

Abstract
ZBP-89 can enhance tumor cells to death stimuli. However, the molecular mechanism leading to the inhibitory effect of ZBP-89 is unknown. In this study, 4 liver cell lines were used to screen for the target of ZBP-89 on cell death pathway. The identified Bak was further analyzed for its role in ZBP-89-mediated apoptosis. The result showed that ZBP-89 significantly and time-dependently induced apoptosis. It significantly upregulated the level of pro-apoptotic Bak. ZBP-89 targeted a region between -457 and -407 of human Bak promoter to stimulate Bak expression based on the findings of Bak promoter luciferase report gene assay and electrophoretic mobility shift assay. ZBP-89-induced Bak increase and ZBP-89-mediated apoptosis were markedly suppressed by Bak siRNA, confirming that Bak was specifically targeted by ZBP-89 to facilitate apoptosis. In conclusion, this study demonstrated that ZBP-89 significantly induced apoptosis of HCC cells via promoting Bak level.
Keywords: ZBP-89, hepatocellular carcinoma, Bak, apoptosis, proliferation
1. Introduction
Despite a great effort has been made to generate new treatments for hepatocellular carcinoma (HCC) for decades, surgical resection of tumor is still the most effective treatment for HCC. However, a significant number of patients with HCC are unresectable at the time of diagnosis. For these patients, antitumor drugs, especially chemotherapeutic agents, will be used to control the tumor growth. Moreover, HCC patients after surgical resection of tumor usually need to undergo chemotherapy to kill tumor cells that may not be completely removed by surgery. It is known that HCC often resists to chemotherapy. Therefore, there is a need to develop a better therapeutic regimen for HCC. Our previous study has suggested that ZBP-89 (also known as ZNF-148, BFCOL1 and BERF-1) might stabilize p53 in HCC cells and can thus sensitize tumor cells to cell death stimulation [1]. Our recent experiments have further shown that ZBP-89 facilitates HCC cell death induced by chemotherapeutic agents such as 5-fluorouracil (5-FU) [2].
ZBP-89 is a Kruppel-type, zinc finger transcription factor that binds to GC-rich elements and activates or represses known target genes. ZBP-89 has been shown to regulate expression of some molecules such as p21Waf1 and metalloproteinase 3, gastrin, ornithine decarboxylase and vimentin [3-8]. By regulating these molecules, ZBP-89 might participate in the control of cell proliferation and growth. In fact, when overexpressed in gastrointestinal cancer cells, ZBP-89 inhibits cellular proliferation and induces apoptosis [9,10]. Moreover, the overexpression of ZBP-89 increases caspase-3 levels, induces TUNEL-positive epithelial cells and abrogates adenoma development in the ApcMin/+ mouse [10], suggesting that ZBP-89 may function as a pro-apoptotic molecule or a tumor suppressor. ZBP-89 is known to influence the cell growth through either p53-dependent or independent mechanisms [9-13]. ZBP-89 might also inhibit the growth of tumors by suppressing angiogenesis [14]. However, the molecular mechanism responsible for the pro-apoptotic effect of ZBP-89 on tumor cells is not understood. In the present study, we explored how ZBP-89 induced apoptosis in HCC cells by studying Bcl-2 family members. We have identified the pro-apoptotic molecule Bak as a target for ZBP-89. ZBP-89 regulated the expression of Bak by directly binding its promoter. A knockdown of Bak significantly suppressed ZBP-89-induced apoptosis in HCC cells.
2. Materials and Methods
2.1 Cell lines and reagents
The human liver Chang cells and three HCC-derived cell lines: Hep3B, PLC/PRF/5 and SK-Hep-1 were obtained from the American Type Culture Collection (Rockville, MD) and were cultured with Dulbecco's modified Eagle medium (Gibco, Grand Island, NY) supplemented with 10% heat inactivated fetal bovine serum (HyClone, Logan, UT) and penicillin/streptomycin. The cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
2.2 Adenovirus ZBP-89 (Ad-ZBP-89) infection
Replication-deficient recombinant Ad-ZBP-89 expressing full-length Flag-tagged human ZBP-89 cDNA was prepared as previous description [11,12]. A wide range of multiplicity of infection (MOI) from 1 to 1000 was tested for virus infection. In our study, the cytotoxicity became apparent when MOI was over 100. We therefore chose MOI 100 as a standard dose for virus infection. Cells infected with Ad5 vector alone were set up as control for each experiment [11,12].
2.3 Cell viability assay
Cell death reflected by cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay [2]. Briefly, cells were grown in 96-well plates (1000 cells per well). After 24 h, cells were treated with different concentrations of Ad-ZBP-89, while control cells were treated with empty Ad vector. After 10 to 72 h, the cells were incubated with MTT solution (Sigma, St Louis, MO) for another 4 h at 37°C. The medium was then replaced with 200μl dimethyl sulfoxide. Reduced MTT was measured spectrophotometrically in a dual beam microtiter place reader at 560nm with a reference wavelength of 670nm.
2.4 Detection of apoptosis
Cells seeded onto 6-well plates were treated by MOI 100 Ad-ZBP-89 and incubated for 10, 24, 48 and 72 h. The control cells treated with empty Ad vector were included in every experiment. After the indicated time periods, cells were collected and the number of apoptotic cells was determined by TUNEL assay and quantified by flow cytometry. The results were plotted as a percent of the 0 h control conditions.
2.5 Western blot
Western blots were performed according to the standard methods published elsewhere [1-4]. Briefly, cells were lysed by incubation on ice for 15 min in RIPA buffer to isolate total proteins. The protein extracts (30μg) were separated by 10-15% SDS (sodium dodecyl sulphate)-polyacrylamide gel electrophoresis and transferred onto Hybond ECL nitrocellulose membrane for electoblotting. The primary antibodies used were anti-ZBP-89 (Abnova Corporation), anti-Bak (Cell Signalling Technology), anti-Bcl-2, (Santa Cruz Biotechnology) and anti-cytochrome c (ZYMED Laboratories). The primary reaction was followed by incubation with HRP-conjugated secondary antibodies and the blots were developed by ECL enhanced chemiluminescence system (Amersham Biosciences, Buckinghamshire, UK) and exposed to X-Ray film. As a control for sample loading, the blot was stripped and reprobed with anti-actin polyclonal antibody (Santa Cruz Biotechnology). The density of the protein bands was quantified using a GSP-700 scanner and Quantity One Image software (Bio-Rad, Hercules, CA).
2.6 Luciferase activity assay
Ten different Bak promoter fragments (181 – 1965pb) were prepared by the sequential 5′ deletion of the human Bak promoter (-1965, to -1) (Figure 3C) [15]. After the initial analysis of these 10 Bak promoter fragments, five truncated (about 100 bp each) Bak fragments were further generated from the Bak promoter region between -706 and -287 (Figure 3D). These fragments were cloned into pGL3 (Promega, Madison, WI). The sequence of different Bak fragments was confirmed by sequencing (ABI PRISMTM, Applied Biosystems, Foster City, CA). Cells were transfected with the Bak promoter plasmid, pGL3-basic plasmid or pGL3-control plasmid. After 6h of transfection, the transfected cells were infected with a MOI of 100 Ad-ZBP-89, then incubated for an additional 24h. After incubation, the lysate was isolated. Renilla luciferase assay was then performed on cell lysates using the Dual-Lucifearse Reporter Assay System (Promega, Madison, WI) according to the manufactures's instructions. Control Renilla luciferase activity was used to determine transfection efficiency. The activation ratio (fold activation) was determined by dividing the normalized firefly luciferase counts in the presence and absence of Ad-ZBP-89.
Figure 3. ZBP-89 enhanced the expression of Bak.
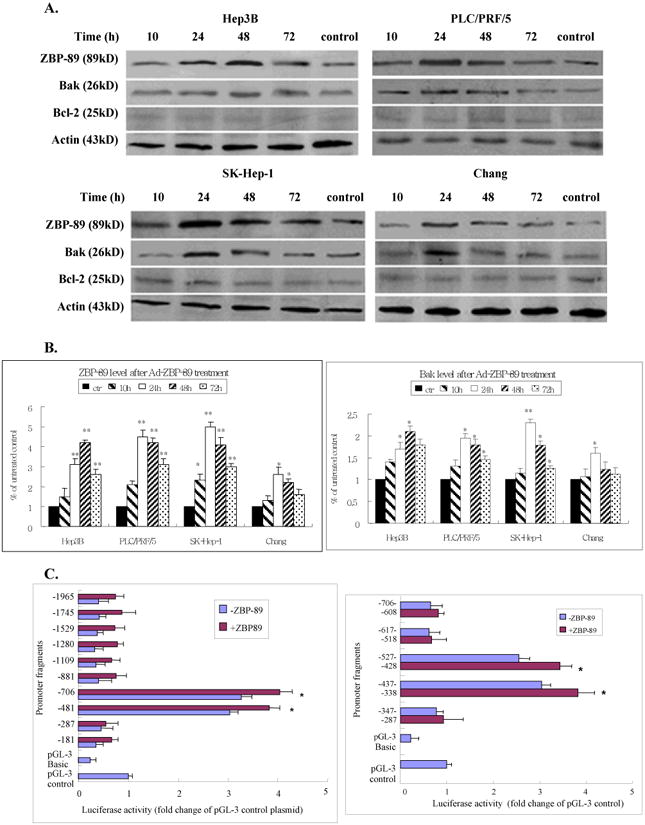
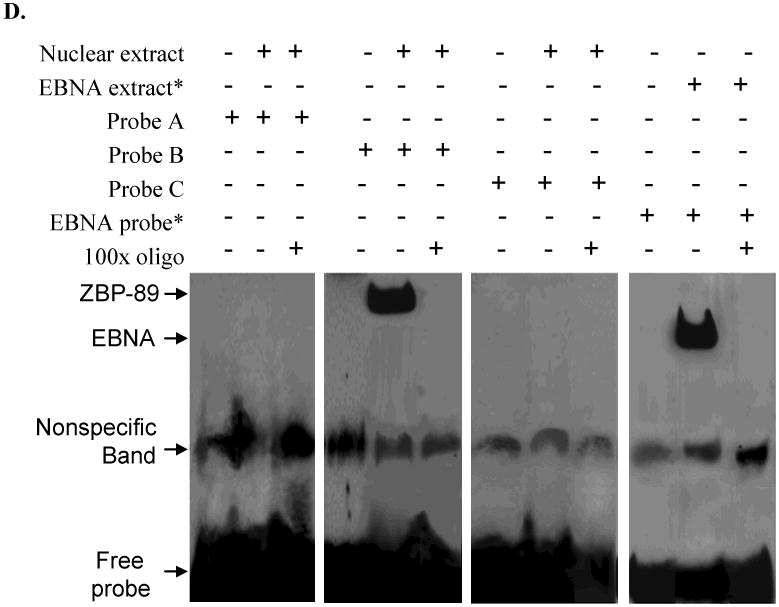
Hep3B, PLC/PRF/5, SK-Hep-1 and Chang cells were treated with MOI 100 Ad-ZBP-89 for 10-72 h. Cells treated with empty Ad5 vector were served as control. Total cell lysates were isolated and subjected to Western blot analysis using ZBP-89, Bak and Bcl-2 antibodies. An apparent increase of ZBP-89 and Bak was observed in Ad-ZBP-89-treated cells. Actin level was also examined to serve as a control of protein loading. A: representative Western blots. B: density analysis of ZBP-89 and Bak Western blot bands. Compared with the control: *p<0.05 and **p<0.01. C: SK-Hep-1 cells were transfected with individual Bak promoter luciferase construct, pGL3 Basic or pGL3 control plasmids. After 6h of transfection, the cells were infected with MOI 100 Ad-ZBP-89 for 24h. The c/ells were lysed for determination of relative firefly lucifease activity and fold activation was calculated. The results were obtained from 3 independent experiments, and the values are expressed as mean±SD (*p<0.05). D: SK-Hep-1 cells were infected with MOI 100 Ad-ZBP-89 for 24h. EMSA was carried out using the LightShift chemiluminescent EMSA kit (Pierce, Rockford, IL). The nuclear extract was incubated with oligonucleotide probe A 5′-AATACATGAAATATACTTAATAAAAATTCAAATGTTATAGAACTGAAAAA-3′ (-507 to -458), probe B 5′-GATGAAAAGTAAAAACAACCTATTCCCCAGAGGTAGCCACTGTCCATAGT-3′ (-457 to -408) and probe C 5′-TTCTATTTTAGATTCTTTCCTTTATACAAGATTATTATAGCTTCTATTTT-3′ (-407 to -357) respectively. The binding reactions were analyzed by electrophoresis using a native 6% polyacrylamide gel. EBNA (Epstein-Barr Nuclear Antigen) extract and EBNA probe were used as positive control and provided by the LightShift chemiluminescent EMSA kit (Pierce, Rockford, IL).
2.7 Electrophoretic mobility shift assay (EMSA)
A standard EMSA was used to further examine whether ZBP-89 protein from the human liver nuclear extracts binds to the Bak promoter region -457/-407. Cells were plated on a 6-well plate and transfected with pGL3-Bak promoter for 6h. Cells were then treated with MOI100 Ad-ZBP-89 for another 24h. At the end of the incubation, the nuclear extract was prepared. Three pairs of Bak Gel-shift complementary oligonucleotides (50bp each) were annealed and biotinylated using biotin 3′ end-labeling kit (Pierce, Rockford, IL). EMSA was carried out with the Light Shift chemiluminescent EMSA kit (Pierce, Rockford, IL) according to the manufacturer's instruction. Briefly, the binding reactions were analyzed by electrophoresis through a native 6% polyacrylamide gel. After transferring the binding reactions to the nylon membrane, it was cross-linked in a transilluminator and passed several chemiluminescence steps before being exposed to x-ray film to visualize the DNA-protein complexes.
2.8 RNA interference
For siRNA-mediated knockdown of ZBP-89 and Bak genes, the pre-designed siRNA targeting the exon 5 of the human ZBP-89 (NM_021964) was used to block of ZBP-89, and the target-specific 20-25nt siRNA was employed to knockdown the Bak gene in cells. ZBP-89 siRNA and its negative control were synthesized byAmbion Silencer Select Pre-designed siRNA (Ambion, Applied Biosystems). Bak siRNA and its negative control were obtained from Santa Cruz Biotechnology. Their respective complementary oligonucleotides were also synthesized, annealed to form double-stranded siRNA and were used in transfection at a final concentration of 50nM. They were mixed with 175μl of OPTI-MEM (Gibco) and incubated for 10 min. A mixture of 3μl of lipofectamine 2000 reagent (Invitrogen) and 12μl of OPTI-MEM was then added to the above and incubated for 20 min. Medium was removed from adherent HCC cells, they were then plated in 800μl of OPTI-MEM overlaid with 200μl of the siRNA mixture in a 6-well dish.
2.9 Data analysis
All results are expressed as mean±SD of at least three independent experiments. Differences between groups were examined for statistical significance using Student's t-test (paired, two sided) or one way ANOVA followed by Student's t-test. p<0.05 was used to indicate a statistically significant difference.
3. Results
3.1 Overexpression of ZBP-89 induces liver cell death
To examine the effect of Ad-ZBP-89 on cell death of liver cells, Hep3B, PLC/PRF/5, SK-Hep-1 and Chang cells were infected with 5 different concentrations of Ad-ZBP-89. The cells were collected at 10, 24, 48 and 72 h after infection to measure cell death which was reflected by the reduced cell viability. As shown in Figure 1, cell death induced by Ad-ZBP-89 appeared to be time- and dose-dependent. It was noted that the concentration of Ad-ZBP-89 at or higher than a MOI 100 caused apparent cell death. Therefore we choose a MOI 100 as the standard dose for the subsequent experiments. Compared with Chang cells, cell death induced by Ad-ZBP-89 was greater in the 3 HCC cells tested.
Figure 1. Celldeath induced by ZBP-89.

Cell death, reflected by the reduction of cell viability, was determined by MTT assay. Hep3B, PLC/PRF/5, SK-Hep-1 and Chang cells were culture in 96 well plates and infected with Ad-ZBP-89 at different concentrations. Cells infected with empty Ad5 vector were set up as control for each experiment. The infected cells were incubated for 10, 24, 48 and 72 huntil the MTT assay was performed. The viability of the cells with treatment was expressed as a relative value to that of the control. Data were reported as the mean±SD of three separate experiments. The significant difference in cell viability was observed between the cells without Ad-ZBP-89 treatment and those with Ad-ZBP-89 at indicated time points (*p<0.05 and **p<0.01).
It was not clear whether cell death induced by Ad-ZBP-89 was due to apoptosis or another mechanism. We therefore employed two methods to determine whether apoptosis occurred, the TUNEL assay and cytochrome (cyto) c release. The TUNEL assay showed that Ad-ZBP-89 significantly induced apoptosis of all cells tested in a time-dependent manner (Figure 2A). The SK-Hep-1 cells were the most sensitive to Ad-ZBP-89 while the Chang cells were the least sensitive to Ad-ZBP-89. The cyto c release experiments indicated that upon Ad-ZBP-89 treatment, cyto c was released from the mitochondria into the cytosol, resulting in an increase in the level of cytosolic cyto c and a decrease in mitochondrial cyto c (Figure 2B). The finding confirmed the result obtained by TUNEL assay and also suggested that the apoptosis induced was mediated via a mitochondrial-related pathway. The results of the TUNEL assay and cyto c release were in agreement with the cell death detected by the MTT assay. Therefore, we concluded that cell death induced by Ad-ZBP-89 was mainly due to apoptosis.
Figure 2. Ad-ZBP-89 induced apoptosis in liver cells.
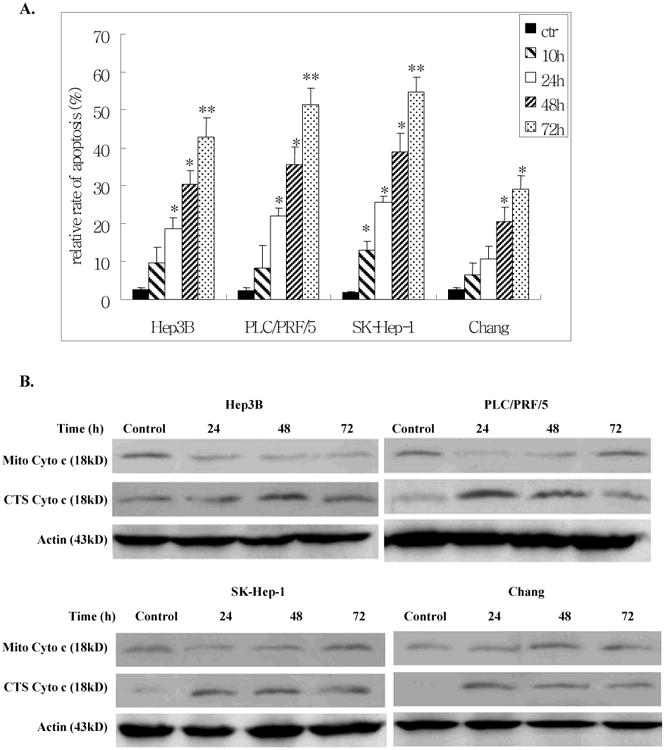
Hep3B, PCL/PRF/5, SK-Hep-1 and Chang cells were infected with MOI 100 Ad-ZBP-89 or with empty Advector as control. After 10, 24, 48 and 72 h, cells were harvested and analyzed by TUNEL for apoptosis (A). For the cytochrome (cyto) c release experiment, the mitochondria (Mito) and cytosolic (CTS) fractions were isolated and subjected to Western blot analysis to measure the level of cyto c in these two fractions (B). Actin was used as the loading controls. Compared with the control: *p<0.05 and **p<0.01.
3.2 ZBP-89 induces Bak expression in liver cells
To further investigate the mechanism of ZBP-89-induced apoptosis, we infected the liver cell lines with a MOI of 100 Ad-ZBP-89. The cells were collected at different time points between 10 and 72 h after virus infection. Total protein was extracted and Western blot was performed to examine the expression of ZBP-89, Bcl-2 and pro-apoptotic Bcl-2 family members including Bax, Bid and Bak. As shown in Figure 3, infection with the Ad-ZBP-89 dramatically increased ZBP-89 levels. Ad-ZBP-89 did not obviously affect Bcl-2 in cell lines tested (Figure 3A), suggested that Bcl-2 might not contribute to the apoptosis induced by Ad-ZBP-89. Among pro-apoptotic Bcl-2 family members, Ad-ZBP-89 significantly up-regulated the level of Bak at 24 or 48 h after the infection (Figures 3A & 3B) but did not obviously affect Mcl-1, Bax and Bid (data not shown). Similar to changes in the expression of Bak protein, the expression of Bak mRNA was also increased in cells infected with Ad-ZBP-89 (data not shown).
To explore the mechanism of Bak gene activation by ZBP-89, SK-Hep-1 cells were selected for the further investigation using Bak reporter constructs. Ten different Bak promoter fragments (181 – 1965bp) were prepared by the sequential 5′ deletion of the human Bak promoter (-1965, to -1) (Figure 3C) [15]. After the initial test with these 10 Bak promoter fragments, five shorter (about 100 bp each) Bak fragments were generated from the Bak promoter region between -706 and -287. We found that ectopic expression of ZBP-89 significantly activated the -706 and -288 Bak promoters in SK-Hep-1 cells (Figure 3C), suggesting that the protein-DNA binding site of ZBP-89 at the Bak promoter was within a 419bp region between -706 and -288. We further divided this 419bp fragment into five ∼100bp fragments with an ∼10bp overlap between segments. Luciferase assays were then performed to further identify the potential Bak-ZBP-89 binding site within the five shortened Bak promoter fragments. The result showed that ZBP-89 induced the Bak promoter activity through DNA elements residing at -527-428 and -437-338 (Figure 3C). The finding suggested that ZBP-89 inducible element resides within a 189 bp fragment between -527 and -338. In order to test whether the candidate protein ZBP-89 binds to the putative regulatory elements, we randomly generated three pairs of gel shift probes from sequences within the 189bp response element (Figure 3D). The results showed a clear shifted complex when the 50bp oligonucleotide B probe containing the Bak promoter sequence from -457 to -408 was incubated with Ad-ZBP-infected cells. Unlabeled probe competed with labeled probe for the shifted complex and thus blocked formation of the shifted complex. The finding confirmed the specificity of binding and demonstrated that a protein:DNA binding/interaction existed between ZBP-89 protein and the Bak promoter at -457 to -407 region.
3.3 Knockdown of ZBP-89 prevents Bak expression
In order to confirm ZBP-89 induction of Bak promoter, ZBP-89 siRNA was used to block ZBP-89 expression in liver cells. Double stranded ZBP-89 siRNA was transfected into cells and reduced ZBP-89 protein levels within 24 and 48 h after treatment (Figure 4). As expected, suppression of ZBP-89 expression by its siRNA effectively reduced the Bak expression in PLC/PRF/5 and SK-Hep-1 (Figure 4). These results convincingly showed that ZBP-89 was a positive regulator of the Bak promoter. Compared with no treatment, knockdown of ZBP-89 increased the level of Bcl-2 in HCC cells, especially in SK-Hep-1 and Hep 3B, but had no effect in Chang cells (Figure 4).
Figure 4. Effect of ZBP-89 on the expression of Bak and Bcl-2.
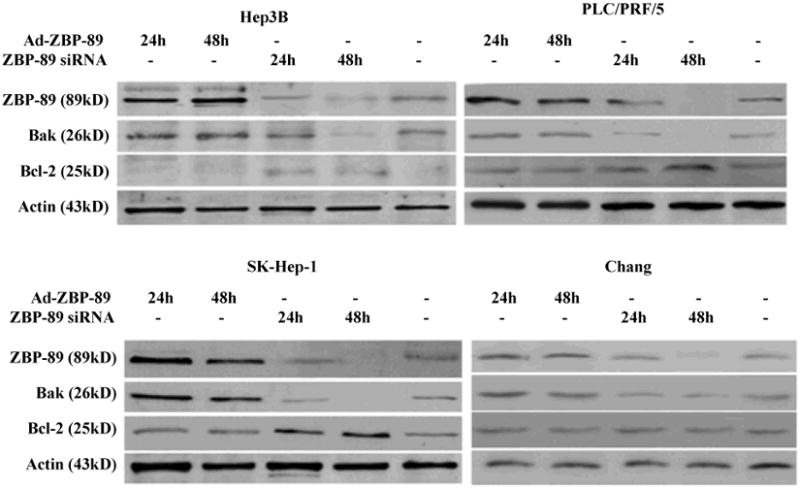
Whole cells lysates of HCC cells were harvested at 24 and 48 h after infection with Ad-ZBP-89 (MOI 100) or treatment with ZBP-89 siRNA (40nM). Bak and Bcl-2 proteins were measured by Western blot analysis. Actin was used as the control to ensurethe equal loading of lysates in each lane.
3.4 Knockdown of ZBP-89 increases cell growth
Having confirmed the apoptotic effect of ZBP-89 by TUNEL and cyto c release assays and the promoting effect of ZBP-89 on Bak by ZBP-89 siRNA, we further tested whether the inhibition of ZBP-89 had a positive effect on the growth of liver cells. We therefore treated the liver cells with ZBP-89 siRNA, then measured the cell death using the MTT viability assay. The findings showed that ZBP-89 siRNA significantly enhanced the growth of liver cells, compared with the mock control (Figure 5). The effect of ZBP-89 siRNA was time-dependent. Among the 4 types of cells tested, it appeared that SK-Hep-1 cells were most sensitive to ZBP-89 siRNA treatment, which correlated with the effect of Ad-ZBP-89 on the apoptosis in these cells (Figure 2).
Figure 5. Knockdown of ZBP-89 promoted the growth of cells.

Hep3B, PLC/PRF/5, SK-Hep-1 and Chang cells were transfected with ZBP-89 siRNA. MTT assay was used to measure the growth of cells which was reflected by the cell viability after 24-, 48- and 72-h cultures. Results were expressed as percentage of the cell viability with reference to the untreated control. Data were reported as the mean±SD of three separate experiments. Compared with the controls (no treatment or mock): *p<0.05 and **p<0.01.
3.5 Knockdown of Bak expression prevented ZBP-89-mediated induction of Bak and apoptosis
Bak siRNA was employed to confirm whether Bak was the target of ZBP-89. We first measured Bak level in HCC cells transfected with Bak siRNA and the result showed that the level of Bak was inhibited by its siRNA in liver cells and the level was completely blocked in Hep3B, PLC/PRF/5 and SK-Hep-1 cells at 48 h after transfection (Figure 6A). Bak siRNA blocked Bak expression in Chang cells at 72 h after transfection. These findings indicated that Bak siRNA was sufficient to inhibit Bak expression in liver cells. Pre-treatment with Bak siRNA reduced the ZBP-89-mediated expression of Bak, especially at 24 h point and in Hep3B, SK-Hep-1 and Chang cells (Figures 6A & 6B). The result again confirmed that ZBP-89 induces Bak expression.
Figure 6. Bak siRNA counteracted ZBP-89-mediated increase of Bak (A) and reduced ZBP-89-induced apoptosis (B).
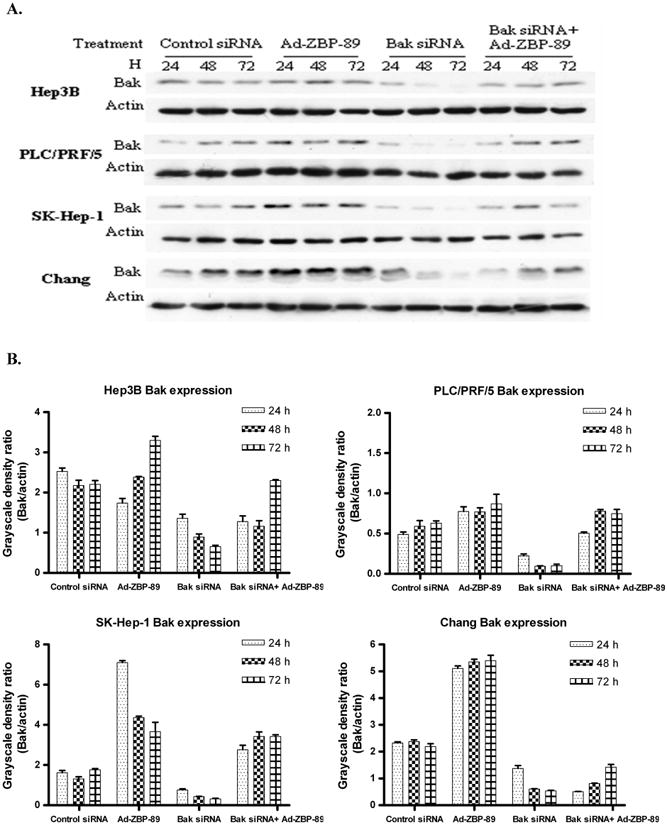

A. HCC cells were pre-transfected with 50nM Bak siRNA or non-specific control siRNA. 6 h later the cells were infectedwith MOI100 Ad-ZBP-89. The total cell lysate wasprepared at 24, 48 and 72 h after infection. Western blot was performed to detect Bak protein (26kD). Actin (43kD)was measured as a control to ensure the equal loading. B. The density of Western blot bands was determined and the ratio of Bak to actin was calculated. C. HCC cells were transfected with Bak siRNA or control siRNA. 24h after transfection, cells were treated with MOI 100 Ad-ZBP-89 for 48 h. Cells were harvested and analyzed by TNUEL assay for apoptosis induction. Assays were performed in triplicates and are representative for at least three independent experiments. Data were reported as the mean±SD. Compared with the controls (mock control or Bak siRNA): *p<0.05 and**p<0.01. Compared with Ad-ZBP-89: ++p<0.01.
The above experiments demonstrated that Bak siRNA blocks Ad-ZBP-89-mediated Bak expression and that ZBP-89 can induce apoptosis. We therefore queried whether suppression of Bak using siRNA prevented ZBP-89-mediated apoptosis. A TUNEL assay was performed to demonstrate whether ZBP-89-induced apoptosis could be reduced by knockdown of Bak expression in liver cells. The result showed that the ZBP-89-mediated apoptosis was significantly reduced by Bak siRNA in Hep3B, PLC/PRF/5 and SK-Hep-1 cells (Figure 6C). Among these three types of cells, the reduction of apoptosis by Bak siRNA was greatest in SK-Hep-1. The apoptosis in Chang cells was only slightly reduced by Bak siRNA. These findings were consistent with the fact that three HCC cell lines were much more sensitive to ZBP-89/Bak-mediated apoptosis than Chang cells (Figures 2 and 6B).
4. Discussion
In this study we have provided evidence that ZBP-89 induces apoptosis of HCC cells by inducing Bak. First, ZBP-89 can significantly enhance the protein level of Bak, which can be blocked or significantly inhibited by either ZBP-89 siRNA or Bak siRNA. Second, ZBP-89-induced apoptosis is markedly inhibited by Bak siRNA. To the best of our knowledge, there are no publications addressing the relationship between Bak and ZBP-89. Bak is a well known cell death initiator in the apoptotic signaling cascade [16,17]. Upon stimulation, Bak undergoes some conformational changes, leading to formation of higher order Bak multimers, mitochondrial membrane permeabilization and the release of pro-apoptotic factors such as cytochrome c into the cytoplasm. Consistent with this known concept, we have observed a significant increase in cytochrome c in the cytoplasm upon ZBP-89 treatment. The role of Bak in the hepatocarcinogensis or HCC treatment has also been documented. The level of Bak is reduced [18,19] or even non-detectable in HCC cells [20]. The pro-apoptotic role of Bak in HCC cells is further supported by several experiments in which different agents induce apoptosis in HCC cells by stimulating Bak expression [21-23]. The overexpression of Bak may sensitize HCC cells to apoptosis induced by the chemotherapeutic agent doxorubicin [24]. It is well known that therapy resistance is a common clinical problem in HCC management. Resistance to apoptosis is a principal mechanism through which HCC cells escape from various cell death inducers [25]. Therefore, the finding that ZBP-89 induces apoptosis by promoting Bak is therapeutically significant for HCC management.
ZBP-89 did not obviously affect the level of Bcl-2 in all four cell lines tested, indicating that Bcl-2 dose not appear to play a role in ZBP-89-mediated apoptosis of liver cells. Bai et al. previously examined several anti- and pro-apoptotic molecules regulated by ZBP-89 [13]. Indeed, they found that ZBP-89 siRNA induced Bcl-xL in gastrointestinal cancer cells, indicating that this anti-apoptotic molecule is regulated by ZBP-89. The finding suggests that Bcl-xL might be the preferred anti-apoptotic protein targeted in those cells treated by ZBP-89. However, whether this is the case in HCC cells remains further investigation.
By using RT-PCR and Western blot analyses, we showed that all cells tested (Hep3B, PLC/PRF/5, SK-Hep-1 and Chang) are able to constitutively express endogenous Bak. The base level of Bak can be greatly enhanced by ZBP-89 treatment and the increase in Bak is functional in terms of apoptotic induction in HCC cells. In order to better understand the role of ZBP-89, a transcriptional factor, in the induction of Bak, we isolated overlapping Bak promoter fragments. Once a 189 bp element was identified using the reporter constructs, EMSAs were used to narrow the regulatory element to a 50 bp fragment between -457 and -407. This finding was consistent with in silico analysis of the Bak promoter predicting a ZBP-89 binding site between -435 and -426 using TFSEARCH algorithms (http://motif.genome.ad.jp/). Therefore, all these results support that the human Bak promoter contains the binding site for ZBP-89 and that ZBP-89 is able to bind the promoter to induce Bak expression.
Therefore, we conclude that ZBP-89 markedly induced Bak expression by interacting with its promoter. Subsequently, Bak induction causes apoptosis of HCC cells. We have noted that factors other than Bak are likely to also contribute to the apoptosis induced by ZBP-89 because Bak siRNA does not completely prevent the ZBP-89-mediated apoptosis. Nevertheless, our finding contributed to understanding how ZBP-89 induces apoptosis in HCC, which may provide some useful information for us to improve the efficacy of anti-HCC chemotherapy.
Acknowledgments
This work was supported by the Research Grants Council of the Hong Kong SAR. No: CUHK4551/05M and CUHK462009 to PBSL and GGC, and Public Heath Service Grant R01 DK-55732 to JLM.
References
- 1.Chen GG, Merchant JL, Lai PBS, Ho RL, Hu X, Okada M, Huang SF, Chui AK, Law DJ, Li YG, Lau WY, Li AK. Mutation of p53 in recurrent hepatocellular carcinoma and its association with the expression of ZBP-89. Am J Pathal. 2003;162:1823–1829. doi: 10.1016/S0002-9440(10)64317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen GG, Chan UPF, Bai L, Fung KY, Tessier A, To AK, Merchant JL, Lai PB. ZBP-89 reduces the cell death threshold by increasing caspase-6 and S phase arrest in hepatocellular carcinoma. Cancer Letters. 2009;283:52–58. doi: 10.1016/j.canlet.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai L, Merchant JL. ATM phosphorylates ZBP-89 at Ser202 to potentiate p21waf1 induction by butyrate. Biochem Biophys Res Commun. 2007;359:817–821. doi: 10.1016/j.bbrc.2007.05.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bai L, Kao JY, Law DJ, Merchant JL. Recruitment of ataxia-telangiectasia mutated to the p21(waf1) promoter by ZBP-89 plays a role in mucosal protection. Gastroenterol. 2006;131:841–852. doi: 10.1053/j.gastro.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Borghaei RC, Rawlings PL, Jr, Javadi M, Woloshin J. NF-kappaB binds to a polymorphic repressor element in the MMP-3 promoter. Biochem Biophys Res Commun. 2004;16:182–188. doi: 10.1016/j.bbrc.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 6.Merchant JL, Iyer GR, Taylor BR, Kitchen JR, Mortensen ER, Wang Z, Flintoft RJ, Michel JB, Bassel-Duby R. ZBP-89, a Krüppel-like zinc finger protein, inhibits epidermal growth factor induction of the gastrin promoter. Mol Cell Biol. 1996;16:6644–6653. doi: 10.1128/mcb.16.12.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Law GL, Itoh H, Law DJ, Mize GJ, Merchant JL, Morris DR. Transcription factor ZBP-89 regulates the activity of the ornithine decarboxylase promoter. J Biol Chem. 1998;273:19955–19964. doi: 10.1074/jbc.273.32.19955. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Zhang X, Salmon M, Zehner ZE. The zinc finger repressor, ZBP-89, recruits histone deacetylase 1 to repress vimentin gene expression. Genes Cells. 2007;12:905–918. doi: 10.1111/j.1365-2443.2007.01104.x. [DOI] [PubMed] [Google Scholar]
- 9.Okada M, Tessier A, Bai L, Merchant JL. p53 mutants suppress ZBP-89 function. Anticancer Res. 2006;26:2023–2028. [PubMed] [Google Scholar]
- 10.Law DJ, Labut EM, Merchant JL. Intestinal overexpression of ZNF148 suppresses ApcMin/+ neoplasia. Mamm Genome. 2006;17:999–1004. doi: 10.1007/s00335-006-0052-4. [DOI] [PubMed] [Google Scholar]
- 11.Bai L, Merchant JL. Transcription factor ZBP-89 cooperates with histone acetyltransferase p300 during butyrate activation of p21waf1 transcription in human cells. J Biol Chem. 2000;275:30725–30733. doi: 10.1074/jbc.M004249200. [DOI] [PubMed] [Google Scholar]
- 12.Bai L, Merchant JL. ZBP-89 promotes growth arrest through stabilization of p53. Mol Cell Biol. 2001;21:4670–5683. doi: 10.1128/MCB.21.14.4670-4683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai L, Yoon SO, King PD, Merchant JL. ZBP-89-induced apoptosis is p53-independent and requires JNK. Cell Death Differ. 2004;11:663–673. doi: 10.1038/sj.cdd.4401393. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Xiong JW, Shelley CS, Park H, Arnaout MA. The transcription factor ZBP-89 controls generation of the hematopoietic lineage in zebrafish and mouse embryonic stem cells. Development. 2006;133:3641–3650. doi: 10.1242/dev.02540. [DOI] [PubMed] [Google Scholar]
- 15.Liu HC, Chen GG, Vlantis AC, Chan ATC, van Hasselt CA. Inhibition of apoptosis in human laryngeal cancer cells by E6 and E7 oncoproteins of human papillomavirus 16. J Cell Biochem. 2008;103:1125–1143. doi: 10.1002/jcb.21490. [DOI] [PubMed] [Google Scholar]
- 16.Cory S, Adam JM. The Bcl-2 family: regulators of the cellular life-or-death switch. Nature Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, Dive C, Hickman JA. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol. 1999;144:903–914. doi: 10.1083/jcb.144.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu LX, Jiang HC, Liu ZH, Zhu AL, Zhou J, Zhang WH, Wang XQ, Wu M. Gene expression profiles of hepatoma cell line BEL-7402. Hepato-Gastroenterol. 2003;50:1496–1501. [PubMed] [Google Scholar]
- 19.Rousseau B, Ménard L, Haurie V, Taras D, Blanc JF, Moreau-Gaudry F, Metzler P, Hugues M, Boyault S, Lemière S, Canron X, Costet P, Cole M, Balabaud C, Bioulac-Sage P, Zucman-Rossi J, Rosenbaum J. Overexpression and role of the AT Pase and putative DNA helicase RuvB-like 2 in human hepatocellular carcinoma. Hepatology. 2007;46:1108–1118. doi: 10.1002/hep.21770. [DOI] [PubMed] [Google Scholar]
- 20.Madesh M, Hajnoczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol. 2001;155:1003–1015. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu R, Zhai Q, Liu W, Liu X. An insight into the mechanism of cytotoxicity of ricin to hepatoma cell: roles of Bcl-2 family proteins, caspases, Ca(2+)-dependent proteases and protein kinase C. J Biol Chem. 2001;81:583–593. doi: 10.1002/jcb.1076. [DOI] [PubMed] [Google Scholar]
- 22.Wang QF, Chen JC, Hsieh SJ, Cheng CC, Hsu SL. Regulation of Bcl-2 family molecules and activation of caspase cascade involved in gypenosides-induced apoptosis in human hepatoma cells. Cancer Lett. 2002;183:169–178. doi: 10.1016/s0304-3835(01)00828-x. [DOI] [PubMed] [Google Scholar]
- 23.Li MY, Deng H, Zhao JM, Dai D, Tan XY. PPARgamma pathway activation results in apoptosis and COX-2 inhibition in HepG2 cells. World Journal of Gastroenterol. 2003;9:1220–1226. doi: 10.3748/wjg.v9.i6.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Wang WL, Yang XK, Yu XX, Hou YD, Zhang J. Inducible overexpression of Bak sensitizes HCC-9204 cells to apoptosis induced by doxorubicin. Acta Pharmacologica Sinica. 2000;21:769–776. [PubMed] [Google Scholar]
- 25.Fisher DE. Apoptosis in cancer therapy: crossing the threshold. Cell. 1994;78:539–542. doi: 10.1016/0092-8674(94)90518-5. [DOI] [PubMed] [Google Scholar]