a, the Cdkn2a locus encodes for another cell-cycle inhibitor, p19Arf.14 Both these regulators play a pivotal role in the generation of senescent cells. Under conditions of stress, p16Ink4a inhibits the formation of active cyclin D–CDK complexes, preventing RB phosphorylation and subsequent cellcycle progression through the G1-phase restriction point into S phase.14 Chronic long-term expression of p16Ink4a in stressed cells leads to cellular senescence.5 In contrast, p19Arf acts to sequester Mdm2, allowing for stabilization of p53 which, in turn, leads to apoptosis or arrest of growth.14 Apoptosis occurs when cells experience irreparable damage and p53 transcriptional activity is robust.15 Growth arrest is established through p53-mediated transcriptional activation of p21. This occurs in response to moderate stress-related damage in order to allow time for repair. In instances when repair is unsuccessful, p21 inhibits the formation of active cyclin E–Cdk2 complexes, imposing a more permanent cell-cycle arrest by keeping RB hypophosphorylated.5 This particular senescent state differs from p16Ink4a-mediated senescence in that it is reversible upon the deactivation of p53 or p21.5
Most of the pioneering mechanistic work on cellular senescence was carried out on cultured cells, leaving open the question of whether the same molecular pathways apply to senescence in vivo.16 Furthermore, although the identification of novel senescence markers revealed that senescent-like cells accumulate in tissues of aged humans, mice, and rats, as well as at sites of age-related disease pathology, whether senescent cells play a causative role in aging or diseases associated with aging remained unknown. A critical barrier to progress was that key members of the p19Arf–p53 and p16Ink4a–RB pathways are tumor suppressors whose systemic inactivation in mice leads to death from cancer before the onset of age-related tissue deterioration. 14 In addition, p19Arf and p53 control two distinct cell fates, apoptosis and senescence, respectively. Therefore, in mice that lacked these tumor suppressors, it was difficult to assign any potential delays in onset of age-related tissue dysfunction to loss of senescence. Studies with p16Ink4a do not have this problem, given that p16Ink4a is involved exclusively with senescence.16,17
The first direct in vivo evidence for a causal link between p16Ink4a-positive senescent cells and the development of age-related pathologies was obtained from a study using the BubR1 progeroid mouse model in combination with p16Ink4a inactivation. 18 Mutant mice with low levels of BubR1, a key inhibitor of the E3 ubiquitin ligase APC/CCdc20, which controls diverse biological processes such as chromosome segregation, differentiation of postmitotic neurons, and ciliogenesis,19,20 developed progeroid phenotypes at an early age in a broad spectrum of tissues, including skeletal muscle, fat (hypodermal fat and subcutaneous fat depots), eye, artery, heart, and brain (Figure 2).21 Importantly, in skeletal muscle, fat, and eye, but not in other tissues with age-related pathologies, p16Ink4a was expressed at chronically elevated levels, making this progeroid model particularly suitable for the study of the causal relationship between in vivo senescence and aging.18 Age-related pathologies in other progeroid mouse models are either driven by p5322–24 or are not known to involve p16Ink4a engagement.25–28 Two key findings that causally linked in vivo senescence with aging-associated dysfunction were that (i) premature aging in BubR1-p16Ink4a double-mutant mice was delayed exclusively in progeroid tissues with chronic p16Ink4a expression and (ii) delayed aging in these same tissues coincided with a markedly reduced accumulation of senescent cells (Figure 2).18
Figure 2.
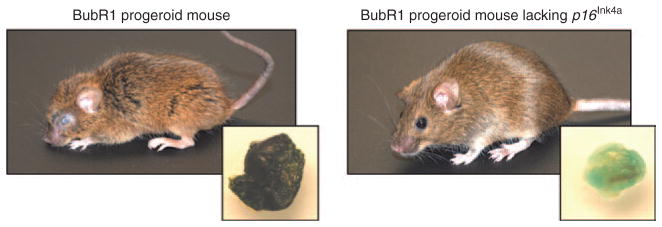
Germline deletion of p16Ink4a in BubR1 progeroid animals dramatically impacts overt age-associated phenotypes and delays cellular senescence. Representative images of 5-month-old mice are shown. The inset images are SA-β-Gal-stained fat depots, demonstrating the dramatic attenuation of cellular senescence. SA-β-Gal, senescence-associated-β-galactosidase.
Senescent cell clearance in mice
Genetic inactivation of p16Ink4a in BubR1 progeroid mice delayed aging by preventing the formation of p16Ink4a-positive senescent cells,18 thereby identifying senescent cells as a therapeutic target for aging and age-related disease. Inactivation of the p16Ink4a gene in humans is an impracticable therapeutic approach because it would also eliminate a critical tumor-suppressive mechanism, thereby resulting in cancer. Therefore, the most logical extension of the studies on BubR1-p16Ink4a double-mutant mice was to try and phenocopy these mice by using a drug to selectively remove p16Ink4a-positive cells from BubR1 progeroid mice. For these proof-of-concept experiments, a transgene called INK-ATTAC was designed to make stressed cells that express the p16Ink4a gene in the context of cellular senescence susceptible to drug-induced apoptosis. INK-ATTAC was modeled after the FAT-ATTAC (fat apoptosis through targeted activation of caspase) transgene used earlier by the Scherer lab to inducibly clear adipocytes from mice upon administration of AP20187.29 FAT-ATTAC is driven by the Fabp4 promoter and produces a membrane-bound Casp8-FKBP fusion protein specifically in adipocytes. INK-ATTAC was created by replacing the Fabp4 promoter with a 2,617 bp fragment of the p16Ink4a gene promoter that is transcriptionally active in senescent but not in nonsenescent cultured fibroblasts.30 An added feature of INK-ATTAC was the insertion of an internal ribosome entry site–containing EGFP open reading frame downstream of Casp8-FKBP to allow for detection and FACS sorting of p16Ink4a-positive cells.
Two independently generated transgenic strains of mice specifically expressed INK-ATTAC in p16Ink4a-positive senescent cells when bred onto a BubR1 progeroid background, and efficiently eliminated these cells upon AP20187 administration. Using these strains, two distinct senescent cell clearance strategies were studied: lifelong clearance and late-life clearance. Lifelong treatment with AP20187 every third day from weaning onward was intended to clear senescent cells immediately after their genesis, preventing any potential irreversible tissue damage. Late-life treatment, starting when BubR1 progeroid mice had developed established age-related phenotypes in skeletal muscle, fat, and eye, was designed to test the extent to which removal of senescent cells can rejuvenate aged tissues. Lifelong removal of p16Ink4a-positive senescent cells delayed the onset of fat loss, skeletal muscle deterioration, and cataract formation in BubR1 progeroid mice,31 just as genetic inactivation of p16Ink4a had done earlier (Figure 3).18 Although late-life clearance attenuated the progression of already established age-related disorders in skeletal muscle, fat, and eye, it did not revert these phenotypes. Neither lifelong nor late-life drug treatment was able to substantively prolong life span, most likely because accelerated cardiovascular dysfunction in the BubR1 progeroid mice, which develops independently of p16,31 was not corrected with AP20187 treatment. However, the fact that heart and blood vessel dysfunction in BubR1 progeroid mice do not involve p16Ink4a-positive senescent cells does not necessarily imply that elderly individuals with cardiovascular diseases might not benefit from senescent cell clearance. In short, both human and mouse heart tissue are known to accumulate p16Ink4a-positive senescent cells with aging.3
Figure 3.

Proposed mechanism through which removal of senescent cells from aged tissues benefits tissue function. Senescent cells, through their SASP, disrupt the functionality of neighboring cells. When the senescent cells are selectively cleared, tissue function improves. The aged cells present retain whatever intrinsic damage they had accrued before the removal of the senescent cells, demonstrating that tissues are not likely to revert to a pre-aging condition.
The senescent cell clearance experiments strengthen the earlier conclusion (from studies on BubR1 hypomorphic mice lacking p16Ink4a) that senescent cells drive aging-related pathologies. 18 In addition, they indicate that removal of senescent cells can delay tissue dysfunction and extend health span. The apparent absence of any overt negative side effects of senescent cell clearance underscores the fact that senescent cells represent a promising new therapeutic target for delaying age-associated diseases and improving quality of life in the elderly. Elimination of senescent cells by genetic or therapeutic means results in delay rather than a complete block or reversal of age-related tissue dysfunction.18,31 The most straightforward explanation for this observation is that there are at least two distinct phenomena driving age-related dysfunction, one constituting cell-intrinsic functional decline, and the other, deterioration of the cellular milieu (Figure 3). Cell-intrinsic decline, which almost all cells seem to experience over time, involves progressive damage to DNA, proteins, and lipids.32,33 Senescent cells add to the intrinsic dysfunction by secreting proinflammatory cytokines, proteases that disintegrate the extracellular matrix, and growth factors that provide aberrant mitogenic signals. Clearance of senescent cells is expected to improve the tissue milieu, thereby improving the function of the remaining nonsenescent cells. However, this is unlikely to restore aging-related cell-intrinsic macromolecular damage to nonsenescent cells. True rejuvenation would probably require extensive replacement of nonsenescent damaged cells through stem cell–mediated regeneration. This apparently does not occur with mere elimination of senescent cells, at least not at a tissue-wide level.
SENESCENT CELL CLEARANCE THERAPY: APPLICATIONS
Common age-related features
Aging is the result of progressive changes in biological systems over time, and is experienced by most individuals, although at different rates. Aging is not considered a disease; however, disease prevalence and age are closely related. Putative features of “normal” aging include, but are not limited to systemic decline of the immune system, muscle atrophy and decreased muscle strength, decreased skin elasticity, delayed wound healing, retinal atrophy, and reduced lens transparency (Figure 4). Given that p16Ink4a expression increases across a broad spectrum of tissues in mice and humans,3 we discuss here the potential role and therapeutic implications for senescent cells in universal features of normal aging and in age-related diseases that only a subset of the elderly population will experience.
Figure 4.

Normal, age-related, nearly universal changes (left) and age-associated diseases (right) that have senescent cells potentially underlying their incidence, progression, and/or severity. For further details, see individual sections within the text. COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis.
Therapeutic intervention in normal aging may prevent comorbidity and delay mortality in the elderly. In this way, targeting of senescent cells during the course of normal aging would be a preventative strategy rather than a treatment, like in the context of immunosenescence. Immunosenescence diminishes adaptive immune function in the elderly while increasing their susceptibility to infection and malignancy as well as increasing the concentration of proinflammatory cytokines in their blood.34 Centenarians—individuals who live to be more than 100 years of age—have low levels of senescent CD8+ T-cells (CD8+CD28−).35 Removal of senescent immune cells in the elderly may significantly boost their immune systems, extend their health spans, improve their quality of life, and reduce health-care costs. These are the benefits often observed in centenarians.
Another area in which therapeutic prevention would be beneficial is sarcopenia. Muscle atrophy and diminished strength contribute to frailty in the elderly, thereby increasing their risk for falls and fractures, functional disability, and death. Sarcopenia is a complex process involving denervation of neuromuscular junctions and dysfunction of skeletal muscle mitochondria. Mitochondrial dysfunction can lead to muscle fiber apoptosis and generate high levels of reactive oxygen species, which can conceivably invoke a senescence response. Curiously, mice with high levels of senescent cells in skeletal muscle were shown to be prone to sarcopenia, which was attenuated upon inactivation of p16Ink4a-positive senescent cells in BubR1 mutant mice.18
One of the prominent characteristic features of aging is skin wrinkling caused by loss of subdermal adipose tissue. In addition to wrinkles, aged skin also has decreased immune barrier function and an increased susceptibility to skin cancer and trauma. Human skin accumulates senescent cells over time;4 also, mice with accelerated senescence were demonstrated to have thinner skins and reduced subdermal adipose tissue.36 Given these findings, the targeting of senescent cells may restore the skin’s protective function and provide cosmetic enhancement. It is not known, however, what effect this clearance would have on cutaneous wound healing, given that myofibroblast senescence has been shown to have a beneficial role in limiting fibrogenesis during wound healing,37 although this may be a transient process that is minimally affected by senescence-targeting therapeutics.
Age-related diseases
Senescent cells are often present at sites of chronic diseases in humans. They are known to develop because of certain treatments, including chemotherapy and radiation therapy (Figure 4). However, whether senescent cells are causally implicated in the pathogenesis of age-related disorders or merely bystanders in the aging process is not known. The development of the INK-ATTAC mouse model25 now enables researchers to determine the contributory effects of senescent cells to human disease and to explore the preventive and therapeutic effects of clearing senescent cells by crossing well-established mouse models for chronic age-related human diseases with INK-ATTAC mice.
Cancer
Age is the single greatest risk factor for developing cancer, with an incidence that rises exponentially during midlife in humans and in mice.38 However, little is known about the underlying molecular and cellular bases for this dramatic increase. Because sequential mutations in cancer-critical genes are known to drive tumorigenesis, it has long been suspected that cancer incidence increases with age simply because it takes time to accumulate sufficient mutations. However, it is now clear that malignant transformation also requires a tissue microenvironment that permits survival, proliferation, and dissemination of mutant cells.39 One emerging idea is that senescent cells create such an environment through their SASP (Figure 5).5
Figure 5.

Proposed contribution of senescent cells to tumorigenesis and cancer therapy–induced side effects. Senescent cells stimulate tumor cell growth via various growth factors, and tissue disorganization via the components that they secrete. Once a tumor is removed by systemic radiation or chemotherapy, senescence is triggered in a variety of other organs, leading to long-term ramifications for the patient.
In support of this theory, various components of the SASP, such as GROα, have been demonstrated to stimulate proliferation of premalignant cells in culture. In addition, other SASP components, such as interleukin (IL)-6, IL-8, Pai1, and various matrix metalloproteinases, were shown to facilitate tumor cell invasion.6,10 Further proof that senescent cells enhance nearby tumor cells and are causally implicated in malignant tumorigenesis was provided by the observation that co-injection of senescent fibroblasts with preneoplastic SCp2 mammary epithelial cells into immunocompromised mice formed tumors, whereas injection of SCp2 cells alone or with presenescent fibroblasts did not.40 Furthermore, senescent fibroblasts also facilitated tumor cell dedifferentiation, a hallmark of invasive disease also facilitated tumor cell dedifferentiation, a hallmark of invasive disease. One limitation of these studies, however, is that they were performed either in vitro or in mouse xenograft models, neither of which accurately reflects the physiologic conditions of cancers in mice or humans. Therefore, the key questions are whether senescent cells promote tumorigenesis under physiologic conditions and, if so, what mechanism is involved. These can now be addressed, given the availability of the INK-ATTAC model from which senescent cells can be selectively cleared.
The finding that senescent cells have tumor-promoting properties raises the question as to what functional advantage senescence might have over apoptosis. If both senescence and apoptosis are intrinsic tumor suppressive-mechanisms, with senescence having extrinsic tumor-promoting properties, why don’t irreparably damaged cells always undergo apoptosis? One possibility is that SASP components, in addition to facilitating late-life tumorigenesis, play a critical role in antitumor immune surveillance. Indeed, recent studies have implicated senescent cells in immune-mediated clearance of premalignant and neoplastic cells.8,9 How, then, might one reconcile the tumor-protective and tumor-promoting effects of senescent cells in terms of their SASP? We propose a mechanism whereby senescent cells become increasingly tumor-promoting as the ratio of functional immune cells to senescent cells decreases. This can happen in two clinically relevant ways: (i) the combination of decrease in immune function and increase in senescent cells in the elderly and (ii) the accelerated accumulation of senescent cells after a major cytotoxic event such as cancer treatment (see below). We do not consider decreased immune function by itself to be relevant to this discussion, given that the primary concern in immunocompromised patients is their increased susceptibility to life-threatening infections.
Cancer therapy–related disability
There are an estimated 10 million cancer survivors in the United States alone. With presumably millions more worldwide, the consequences of receiving toxic cancer treatments have far reaching implications for patients and global health-care systems. Short-term side effects of chemotherapy and radiation therapy are thought to be due primarily to the mass destruction of rapidly dividing cells, for instance in the bone marrow, gastrointestinal tract, and hair follicles. Although the cause is unknown, long-term side effects from these toxic therapies can reduce the quality of life of cancer survivors and predispose them to disabilities and comorbidities, thereby creating a link between cancer treatment and accelerated aging. For instance, some survivors of breast cancer experience a disproportionate decline in physical function, such as the inability to walk up stairs or to reach up to put things onto shelves. These patients may also experience increased functional disabilities such as difficulty in eating, dressing, and maintaining adequate hygiene, as compared with cancer-free women (reviewed in ref. 41). Of note, these are the same standards by which clinicians gauge functional status in the elderly. The link between accelerated aging and cancer treatment is further strengthened by reports that correlate elevated levels of inflammatory cytokines with decreased physical function in a subset of breast cancer survivors and also in the elderly.42,43
Although a correlation between cancer treatment and declining physical function has been established, the cellular mechanisms linking these remain unknown.41 Based on the observation that ionizing radiation and various chemotherapeutic agents elicit a marked senescence response in vivo (Figure 5),44,45 senescent cells have recently been implicated as a cause of long-term complications after cancer therapy.45 Therefore, we hypothesize that the systemic accumulation of therapy-induced senescent cells results in a robust induction of proinflammatory SASP components and that this, in turn, drives accelerated physical decline in cancer survivors. Of particular interest is the cytokine IL-6, a highly expressed component of the SASP implicated in age-associated, and presumably therapy-associated, physical decline.43
In addition to declining physical function, cancer survivors are highly susceptible to developing a number of severe or life-threatening health conditions.46 For example, it has been discovered that survivors of childhood cancer were nearly 15 times more likely to develop a secondary malignancy as compared with the likelihood of their sibling(s) developing a first cancer.46 Although the role of an inherited or acquired genetic predisposition to cancer in these patients cannot be ignored and should not be trivialized, it is worth considering that the accumulation of therapy-induced senescent cells permits the transformation of preneoplastic cells into malignant ones by creating a protumorigenic microenvironment via their SASP. Unfortunately, secondary cancer is not the only serious medical condition these patients are at risk of developing. In the same study, childhood cancer survivors were also found to be at significantly greater risk for developing other diseases thought to involve senescent cells, including coronary artery disease and cerebrovascular disease. These topics are discussed later in this review.
Type 2 diabetes
Type 2 diabetes (D2M), characterized by “a progressive insulin-secretory defect on the background of insulin resistance,”47 is quickly becoming a global epidemic with grave implications for our health-care systems. D2M is a leading risk factor for coronary artery disease and myocardial infarction, cerebrovascular accidents, neuropathy, retinopathy, nephropathy, and many other disabling conditions. Obesity and increasingly sedentary lifestyles are the primary risk factors responsible for this disturbing trend. The role of senescent cells in obesity and D2M has already been extensively reviewed.48 Briefly, fat tissue from obese mice showed induction of the senescence markers SA-β-Gal, p53, and p21.48,49 A concomitant upregulation of proinflammatory cytokines—specifically, tumor necrosis factor-α and Ccl2/MCP1—was observed in the same tissue.49 A significant induction of senescent cells in obesity has potentially significant clinical implications because proinflammatory SASP components are thought to contribute to D2M.48 Indeed, a similar pattern of upregulation of senescence markers and SASP components was found to be associated with diabetes, both in mice and in humans.49 As a demonstration of the potential therapeutic benefits of targeting senescent cells in prediabetic patients as well as in those with diabetes, inhibition of p53-mediated senescence in murine fat tissue reduced the expression of inflammatory mediators and significantly ameliorated insulin resistance.49
Atherosclerosis
Acute coronary syndrome, myocardial infarction, and stroke from atherosclerosis are leading causes of death and morbidity, particularly in Western countries.50 In addition to hypertension, diabetes, hypercholesterolemia, and smoking, aging is one of the major risk factors for atherosclerosis. A likely mechanism by which aging and some of the lifestyle-related factors might advance atherogenesis is through cellular senescence. Senescent cells have long been linked to atherosclerosis. For instance, atherogenic plaques contain SA-β-Gal-positive vascular smooth muscle cells, endothelial cells, and inflammatory cells,51 as well as high levels of p16Ink4a and p19Arf.52 One hypothesis is that cytokines released by senescent cells amplify the proatherogenic inflammatory environment created by macrophages infiltrating the subendothelial vascular space.53 Other SASP components may further boost the atherogenic process by promoting dysfunction of endothelial cells and vascular smooth muscle cells. Senescence-induced fat tissue dysfunction might disturb proper lipid metabolism and storage and cause accumulation of atherogenic lipids within the arterial wall,48 leading to systemic changes.
Hypertension
Hypertension is extremely prevalent and has far-reaching implications for patients and health-care systems. It is one of the primary risk factors for cardiovascular disease, stroke, and kidney disease, and is often suboptimally managed. Interestingly, hypertension has been shown to induce senescence in a pattern consistent with targeted end-organ damage, namely in the kidney and heart, indicating a contributory role for senescent cells in this process.54 It is reasonable to theorize that various SASP components may inhibit tissue regeneration and promote fibrotic remodeling of end organs. If senescent cells do, in fact, contribute to hypertensive end-organ damage, this opens up a new dimension of potential therapeutic interventions for the millions of patients with a significant history of long-standing, poorly controlled hypertension.
Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease (COPD) is a major form of pulmonary emphysema, in which elastase derived from cigarette smoke-activated neutrophils and macrophages disintegrate the extracellular matrix of alveolar structures, resulting in enlarged air spaces and loss of respiratory capacity.55 COPD affects ~14 million individuals in the United States alone and is one of the leading causes of death. Besides cigarette smoke, aging is a major risk factor for COPD.55 Both aging and cigarette smoke result in elevated p16Ink4a expression in lung tissue,3,56,57 raising the possibility that senescent cells play a role in both the smoke-related and the age-related pathogenic mechanisms of COPD. Consistent with this, cultured alveolar epithelial cells exposed to cigarette smoke extracts exhibit various markers of cellular senescence, including increased SA-β-Gal activity, elevated levels of p21 and lipofuscin, and growth arrest.57 One emerging hypothesis is that alveolar and airway cell senescence from cigarette smoke or aging contributes to destruction of alveoli by limiting the proliferative capacity necessary for tissue repair and promote chronic inflammation, both of which are hallmarks of COPD.56
Idiopathic pulmonary fibrosis
Cellular senescence has also been implicated in idiopathic pulmonary fibrosis (IPF),58 a progressive respiratory condition in which the walls of the alveoli accumulate excessive amounts of collagen, causing lung stiffness and ventilator restriction.59 As the name indicates, the etiology of IPF is unknown. The incidence of IPF is ~7 per 100,000 persons, and the median survival is 3–5 years after diagnosis.60 As with COPD, age is a major risk factor for the development of IPF, with most patients being first diagnosed at the age of 50 years or older.60 Unlike COPD, IPF is not clearly linked to cigarette smoking. The lung tissue in IPF patients is enriched for SA-β-Gal-positive cells and contains elevated levels of the senescence marker p21.58 In addition, short telomeres are a risk factor for both IPF and cellular senescence.61 One hypothesis that is emerging from these data is that SASP components of senescent cells, such as IL-6, IL-8, and IL-1β, promote fibroblast-to-myofibroblast differentiation and epithelial– mesenchymal transition, resulting in extensive remodeling of the extracellular matrix of the alveolar and interstitial spaces.58
Osteoarthritis
Osteoarthritis (OA) is a complex degenerative joint disease characterized by fibrillation of the cartilage at sites of high mechanical stress, bone sclerosis, and thickening of the synovium and the joint capsule. It is the most common cause of chronic disability in the elderly. Aging is a prominent risk factor for the development of OA but by itself is insufficient to induce progression to symptomatic disease. Rather, aging, together with other risk factors such as joint overuse and obesity, seems to promote OA. Chronic inflammation is thought to be the main age-related driver of OA. Many of the inflammatory cytokines found to be elevated in arthritic joint tissues, including IL-1β, IL-6, and IL-8, are key components of the SASP.62 This holds true for extracellular matrix-remodeling proteins such as matrix metalloproteinase 3 and 13, which are thought to contribute to architectural anomalies in atherogenic joints. This, together with the observation that senescent cells with SA-β-Gal activity, telomere shortening, and elevated p16Ink4a and p21 expression accumulate in articular cartilage, suggests that senescence may play a causal role in promoting OA via proinflammatory factors.63
Alzheimer’s disease
“Dementia” is an umbrella term used to describe diseases that cause dysfunction or death of neurons. Alzheimer’s disease (AD), which is characterized by the presence of neurofibrillary tangles and amyloid (senile) plaques in histological specimens, is the leading cause of dementia in the elderly. Age is the single greatest risk factor for developing AD. An estimated 5.4 million Americans have AD, with 95% of them being >65 years of age.64 Although it is not considered to be part of the normal aging process, approximately half of all individuals over the age of 85 years have AD.64 Early clinical symptoms show remarkable similarity to mild cognitive impairment (see below), which is characterized by difficulty in remembering recent life experiences or people’s names. Because of these lapses in memory, depression and apathy are also commonly observed early in AD. As the disease progresses, impaired judgment, confusion, behavioral changes, disorientation, and difficulty in walking and swallowing occur. In the final stages of AD, people require assistance with all basic activities of daily living. Unfortunately, no treatment is available for AD, which is ultimately fatal.
Interestingly, both neurofibrillary tangles and neuritic components of the plaques of patients with AD show strong immunoreactivity for p16Ink4a but not for other members of this cell-cycle regulatory family.65 This biomarker of aging is not expressed in terminally differentiated neurons, demonstrating that the diseased neurons have acquired the expression of at least one senescence-associated protein.
Parkinson’s disease
Parkinson’s disease (PD) is the second most common form of neurodegeneration in Americans, affecting ~1 million individuals, the majority being >50 years of age. Resting tremor and shuffling gait are commonly observed in patients with PD. In this disease, dopamine-producing neurons located in the substantia nigra pars compacta are believed to degenerate because of the aggregation of α-synuclein, a protein with unknown function that is abundantly detected in neuronal tissue. There is no known cure for PD, but medications can be used to increase the levels of dopamine in the brain to help control the symptoms. If left untreated, the disorder will get progressively worse until the individual becomes completely disabled. The senescence of dopamine-producing neurons is thought to contribute to the observed cell death in PD through the production of reactive oxygen species.66 Alleviation of these effects may enable attenuation of the severity of PD and its progression, particularly early in the course of the disease.
Cataracts
Several disorders of the eye are associated with increasing age. One of the most common disorders observed is cataract, a clouding of the lens of the eye, resulting in blurred vision and eventual blindness if left untreated. More than 22 million individuals >40 years of age are affected with cataracts. It is estimated that by the time they are 80 years of age, more than half of all persons in the United States have cataracts or have had corrective surgery (National Eye Institute). The lens of the eye must remain completely clear and transparent to enable proper focusing of light onto the retina to obtain sharp images. As we age, the proteins that make up the lens lose their organization, form clumps, and impair transmission of the light. Fortunately, surgery to treat cataract is one of the most common operations, and it is safe and extremely effective. Patients who undergo cataract surgery have better vision after the procedure because the artificial lens that has been placed in the space previously occupied by the diseased lens properly focuses light onto the retina. Senescence has not been implicated in cataract formation in humans; however, there is evidence that BubR1 hypomorphic mice develop posterior subcapsular cataracts bilaterally early in life, suggesting that senescence may play a key role. For instance, there appears to be a positive correlation between accelerated senescence and cataract formation in BubR1 hypomorphic mice.18 Furthermore, by preventing the development of senescence in mice through ablation of p16Ink4a, the appearance of cataracts is significantly delayed.18 In addition, if started before cataract formation, targeted removal of p16-expressing senescent cells from BubR1 mutant mice also delays its onset,31 further suggesting that senescence plays a role in the process of cataractogenesis in mice.
Glaucoma and age-related macular degeneration
Several other diseases of the eye increase in incidence with advancing age, including glaucoma and macular degeneration. Glaucoma affects nearly 2.3 million Americans >40 years of age (National Eye Institute). Normally, clear fluid flows into and out of the front part of the eye, known as the anterior chamber. In individuals who have open/wide-angle glaucoma, this fluid drains too slowly, leading to increased pressure within the eye. If left untreated, this high pressure subsequently damages the optic nerve and can lead to complete blindness. Using SA-β-Gal staining, a fourfold increase in senescence has been observed in the cellular network required for the outflow of fluid in glaucoma patients.67 The proliferation rate of trabecular meshwork cells is normally quite low, suggesting that the senescence observed in this disorder is not likely due to an exhaustion of replication potential but could be another example of the effect of senescent cells (Figure 1).
Age-related macular degeneration (AMD) is the leading cause of vision loss in older adults. Gradually, the macula, the most sensitive part of the retina that is required for providing sharp, central vision, is destroyed, leaving peripheral vision intact. It is estimated that in the United States more than 2 million individuals >50 years of age have advanced AMD (National Eye Institute). There are two forms of AMD: dry and wet. Dry AMD, the more common form, is defined by the death of cells located in the macula. Currently, no treatments are available for this form of AMD. In wet AMD, blood vessels located behind the retina begin to grow. These vessels are fragile, leading to leakage of blood and fluid. This, in turn, causes the macula to swell quickly, resulting in very rapid damage. Although it has not been experimentally demonstrated, senescence of retinal pigment epithelial cells has been hypothesized to underlie the etiology of AMD.68
SENESCENCE AND STEM CELLS
As mammals age, their capacity to regenerate damaged and atrophic tissue declines as a result of stem cell loss and progressive loss of function. Stem cells and pluripotent cells are progressively lost over time through a variety of mechanisms including apoptosis, replicative or cellular senescence, and transdifferentiation. However, recent evidence suggests that aged stem cells with diminished proliferative capacity can be “rejuvenated” by manipulation of their anatomic location or systemic environment.69,70 For example, selective depletion of testosterone-producing cells in rat testes induced the normally quiescent stem cells to regenerate Leydig cells within 2 weeks.70 Likewise, muscle stem cells (satellite cells), hepatic progenitor cells, and neural stem cells with diminished regenerative potential in aged mice can be enticed to proliferate upon exposure to a young systemic environment through parabiotic pairing.69 Taken together, the findings from these studies suggest that the cellular microenvironment, or niche, in which a stem cell resides, can negatively regulate stem cell function with advancing age.
This intriguing putative role of senescent cells in the aging niche has not been directly studied. Selective inactivation of p16Ink4a has been shown to delay aging in skeletal muscle and fat in BubR1 hypomorphic mice.18 In this model, satellite cells demonstrated an improved regenerative potential when p16Ink4a was ablated, implying that improved functionality of muscle stem (satellite) cells contributed to this restoration. This interpretation is consistent with earlier observations that stem cell proliferation at advanced age is improved in certain tissues of mice lacking p16Ink4a.71 Consistent with these results, p16Ink4anull mice exposed to ischemic injury demonstrated increased renal tubular epithelial cell regeneration as compared with wildtype mice. Similarly, mice transplanted with p16/p19-deficient kidneys had significantly improved allograft survival compared with wild-type kidneys, owing to increased allograft regenerative capacity in the absence of p16-mediated senescence.72 These studies, while important, do not provide direct evidence that the observed improvement in stem cell proliferation was the cause of the delay in age-related tissue deterioration. Nonetheless, they do suggest a role for senescent cells in establishing and maintaining a suppressive stem cell niche. Overall, senescent cells are emerging as an important target in stem cell–based therapies, especially in the aged population.
OPEN QUESTIONS ABOUT SENESCENT CELLS
Senescent cells are found at an increased frequency during the normal aging process and in various diseases. In many instances, it is not known whether these cells contribute to tissue dysfunction and, if they do contribute, what the underlying mechanism is. Although a common SASP signature has been identified in a variety of senescent cell types in vitro, it is not known whether a similar SASP exists in vivo. Given the complexity of tissues and organs, it is unlikely that all senescent cells in vivo express identical SASPs, and there are potentially tissue-specific SASP hallmarks. It is possible that certain SASP components are universally elevated in all tissues; this would facilitate therapeutic intervention (see section below) but has yet to be established. One could envisage that if a similar SASP profile is seen in all tissues, there would be negative systemic consequences due to the proteases, cytokines, chemokines, and growth factors secreted. Furthermore, it is not known whether the SASP of cells in vivo changes over time. There may be an early SASP signature as cells enter into senescence, different from the secretome of cells with long-standing cell-cycle arrest. By exploiting the fluorescent tag engineered to coincide with senescence in vivo in the INK-ATTAC mouse model, the molecular signature of senescent cells can be revealed, and these critical questions can begin to be answered experimentally.
It is difficult to determine the specific cell or cells that senesce in aged tissues. In addition, it is unclear whether aging of tissues results from senescence of stem cells or differentiated cells or both. Interestingly, terminally differentiated, postmitotic cells, which cannot reenter the cell cycle, have been found to acquire a senescence-like phenotype in several diseases. For instance, mature adipocytes, cardiac myocytes, and neurons express p16Ink4a in obesity/diabetes, hypertension, and AD/PD, respectively. How/why does postmitotic senescence occur, and what are its consequences? We have termed this process as “senescence after differentiation” and propose that these cells significantly contribute to age-associated disease (Figure 1).
The INK-ATTAC mouse model has proven extremely useful for dissecting the involvement of p16Ink4a-expressing senescent cells in BubR1 hypomorphic mice. However, a variety of aging phenotypes of BubR1 mutant mice that are not dependent on p16Ink4a expression are not affected, such as those of the heart and vasculature. Does this mean that cell populations other than those expressing p16Ink4a contribute to aging? Perhaps these results already point toward evidence of tissue-specific aging signatures of senescent cells in different tissues, suggesting that p16Ink4a is not one of the “universal” hallmarks of senescent cells in vivo. Furthermore, it is unclear whether all senescent cell populations, when stably maintained in tissue, result in negative effects on tissue function. Perhaps certain populations observed in aged tissues are truly innocent bystanders in the aging process.
Increased expression levels of p16Ink4a, p19Arf, and p21 are observed with aging across a broad spectrum of tissues, both in mice and humans.3 As fibroblasts begin to enter into senescence in vitro, p21 levels are elevated before those of p16Ink4a.73 Once these cells have established permanent arrest due to high levels of p16Ink4a, the expression of p21 drops dramatically. This suggests that these two cell-cycle regulators are not always coexpressed in senescent cells. As with the observations of delayed aging in BubR1 mutant mice lacking p16Ink4a, p21 drives age-related pathology in a progeroid mouse model for telomere dysfunction.74 Stem and progenitor cells of the intestine and hematopoietic system are functionally rescued by the deletion of p21, demonstrating that, in this model, p21 promotes the aging process of these tissues. However, it is unclear whether this pro-aging effect is due to accelerated cellular senescence or to an increased SASP.
Clearly, cellular senescence does have beneficial effects, including its potent anticancer mechanism. Furthermore, senescent cells are important for efficient tissue repair and wound healing. 17 Senescent cells are quickly established near wounds to help mount an inflammatory response that initiates the process of healing during the proliferation phase.37 This rapid boost in senescence attracts and activates immune cells to fight infection and clear dead cells and debris. During the remodeling phase, senescent cells play a role in dissolving the fibrous proteins laid down during the proliferative phase and limit the formation of scars. Consequently, not all circumstances involving senescence can be thought of as having a negative effect on tissue function. A key question is whether these acutely formed senescent cells are synonymous with senescent cells that accumulate with aging. Older persons have more senescent cells present in the skin than young persons, yet they have delayed wound-healing responses by comparison. This suggests that the maintenance of high amounts of senescent cells may prevent the required boost necessary for efficient wound healing. Experiments involving selective removal of these cells would be able to dissect these differences.
THERAPEUTIC STRATEGIES
The relative success of targeted cancer therapies predicts that analogous approaches to killing senescent cells are likely to be highly effective. Unlike tumor cells, senescent cells, by definition, are those that have lost their ability to divide in order to replenish themselves. Given that senescent cells are present in numerous age-related pathologies and diseases, selective removal of these cells represents a novel avenue to prevent the accumulation of a broad range of phenotypes affecting a variety of tissues. Therapeutic interventions, either through drugs or through viruses designed to kill these cells, should prove to be much more effective, because the target cells are able to acquire drug resistance via mutational adaptation. If a common signature is identified for senescent cells in vivo, strategies to alleviate these effects with compounds or drugs, whether by dampening the SASP profile or by completely removing the senescent cells, can begin to be elucidated.
There are potentially many other ways to remove senescent cells or delay their accumulation. Senescent cells express several ligands needed for natural killer immune cell recognition and subsequent cytotoxity.8 It is believed that this mechanism is attenuated with advancing age, which would explain why senescent cells are rare in young individuals but become increasingly more common as we age. Therefore, strategies to boost the body’s natural ability to selectively remove senescent cells may prove to be highly effective. A potential problem with this strategy is that the beneficial effects of senescence, such as proper wound healing, would also probably become attenuated if the immune system has been primed to target senescent cells as they arise.
Many of the diseases associated with increased rates of cellular senescence can potentially be modulated by changes in lifestyle. It is consistently found that one of the best things that people can do to promote health is to exercise. In accordance with an observed lower rate of accumulation of senescent cells, individuals who exercised were found to have consistently lower levels of p16Ink4a in peripheral T cells than sedentary individuals did.75 Staying active and choosing to avoid senescence-inducing activities will have beneficial effects in the short term until therapeutic strategies to eliminate senescent cells are developed and proven to be safe and effective.
Footnotes
CONFLICT OF INTEREST
The authors declared no conflict of interest.
References
- 1.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 2.Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol. 2000;1:72–76. doi: 10.1038/35036093. [DOI] [PubMed] [Google Scholar]
- 3.Krishnamurthy J, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dimri GP, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 6.Coppé JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 8.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang TW, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 10.Coppé JP, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE. 2010;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodier F, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–36403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999;9:22–30. doi: 10.1016/s0959-437x(99)80004-5. [DOI] [PubMed] [Google Scholar]
- 15.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 16.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker DJ, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–836. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyamoto T, et al. Insufficiency of BUBR1, a mitotic spindle checkpoint regulator, causes impaired ciliogenesis in vertebrates. Hum Mol Genet. 2011;20:2058–2070. doi: 10.1093/hmg/ddr090. [DOI] [PubMed] [Google Scholar]
- 20.Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet. 2008;24:457–466. doi: 10.1016/j.tig.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker DJ, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 22.Chin L, et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 23.Varela I, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- 24.Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201–213. doi: 10.1101/gad.1050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Vogel H, Holcomb VB, Gu Y, Hasty P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol Cell Biol. 2007;27:8205–8214. doi: 10.1128/MCB.00785-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci USA. 1999;96:10770–10775. doi: 10.1073/pnas.96.19.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niedernhofer LJ, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 28.de Boer J, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–1279. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- 29.Pajvani UB, et al. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005;11:797–803. doi: 10.1038/nm1262. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Wu J, Zhang Z, Tong T. Characterization of regulatory elements on the promoter region of p16(INK4a) that contribute to overexpression of p16 in senescent fibroblasts. J Biol Chem. 2001;276:48655–48661. doi: 10.1074/jbc.M108278200. [DOI] [PubMed] [Google Scholar]
- 31.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–774. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- 33.Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol. 2007;8:715–722. doi: 10.1038/nrm2242. [DOI] [PubMed] [Google Scholar]
- 34.Franceschi C, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 35.Strindhall J, et al. No Immune Risk Profile among individuals who reach 100 years of age: findings from the Swedish NONA immune longitudinal study. Exp Gerontol. 2007;42:753–761. doi: 10.1016/j.exger.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol. 2008;28:4018–4025. doi: 10.1128/MCB.00296-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jun JI, Lau LF. Cellular senescence controls fibrosis in wound healing. Aging (Albany, NY) 2010;2:627–631. doi: 10.18632/aging.100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DePinho RA. The age of cancer. Nature. 2000;408:248–254. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 39.Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitz KH, Cappola AR, Stricker CT, Sweeney C, Norman SA. The intersection of cancer and aging: establishing the need for breast cancer rehabilitation. Cancer Epidemiol Biomarkers Prev. 2007;16:866–872. doi: 10.1158/1055-9965.EPI-06-0980. [DOI] [PubMed] [Google Scholar]
- 42.Bower JE, Ganz PA, Aziz N, Fahey JL. Fatigue and proinflammatory cytokine activity in breast cancer survivors. Psychosom Med. 2002;64:604–611. doi: 10.1097/00006842-200207000-00010. [DOI] [PubMed] [Google Scholar]
- 43.Cesari M, et al. Inflammatory markers and physical performance in older persons: the InCHIANTI study. J Gerontol A Biol Sci Med Sci. 2004;59:242–248. doi: 10.1093/gerona/59.3.m242. [DOI] [PubMed] [Google Scholar]
- 44.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–2715. [PubMed] [Google Scholar]
- 45.Le ON, et al. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell. 2010;9:398–409. doi: 10.1111/j.1474-9726.2010.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oeffinger KC, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355:1572–1582. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 47.Standards of medical care in diabetes--2010. Diabetes Care. 2010;33(suppl 1):S11–S61. doi: 10.2337/dc10-S011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tchkonia T, et al. Fat tissue, aging, and cellular senescence. Aging Cell. 2010;9:667–684. doi: 10.1111/j.1474-9726.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minamino T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15:1082–1087. doi: 10.1038/nm.2014. [DOI] [PubMed] [Google Scholar]
- 50.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 51.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 52.Holdt LM, Sass K, Gäbel G, Bergert H, Thiery J, Teupser D. Expression of Chr9p21 genes CDKN2B (p15(INK4b)), CDKN2A (p16(INK4a), p14(ARF)) and MTAP in human atherosclerotic plaque. Atherosclerosis. 2011;214:264–270. doi: 10.1016/j.atherosclerosis.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 53.Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–259. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 54.Westhoff JH, et al. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension. 2008;52:123–129. doi: 10.1161/HYPERTENSIONAHA.107.099432. [DOI] [PubMed] [Google Scholar]
- 55.Shapiro SD, Ingenito EP. The pathogenesis of chronic obstructive pulmonary disease: advances in the past 100 years. Am J Respir Cell Mol Biol. 2005;32:367–372. doi: 10.1165/rcmb.F296. [DOI] [PubMed] [Google Scholar]
- 56.Aoshiba K, Nagai A. Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:596–601. doi: 10.1513/pats.200904-017RM. [DOI] [PubMed] [Google Scholar]
- 57.Tsuji T, Aoshiba K, Nagai A. Cigarette smoke induces senescence in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2004;31:643–649. doi: 10.1165/rcmb.2003-0290OC. [DOI] [PubMed] [Google Scholar]
- 58.Minagawa S, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L391–L401. doi: 10.1152/ajplung.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swigris JJ, Kuschner WG, Kelsey JL, Gould MK. Idiopathic pulmonary fibrosis: challenges and opportunities for the clinician and investigator. Chest. 2005;127:275–283. doi: 10.1378/chest.127.1.275. [DOI] [PubMed] [Google Scholar]
- 60.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 61.Alder JK, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16:238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Price JS, et al. The role of chondrocyte senescence in osteoarthritis. Aging Cell. 2002;1:57–65. doi: 10.1046/j.1474-9728.2002.00008.x. [DOI] [PubMed] [Google Scholar]
- 64.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 65.McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 66.Cohen G. The pathobiology of Parkinson’s disease: biochemical aspects of dopamine neuron senescence. J Neural Transm Suppl. 1983;19:89–103. [PubMed] [Google Scholar]
- 67.Liton PB, Challa P, Stinnett S, Luna C, Epstein DL, Gonzalez P. Cellular senescence in the glaucomatous outflow pathway. Exp Gerontol. 2005;40:745–748. doi: 10.1016/j.exger.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kozlowski MR. RPE cell senescence: a key contributor to age-related macular degeneration. Med Hypotheses. 2012;78:505–510. doi: 10.1016/j.mehy.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 69.Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 70.Stanley E, et al. Identification, proliferation, and differentiation of adult Leydig stem cells. Endocrinology. 2012;153:5002–5010. doi: 10.1210/en.2012-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Janzen V, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 72.Braun H, et al. Cellular senescence limits regenerative capacity and allograft survival. J Am Soc Nephrol. 2012;23:1467–1473. doi: 10.1681/ASN.2011100967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999;19:2109–2117. doi: 10.1128/mcb.19.3.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choudhury AR, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. 2007;39:99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- 75.Liu Y, et al. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009;8:439–448. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]