

Abstract
Natural products account for a significant proportion of modern day therapeutic agents. However, the discovery of novel compounds is hindered by the isolation process, which often relies upon extraction and chromatographic separation techniques. These methods, which are dependent upon the physicochemical properties of the compounds, have a limited ability to both purify and concentrate the minor components of a biological extract. We have devised an isolation strategy based upon an orthogonal chemical feature, namely, functional group composition. Development of a functional group-targeted method is expected to achieve exceptional resolution given the large number of distinct moieties present in natural product extracts. Here, we describe the generation of controllably reversible covalent enrichment tags for the chemoselective isolation of alcohol-containing natural products from complex mixtures.
Introduction
Natural products have long been recognized as privileged scaffolds due to their high propensity to interact with biological targets.1 Thus, it is not surprising that naturally produced compounds and their derivatives are an essential component of today's pharmaceutical arsenal, with nearly half of the currently available drugs being of biological origin.2 Natural products have also played myriad roles as chemical probes.3,4 The continued investigation of the small molecule repertoire of organisms such as plants and microbes is bound to prove fruitful for the discovery of new classes of bioactive compounds. Efforts to identify new leads, however, are often frustrated by the cumbersome and inefficient process of compound isolation.
Generally, natural products are identified by extraction of biological material (e.g., plant material, microbe pellet) and the crude extract is assayed for a desired activity.5–7 Active extracts are then further purified, either by extraction and/or chromatographic methods. Although considerable advances have been made in separation technology yielding strategies that minimize solvent consumption and show increased resolving power,8–10 purification of the active components of a crude extract, which often represent less than 1% by weight, is still considered a major bottleneck in natural products discovery.6 Thus, the need to develop new isolation technologies is clear.
Current strategies facilitate enrichment based on a restricted set of separation mechanisms that are dependent upon molecular properties such as solubility, charge state, or size. An alternative and complementary approach is to target functional group composition. In the early 1980s, Fr echet and coworkers demonstrated the capture of α,β-unsaturated lactone-containing allergens from natural oils with polymer-supported reagents.11,12 This approach, which targeted a very specific functionality, has not been significantly utilized because a practical natural products discovery toolkit requires the development of strategies to address more prevalent functional groups such as the amine, carboxylic acid and alcohol using highly selective and readily reversible reaction conditions.
We recently reported the development of a functional group-targeted method for selective enrichment and profiling of metabolites for metabolomic studies.13,14 The devised methods facilitate exploration of the amine, thiol, carboxylic acid and ketone/alde-hyde complements of a cell. Inspired by these results, we sought to extend our functional group-based separation methods to the isolation of natural products. However, for metabolomic studies, we utilized a permanent tagging strategy in which the metabolites were covalently altered with a tag that subsequently aids in their detection by mass spectrometry. We anticipated that a permanent tag would be detrimental to natural products discovery efforts because of its unpredictable effect on biological activity. Thus, we aimed to develop a tagging strategy that employs reactions that are controllably reversible to enable covalent capture of small molecules, facilitating their chemoselective enrichment, followed by release of the unaltered chemical structures. With this approach, targeted compounds, including low-abundance molecules, would be enriched independent of their physicochemical properties.
The devised agents, referred to as reversible enrichment tags, are immobilized on solid support to enable the selective isolation of fractions of natural products present in a complex biological matrix (Fig. 1). Following capture, elimination of compounds that do not contain the targeted functional group is accomplished simply by washing of the resin. Finally, enriched compounds are liberated from the resin using gentle conditions that we expect will not interfere with either the structural integrity of the natural products or subsequent bioassays. Here, we describe the development of a reversible enrichment tag for the capture of alcohol-containing natural products.
Fig. 1.
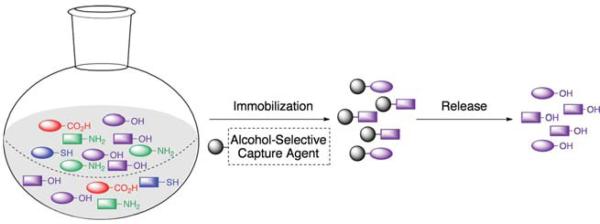
Depiction of the chemoselective enrichment strategy. Compounds from a crude natural product extract are immobilized onto reversible tagging agents. After enrichment, compounds are released from the solid support to enable direct structural and functional characterization.
Results and discussion
To generate a functional group-targeted strategy, a number of specifications must be met. First, not only must the capture reaction be chemoselective, but the devised reagent must also facilitate the immobilization of a breadth of alcohols (e.g., primary, secondary). Additionally, the covalent bond formed between the natural products and the solid support must be sufficiently stable to withstand extensive washing protocols to remove all compounds not containing the targeted functionality. However, the formed bond must be readily cleavable to permit release of the enriched compounds. As mentioned, the conditions for immobilization and release must be gentle; however, the reagents utilized and their resulting byproducts must also enable direct structural and functional characterization of the isolated compounds. Accordingly, only volatile reagents and byproducts should be employed or generated during the course of the reactions to prevent the need for purification after release of the enriched compounds.
Natural products are rich in oxygen atoms, most often found as alcohols or in heterocycles.15,16 To facilitate the enrichment of alcohol-containing natural products, we pursued development of a reversible tagging strategy using a silyl-functionalized solid support (Scheme 1). Alcohol immobilization is accomplished by activation of the resin to generate the resin-bound silyl triflate or chloride, followed by addition of the alcohol and triethylamine. After extensive washing, release is performed using HF-pyridine followed by quenching with TMSOMe. Though some functionalities, such as the epoxide, may be sensitive to HF-pyridine, we found that this reagent provides the best overall cleavage efficiency. Alternative conditions were significantly less general requiring long reaction times, elevated reaction temperatures and/or subsequent purification steps.
Scheme 1.
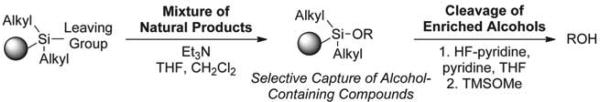
General strategy for enrichment of alcohol-containing natural products.
Although silyl-functionalized resins have been extensively explored for use in synthetic chemistry,17–21 they have not been applied to complex mixtures of compounds. Here, we sought to identify a resin that could not only efficiently capture a breadth of alcohols, but one that could also do so in a chemoselective fashion.
Exploration of a silyl-functionalized resin
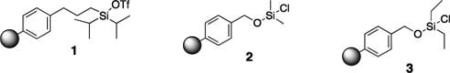
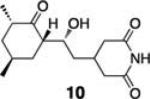
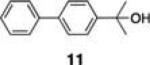
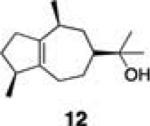
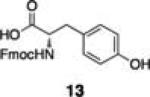
Initial studies examined the utility of a commercially available solid support, (4–methoxyphenyl) diisopropylsilylpropyl polystyrene resin, which is activated as the triflate just prior to use (Table 1, 1).17 To test the proposed strategy, we sought to capture several alcohol standards, followed by resin washing, release and quantification by liquid chromatography-mass spectrometry (LC-MS) analysis. We selected eleven standards with varying physicochemical properties and alcohol accessibility (Table 1, 4–14) and subjected them to the activated resin (0.1 equiv each compared to resin, capture performed as mixtures of 3–4 compounds). Enrichment yields were highly dependent upon the steric accessibility of the alcohol. For example, enrichment of most primary and secondary alcohol- and phenol-containing compounds proceeded well (4–10, 13 and 14; yields indicate the amount of compound detected following immobilization and release). However, tertiary alcohols (11 and 12) were not enriched by resin 1.
Table 1.
Yields of enrichment for alcohol-, amine-, carboxylic acid- or thiol-containing small molecule standards with capture resins. Enrichment was quantified by LC-MS.
|
|||
|---|---|---|---|
|
70% | 0% | 14% |
|
|
63% | 87% | 68% |
|
60% | 0% | 62% |
|
90% | 81% | 92% |
|
83% | 28% | 66% |
|
68% | 97% | 91% |
|
68% | 89% | 29% |
|
0% | 0% | 0% |
|
0% | 53% | 0% |
|
67% | 66% | 79% |
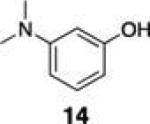
|
53% | 27% | 21% |
| Compounds to Assess Chemoselectivity | |||
|
3% | 0% | 0% |
|
45% | 0% | 0% |
|
46% | 0% | 0% |
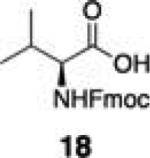
|
38% | 0% | 0% |
|
96% | 0% | 0% |
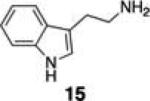
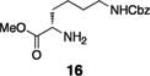
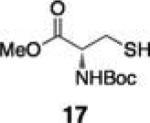
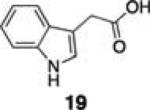
Next, we tested the chemoselectivity of resin 1 by subjecting amine-, thiol- and carboxylic acid-containing compounds (15–19) to the enrichment protocol. All three functional group classes were captured in moderate to high yields clearly demonstrating that exploration of alternative resin architectures would be required to identify an alcohol-selective reagent.
Generation of chemoselective enrichment reagents
Synthesis and evaluation of a library of resins with varying linkers and silyl substitution patterns revealed that a benzyl alcohol-derived siloxyl resin afforded the required chemoselectivity (Table 1, resins 2 and 3; resins found to be non-selective not shown). In addition to alcohol selectivity, these siloxyl-functionalized resins have several other advantages over the silyl-functionalized resin 1. First, synthesis of the activated resins is accomplished in one step from hydroxymethyl polystyrene resin (Scheme 2). As a result, each analog is easy to produce. In general, synthesis of silanefunctionalized resins requires multiple steps rendering the construction of analogs more labor intensive.
Scheme 2.

Synthesis and regeneration of benzyl alcohol-derived resins. Hydroxylmethyl polystyrene resin can be functionalized with a variety of dichlorodialkyl silanes yielding the capture reagents. Following alcohol enrichment and cleavage, the starting resin is regenerated as indicated by the green arrow.
A further benefit of the siloxyl-functionalized resins is their ability to be regenerated (Scheme 2; Fig. S1, ESI†). Upon cleavage of enriched alcohols from these solid supports, the starting resin is reproduced, whereas cleavage of silane-functionalized resins yields an unrecoverable silylfluoride product. As evidence of this advantage, we determined that the loading capacities of resins 2 and 3 do not change after subjecting them to a round of alcohol capture and release followed by reactivation (Table S1, ESI†). Examination of the total ion chromatograms of enriched compounds also illustrated the high level of purity provided by this strategy (Fig. S2, ESI†).
Comparison of the dimethyl- and diethyl-substituted resins (2 and 3, respectively) demonstrated that resin 3 is superior in overall enrichment efficiency. In particular, the less sterically demanding resin 2 shows poor yields for less hindered alcohols as these compounds suffer from premature cleavage off of the resin during the washing protocol (e.g., compounds 4, 6, 8). Additionally, the less hindered resin 2 is significantly more reactive than resin 3 increasing its susceptibility to hydrolysis and/or inconsistent enrichment yields (data not shown). Interestingly, resin 2 promotes recovery of a tertiary alcohol (12) suggesting that this resin may be useful for the isolation of extremely hindered alcohols.
Enrichment of an endogenous natural product
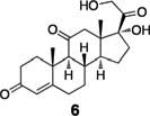
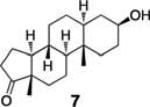
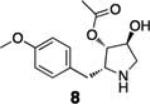
To demonstrate the utility of this strategy in a complex extract, we challenged the technology with the enrichment of an endogenously produced natural product, anisomycin (8). This compound is produced by Streptomyces griseolus22 and is a known inhibitor of protein biosynthesis.23 We subjected the extract of this organism to our capture strategy (resin 3) and successfully isolated the expected natural product with a similar yield to that observed upon capture of the pure standard [82% recovery (31 nmol) compared to 66% with standard; Fig. 2].
Fig. 2.

Extracted ion chromatograms of alcohols captured from Streptomyces griseolus broth. Endogenously-produced anisomycin (8) was captured (30.5 nmol, 82% recovery). Two additional alcohols were spiked into the extract (223 nmol each) and enriched, cortisone (6, 113 nmol, 51% recovery) and trans-androsterone (7, 119 nmol, 53% recovery).
To further establish the alcohol enrichment capabilities and chemoselectivity of this strategy in the context of an extract, we added two alcohol-containing compounds (6 and 7) and one each of an amine-, thiol-, and carboxylic acid-containing standard (15, 17, 18) prior to capture. Both alcohols were detected in the cleavage solution (Fig. 2) providing additional evidence of the utility of resin 3 for alcohol isolation. The amine-, thiol-, and carboxylic acid-containing compounds were not enriched from the extract (Fig. S4, ESI†) validating our initial chemoselectivity results.
Next, we calculated the ratio of enrichment to further illustrate that alcohol-containing compounds are dramatically enriched compared to molecules containing other functional groups. Following the capture and release protocol, at least a 24-fold enrichment of the alcohols was seen in comparison to compounds not containing this functionality in all cases (Table S2, ESI†). Extract material was also subjected to deactivated resin to confirm that the observed alcohol enrichment was only a result of capture by the activated silane and not due to non-specific binding to the resin. These data confirm that unactivated resin does not enable the capture of alcohol-containing compounds (Table S3, ESI†). Thus, the devised strategy readily facilitated chemoselective enrichment of alcohol-containing natural products from a crude biological extract.
Conclusions
Facile identification of new natural products will require the development of novel isolation strategies. Here, we have described the generation of a functional group-targeted approach, providing the first example of a chemoselective method for enrichment of alcohol-containing compounds. In addition to natural products exploration, we expect that the described reagents will find utility in diverse applications such as chemoselective synthetic scavenging and isolation reagents and metabolomic profiling. Future studies will pursue the generation of reversible enrichment tags for additional functional group classes and application of the described alcohol-selective reagent to natural products discovery.
Experimental
General information
Resin reactions were performed in fritted vessels (Biospin vessels from Biorad) under an inert atmosphere of Ar or N2. Resins were purchased from EMD Biosciences. All other chemicals were purchased from VWR or Sigma-Aldrich and used without further purification. Solvents were purchased as anhydrous and not further purified. Triethylamine was distilled over barium oxide.
Sample analysis was performed on an Agilent 1200 LC-MSTOF equipped with a reverse phase column (ZORBAX Eclipse Plus C18, Rapid Resolution HT, 1.8 micron, 2.1 50 mm). All sample and standard curve analysis was performed with the following gradient: isocratic elution of 100% A at 0.5 mL min-1 for 2 min followed by a linear gradient of 0–100% B at 0.5 mL min-1 over 6 min, then an isocratic elution for 2 min at 100% B, and reequilibration with 100% A for 4 min (A: 95 : 5 H2O:CH3CN, 0.1% ammonium acetate; B 95 : 5 CH3CN:H2O, 0.1% ammonium acetate). Fragmentation voltages ranged from 75V to 175V. Gel-phase 13C nuclear magnetic resonance (NMR) spectra20 were recorded on a Varian I500 or a Varian VXR-400 instrument. Chemical shifts are reported relative to residual solvent peaks in parts per million. Infrared (IR) spectra were recorded using a Perkin Elmer Spectrum One FT-IR as a KBr pellet.
(4-Methoxyphenyl)diisopropylsilylpropyl polystyrene resin (1) activation
A 10 mL fritted polypropylene column was charged with 126 mg (0.176 mmol) of (4-methoxyphenyl)diisopropylsilylpropyl polystyrene resin (loading capacity of 1.4 mmol g-1) and equipped with a rubber septum. The resin was flushed with N2 for 10 min. A solution of 94.0 μL trifluoroacetic acid (1.06 mmol, 6.0 equiv relative to resin) and 2.3 mL anhydrous CH2Cl2 was added to the resin. The resin turned red and was agitated at room temperature for 30 min. After activation, the resin was washed with 2 mL anhydrous CH2Cl2 three times and aliquoted into four 2 mL oven dried vials using 1.2 mL of anhydrous CH2Cl2. The vials were capped and placed under N2. Resin was utilized immediately in coupling reactions.
Benzylsiloxane resin (2 and 3) synthesis
A 20 mL scintillation vial was charged with 200 mg (0.22 mmol) of hydroxymethyl polystyrene resin (loading capacity of 1.1 mmol g-1) and equipped with a rubber septum. After flushing the resin with N2 for 10 min, the vial was charged with 4 mL anhydrous CH2Cl2 and the resin was allowed to swell for 5 min. To the swollen resin was added 430 μL (3.1 mmol, 14 equiv relative to Si) of freshly distilled triethylamine, 330 μL (2.2 mmol, 10 equiv relative to Si) of dichlorodiethylsilane or 300 μL dichlorodimethylsilane (2.2 mmol, 10 equiv relative to Si) and 32 mg (0.26 mmol, 1.2 equiv relative to Si) of 4-dimethylaminopyridine. The vial was capped and the reaction was agitated for 4 h at room temperature. The resin was filtered over a 10 mL fritted polypropylene column and washed with 2 × 10 mL andanhydrous CH2Cl2 aliquoted into four 5 mL oven dried vials using 2 mL of anhydrous CH2Cl2. Three aliquoted vials were capped and placed under N2 for subsequent coupling reactions, the fourth vial was transferred back to a biospin vial and rinsed three times with 1 : 1 THF:MeOH to hydrolyze the chlorine and yield deactivated resin to act as a control. Resin was used within 10 min of generation to prevent excessive hydrolysis. Dimethylchloro-benzylsiloxane resin (2): FT-IR (on-bead KBr pellet) νmax: 3059, 1601, 1260, 1017, 909, 733, 538 cm −1; gel-phase 13C NMR (125 MHz, CDCl3) δ: 53.3, 40.3, 2.2. Diethylchloro-benzylsiloxane resin (3): FT-IR (on-bead KBr pellet) nmax: 3060, 1601, 1017, 909, 734, 539 cm −1; gel-phase 13C NMR (125 MHz, CDCl3) δ: 53.3, 40.3, 8.3, 6.2.
Capture of alcohols onto resin
To activated resin was added 50 mL of freshly distilled triethylamine (0.35 mmol, 8.0 equiv relative to Si). A stock solution of model alcohols was prepared in 500 μL of anhydrous THF, which contained three or four compounds. From this solution, 100 μL was added to each reaction vessel (0.1 equiv of each alcohol relative to resin loading capacity). The coupling reactions were gently agitated at room temperature overnigh (~16 h). The resin was transferred to a 2 mL fritted polypropylene column and subjected to the wash protocol described in the Supplementary Information. Resins were dried overnight at room temperature in a vacuum desiccator at 30 mmHg.
Release of alcohols from resin
Coupled resin was transferred to polypropylene vials. To the resin was added 500/50/50 μL (v/v) of a freshly prepared solution of THF/HF$pyridine (70/30 wt%)/pyridine (2.0 mmol of HF, 45 equiv relative to Si) and the reaction was gently agitated at room temperature for 3 h. To this was added 500 μL of TMSOMe (3.6 mmol, 83 equiv relative to Si) to quench excess HF and the resin was agitated for an additional 30 min at room temperature. The resin was washed with THF (3 × 1 mL × 10 min) and filtered over a 1 mL fritted polypropylene column into a 20 mL scintillation vial. The THF wash was concentrated under reduced pressure with no heating and the sample was dissolved in 5 mL of 2 : 1 : 1 H2O/THF/MeOH. Analysis was performed by injection of 1 μL of this solution onto a LC-MS-TOF and comparing the observed peak area to that of standard curve data.
Preparation of Streptomyces griseolus extract
Streptomyces griseolus (Waksman) was purchased from ATCC as a freeze-dried pellet. Cells were grown as described previously.24 Briefly, 20 mL cultures were grown in ISP Medium 2 at 28 °C for ten days yielding white spores. Cells were pelleted by centrifugation at 2000 g for 10 min at 25 °C. The broth was decanted, transferred to a separatory funnel and extracted according to the following protocol: EtOAC (2 × 300 mL), CHCl3 (2 × 300 mL), hexanes (2 × 300 mL), diethyl ether (2 × 300 mL), 3 : 1 CHCl3:MeOH (3 × 300 mL). All organic washes were combined and evaporated under reduced pressure yielding 171 mg of crude material. This material was dissolved in 2 mL of THF and 2 mL of MeOH. From this solution, 5 μL was analyzed on the LC-MS-TOF to quantify the amount of anisomycin present in the crude extract (223 nmol).
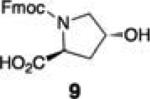
Enrichment of endogenously produced anisomycin (8)
Bacterial extract material (29 mg) was dissolved in 3 mL of anhydrous THF and 2.4 mL of anhydrous DMSO. The chemoselective compound mixture (600 mL solution containing 223 nmol each of cortisone, trans-androsterone, N-Boc-L-Cys-OMe, tryptamine, and Fmoc-Val-OH in THF) was added and the total volume was brought to 6 mL with THF. Six 20 mL scintillation vials were charged with 400 mg (“10 equivalents”; see below) of hydroxymethyl polystyrene and activated with dichlorodiethylsilane as described. This amount of resin was chosen by estimation of the theoretical number of moles of material in the bacterial extract based on the assumption that the average compound molecular weight was 200 Da. Comparable results were obtained with fewer “equivalents” as long as an alcohol capture standard was included to ensure that resin was not the limiting reagent.
After 4 h, each vial of resin was transferred to a 10 mL biospin vessel and washed twice under Ar with 4 mL of anhydrous CH2Cl2. The resin was then transferred back to a freshly dried 20 mL scintillation vial. Five vials were immediately placed under an atmosphere of Ar and the sixth vial was rinsed with MeOH/THF to hydrolyze the activated resin, to yield a deactivated control resin. Next, to all vials was added 83 μL of freshly distilled triethylamine and 1 mL of the mixture of the crude bacteria extract containing the chemoselective set of compounds. The vials were capped and agitated overnight at room temperature. The resin was transferred to 10 mL biospin vessels and subjected to the wash protocol described in the Supplementary Information.† The resin vessels were dried overnight in a vacuum desiccator at 30 mmHg. The dried resin was transferred to 20 mL polypropylene vial and swelled with 3 mL of anhydrous THF. To each vial was added 1 mL of a 50/50 mixture (v/v) of HF$pyridine (70/30 wt%)/pyridine (16 mmol of HF, 28 equiv relative to Si). The vials were capped and agitated for 3 h at room temperature. After this time, 5 mL of TMSOMe (36 mmol, 83 equiv relative to Si) was added to quench excess HF and the resin was agitated for an additional 30 min at room temperature. The resin was washed with THF (3 × 2 mL 10 min) and filtered over a 10 mL fritted polypropylene column into a 20 mL scintillation vial. The THF wash was then concentrated under reduced pressure with no heating and the sample was dissolved in 400 μL of 2 : 1 : 1 H2O/THF/MeOH. Analysis was performed by injection of 5 μL of this solution onto a LC-MS-TOF and comparing the observed peak area to that of standard curve data. Comparison of the TIC of the crude bacterial extract to that of the captured molecules is shown in Fig. S3.† The TIC obtained following extract exposure to deactivated resin is also available (Fig. S3, ESI†) and illustrates that alcohol enrichment results only from specific interactions with the activated siloxyl-functionalized resin. Fig. S4† demonstrates that amine-, thiol-, and carboxylic acid-containing compounds were not enriched in this experiment.
Supplementary Material
Acknowledgements
We thank M. VanNieuwenhze for helpful discussions. This work was supported by NIH R00 GM82983 and a Pew Biomedical Scholar Award (EEC).
Footnotes
Electronic supplementary information (ESI) available: Methods for resin washing, determination of resin loading capacities and compound quantification using mass spectrometry. Characterization data for synthesized resins and mass spectrometry data for enriched alcohols, chemoselective experiments and Streptomyces griseolus enrichment. See DOI: 10.1039/c0sc00620c
Notes and references
- 1.Clardy J, Walsh C. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ, Cragg GM. J. Nat. Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 3.Carlson EE. ACS Chem. Biol. 2010;5:639–653. doi: 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Böttcher T, Pitscheider M, Sieber SA. Angew. Chem. Int. Ed. 2010;49:2680–2698. doi: 10.1002/anie.200905352. [DOI] [PubMed] [Google Scholar]
- 5.Sarker SD, Latif Z, Gray AI, editors. Natural products isolation. Second edn Humana Press; Totowa, New Jersey: 2006. [Google Scholar]
- 6.Koehn FE, Carter GT. Nat. Rev. Drug Discovery. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 7.Butler MS. J. Nat. Prod. 2004;67:2141–2153. doi: 10.1021/np040106y. [DOI] [PubMed] [Google Scholar]
- 8.Sticher O. Nat. Prod. Rep. 2008;25:517–554. doi: 10.1039/b700306b. [DOI] [PubMed] [Google Scholar]
- 9.Månsson M, Phipps RK, Gram L, Munro MH, Larsen TO, Nielsen KF. J. Nat. Prod. 2010;73:1126–1132. doi: 10.1021/np100151y. [DOI] [PubMed] [Google Scholar]
- 10.Araya JJ, Montenegro G, Mitscher LA, Timmerman BN. J. Nat. Prod. 2010;73:1568–1572. doi: 10.1021/np100465h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheminat A, Benezra C, Farrall MJ, Fréchet JMJ. Can. J. Chem. 1981;59:1405–1414. [Google Scholar]
- 12.Fréchet JMJ, Hagen AJ, Benezra C, Cheminat A. Pure Appl. Chem. 1982;54:2181–2188. [Google Scholar]
- 13.Carlson EE, Cravatt BF. Nat. Methods. 2007;4:429–435. doi: 10.1038/nmeth1038. [DOI] [PubMed] [Google Scholar]
- 14.Carlson EE, Cravatt BF. J. Am. Chem. Soc. 2007;129:15780–15782. doi: 10.1021/ja0779506. [DOI] [PubMed] [Google Scholar]
- 15.Gualtieri M, Baneres-Roquet F, Villian-Guillot P, Pugniere M, Leonetti J-P. Curr. Med. Chem. 2009;16:390–393. doi: 10.2174/092986709787002628. [DOI] [PubMed] [Google Scholar]
- 16.Grabowski K, Baringhaus K-H, Schneider G. Nat. Prod. Rep. 2008;25:892–904. doi: 10.1039/b715668p. [DOI] [PubMed] [Google Scholar]
- 17.Tallarico JA, Depew KM, Pelish HE, Westwood NJ, Lindsley CW, Shair MD, Schreiber SL, Foley MA. J. Comb. Chem. 2001;3:312–318. doi: 10.1021/cc000107i. [DOI] [PubMed] [Google Scholar]
- 18.Hu Y, Porco JA, Labadie JW, Gooding OW. J. Org. Chem. 1998;63:4518–4521. [Google Scholar]
- 19.DiBlasi CM, Macks DE, Tan DS. Org. Lett. 2005;7:1777–1780. doi: 10.1021/ol050370y. [DOI] [PubMed] [Google Scholar]
- 20.Meloni MM, White PD, Armour D, Brown RCD. Tetrahedron. 2007;63:299–311. [Google Scholar]
- 21.Boehm TL, Showalter HDH. J. Org. Chem. 1996;61:6498–6499. doi: 10.1021/jo961242d. [DOI] [PubMed] [Google Scholar]
- 22.Sobin BA, Tanner J, F. W. J. Am. Chem. Soc. 1954;76:4053. [Google Scholar]
- 23.Grollman AP. J. Biol. Chem. 1967;242:3226–3233. [PubMed] [Google Scholar]
- 24.Isenberg HD, editor. Clinical Microbiology Procedures Handbook. American Society for Microbiology; Washington, D.C.: 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.