

Abstract
A hyperstable Pin1 WW domain has been circularly permuted via excision of the fold-nucleating turn; it still folds to form the native three-strand sheet and hydrophobic core features. Multiprobe folding dynamics studies of the normal and circularly permuted sequences, as well as their constituent hairpin fragments and comparable-length β-strand-loop-β-strand models, indicate 2-state folding for all topologies. N-terminal hairpin formation is the fold nucleating event for the wild-type sequence; the slower folding circular permutant has a more distributed folding transition state.
Key word phrases: protein folding dynamics, folding landscape analysis, loop closure effects, hairpin models
WW Domains (1, 2) are among the most diverse and abundant independently folding domains in nature, and are popular model systems for examining protein folding pathways and dynamics. They are 34 to 57 residue domains, typically with 29 residues in the required minimal folding motif. WW Domains are characterized by their two conserved, namesake Trp residues; one is essential for forming a hydrophobic core contact with a sequence remote (and essential) proline at the opposite termini, the other is important for function (typically, binding proline-rich sequences)(1, 2). While there have been a priori designs of WW domains (3,4) they have not always (3) retained function and the hydrophobic core features even though they do form the 3-stranded (double hairpin) feature of WW domains. The hairpin nearest the N-terminal is, in the domains that have been studied extensively, fully formed at the folding transition state and is viewed as the folding nucleation site.(5-9) With changes in the [4:6]-turn (10) of this hairpin, re-engineered variants of the pin1 WW domain are among the fastest folding β proteins known. Indeed, some reports (7,8,9) have suggested that the pin1 WW domain is a borderline downhill folding system with a very low barrier. In our minds, this raises an interesting question: what would be the effect on both dynamics and fold stability of eliminating this turn completely. Would the WW fold even form? Circular permutation should provide an answer to these questions. We set the circular permutation of a minimized, sequence-optimized pin1 WW domain, with scission of the folding-nucleating hairpin turn, as our goal. Herein, we report that stable folds retaining all the key features of the normal WW domain topology result from such a circular permutation.
Circular permutation consists of altering the location of the termini of an otherwise preserved chain, and provides a way to separate topology and folding pathway considerations. It can be viewed as a cyclization linking the original termini, followed by scission at another location in the sequence. What folding pathways (if any) are available to a small, compact domain once its known nucleating site is removed? Circular permutation of small domains should be particularly useful, because more investigation and synthesis options are available for small systems; these include more rigorous MD folding simulations and experimental options for labeling and the introduction of unnatural residues. A meaningful exploration of folding landscapes requires that a circular permutant adopt the same structure as the WT sequence. To our knowledge circular permutation had not been reported for proteins smaller than 53 residues (11, 12) prior to our recent report of a 20-residue circular permutant of the Trp-cage fold (13). WW domains have been cyclized (14, 15) but these cyclic constructs have not been cleaved at another point in the sequence.
Our circular permutation strategy for a WW domain is illustrated in cartoon form in Figure 1. It takes advantage of a reported pin1 WW domain with a stability-enhancing cross-strand, turn-flanking Trp/Trp interaction (16) in the nucleating hairpin and our prior development of β-capping units (17) for hairpins which also include a stabilizing Trp/Trp interaction.
Figure 1.
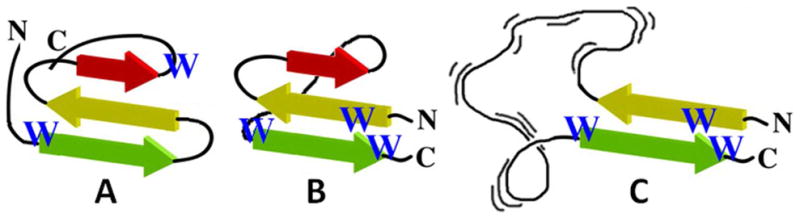
Comparison of various potential WW domain topologies. Throughout this manuscript, beta-strand 1 (the first strand from the N-terminus of the wild-type) is color-coded green, strand 2 is yellow, and strand 3 is red. Panel A represents the wild-type system. Panel B is a circular permutant with the same fold and a Trp/Trp β-capping unit substituting for the turn. Panel C represents an early folding intermediate if the order of interaction formation that has been established for the WT sequences also applies to the circularly permuted sequence.
Edge-to-face (EtF) indole/indole clusters can provide from 5 – 10 kJ/mol of fold stabilization in both Trp-flanked turns and β-capping units.(17-20) We anticipated that a β-cap, with some additional design optimization, would provide a sufficiently stabilizing interaction to allow β-sheet formation at the end of a 22-residue loop as would be the case in Figure 1C. The results reported herein validate this strategy, establishing the use of a β-cap as the extreme N- and C-terminal units of a protein sequence.
Materials and Experimental Methods
Peptide Synthesis and Purification
Hairpin peptides are synthesized on either an Applied Biosystem 433A or Advanced ChemTech 496 synthesizer employing standard Fmoc (9-fluorenyl methoxycarbonyl) solid-phase peptide synthesis methods and purified using RP-HPLC, using C18 and/or C8 stationary phases and a water (0.1% TFA)/acetonitrile(0.085% TFA) gradient as previously described.(18,20) The resins used for the synthesis were Wang resins preloaded with the C-terminal amino acid. Peptides were cleaved from the resin using a 95:2.5:2.5 trifluoroacetic acid (TFA): triisopropylsilane: water mixture. Full-length WW domains were ordered from GenScript as the crude products for subsequent in-house HPLC purification. The sequences of all peptides were confirmed by the molecular ions observed using a Bruker Esquire ion-trap mass spectrometer.
NMR Spectroscopy
Samples for 2D NMR spectral studies consisted of ∼1 mM peptide in 50 mM phosphate buffer at pH 7, 10% D2O, with DSS as an internal chemical shift reference. NMR experiments were collected at 700 or 800 MHz on Bruker DRX and AV spectrometers. Full 1H spectral assignments were made by using a combination of 2D NOESY and TOCSY experiments.
NMR Structure Analysis and Fold Population Analysis
The HN and Hα chemical shift deviations (CSDs) were calculated using the CSDb algorithm.(19, 23) The 100% folded values of CSDs and their temperature dependence are derived from the temperature dependence of these chemical shifts for the hyperstable WW st34 construct (Tm = 94 °C) at temperatures well below the beginning of its melting transition (see Figure S7). The commonality of structural features of the new WW domain constructs and the sequence fragments derived from these was established by CSD comparisons with both a wild-type Pin1 sequence (Figures S1A and S6A) and WW st34 (Figures S1B and S6B). An NMR structure ensemble was calculated for WW cp29 (the detailed procedure appears in the Supporting Information) for direct comparison with a Pin1 WW domain crystal structure. In NMR melts, plots of CSD versus temperature are used to derive fold populations as previously described.(17-22)
Relaxation Dynamics from NMR Linewidths
1D 1H NMR spectra in D2O medium (at 700 or 800 MHz observation) were collected at 5° increments over the maximum useful range for each polypeptide, up to an instrument-maximum of 345K. A peptide's useful range was defined as the temperature range where some peaks exhibited sufficient broadening for lineshape characterizations, but diagnostic peaks were not so broad as to be lost in the baseline and/or grossly overlapped with other peaks (Fig. S3). The folding rates were calculated as previously described (18,21) from kF = 4πχU(χF)2(Δv)2(Δex)−1. Common values of χU and χF (= 1 - χU) were employed for all probes in each system examined; these were the average values from the shift melts of all probes. The temperature-dependent values of Δv were equated with the CSDs displayed by these probes in a hyperstable construct (WW st34) well below its melting transition (Fig. S7). The ΦF values for mutations were calculated as ln(kF/kFref)/ln(kF kUref / kU kFref).
To obtain Δex, we modeled each peak's splitting patterns and coupling constants in an excel program; an intrinsic line width term for each experiment was obtained from resonances that did not display a folding-induced shift change; the additional line width increment required to fit the broadened probe signals was equated with Δex (Fig. 2).
Figure 2.
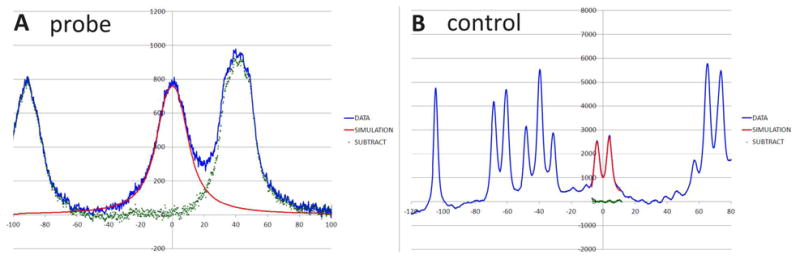
A linewidth comparison for two Trp Hε3 resonances of WW st29 at 325K. The left panel, A, illustrates the signal from the edge-Trp of the turn-flanking Trp/Trp pair which is surrounded by exchange-broadened Hα signals. The right panel, B, illustrates a Trp Hε3 which displays no structuring shift and thus no exchange broadening. The scale in both panels is in Hertz about the peak center. The red lines (both panels) show simulated Trp Hε3 7.8 Hz doublets (including broadening associated with two smaller coupling constants, 0.8 and 1.2 Hz) with different amounts of additional line broadening added. The additional line broadening required to fit the exchange broadened peak (difference between left and right panels) provides Δex (18,21) for the calculation of folding dynamics.
Slopes of the Arrhenius plots (ln k vs. 1/T) were divided by the gas constant in joules to obtain values that serve as surrogates for energies of activation and provide insight into degree of organization at the transition state. In cases where the Arrhenius plots of folding rates appeared curved, the slopes were evaluated at multiple temperatures.
CD spectroscopy
Stock solutions of approximately 200 μM peptide concentration were prepared using 50 mM aqueous pH 7.0 phosphate buffer. Accurate concentrations were determined by UV spectroscopy assuming the standard molar absorptivities for the Trp and Tyr residues present. The samples were typically diluted to obtain circa 30 μM polypeptide solutions, with spectra recorded on a Jasco J720 spectropolarimeter using 0.10 cm pathlength cells over a UV range of 190-270 nm as previously described.(20,22,24) In melting studies, temperatures ranged from 5 to 95 °C in 10° increments. For CD melting temperatures, the folded fraction (χF) was determined by defining the temperature-dependent CD signal of the unfolded and folded states and assuming a linear χF relationship for signals between the two lines. The CD spectrum of the unfolded state is expected to be sequence independent at 228nm, the position of the fold-diagnostic exciton couplet. We employed a molar unfolded baseline of [θ]U = +16000 − 590•T(°C) for species containing a cross-strand Trp/Trp pair and a larger temperature dependence of [θ]F based on the pre-melting slope of the hyperstable species (WW st34).
Results
While we did not view the formation of the high-contact-order β-sheet shown in Figure 1C as the necessary fold-nucleating event for a WW domain circular permutant, the stability of this unit was viewed as key to a successful permutation. This prompted an examination of the N-terminal hairpin of the Pin1 WW domain and its circular permutant, as well as stabilized versions of this fragment with cross-strand Trp/Trp interactions at both turn-flanking and β-capping positions. Alternative β-capping units that would be more readily inserted into proteins were examined. As a test of the feasibility of forming β-sheet structures at the ends of long loops (analogous to the cartoon in Figure 1C), we prepared a series of models employing strands with very high β-propensities at the ends of flexible loops. These constructs were also stabilized by alternative β-capping units. A hydrophilic variant (containing a terminal salt-bridge) proved to be superior to the reported version (17) in both applications. With these studies presented, we move on to the other design aspects for a circular permutation of a WW domain with excision of the nucleating hairpin turn region.
WW Hairpin Fragments
In the case of the hairpin-1 fragment (stHP1wt) analogous to wild type Pin1, sequence truncation was expected (18) to increase the hairpin fold stability due to the attractive Coulombic interaction so introduced. Throughout we employ the “st” designation to indicate standard-topology. Peptide stHP1wt retains the original strands and turn of the wild-type Pin1 WW domain. Removed from the protein context, stHP1wt (as well as its circular permutant) did not form a hairpin fold to any significant extent. In contrast, constructs with the Trp/Trp cross-strand pair introduced, either in the turn-flanking (stHP1) or β-capping (cpHP1) position, proved to be quite stable hairpins. As can be seen below, peptide cpHP1 (shown in two equivalent depictions) is a direct circular permutation of the peptide stHP1 sequence. The cpHP1 diagram illustrates a hydrophilic variant of the β-capping unit containing a terminal salt-bridge as well as the EtF Trp/Trp cluster.

The folded populations for peptide stHP1wt (≤ 0.16 at 280K), and its similarly unstable circular permutant, as well as stHP1 and cpHP1 were based on chemical shift comparisons to folded Pin1 analogs at sites in the associated β strand residues that display large structure-induced shifts, chemical shift deviations (CSDs) greater than 0.4 ppm from statistical coil norms (see Fig. S1, panels A and B). The cpHP1 construct displays some tendency toward aggregation at high concentration. Replacing its DRGSGK turn with higher turn propensity sequences yielded hairpins of greater stability (vide infra) with no observable tendency to aggregate. (25)
In the case of stHP1 and its circular permutant, the CD exciton couplet and ring current shifts reflecting an EtF Trp/Trp geometry, previously observed for both turn-flanking (18, 19, 22) and β-capping (17, 20) Trp/Trp pairs, served as a means for measuring the extent of folding and the melting behavior. In addition, the lineshapes of the 2.5 ppm upfield-shifted edge-Trp-Hε3 (Fig. S3), reflecting exchange broadening due to the folded/unfolded-state dynamics (18, 21), could be used to derive the folding rate constants. In the case of hairpin stHP1, replacing the RGSG turn locus with PATG increased the Tm (as measured by CD) by at least 16 °C. In the case of cpHP1, two fold stabilizing turn replacements were examined: DRGSGK → NPATGK (Tm = 49 °C) and DRGSGK → I-D-Pro-GK, a tight β-turn (Tm = 65 °C).
The improved fold stability of stHP1 can be attributed to the turn-flanking Trp/Trp interaction; however, hairpin stHP1 is, to our knowledge, the slowest folding hairpin (1/kF = 71 μs at 300K) with an aryl/aryl flanked [4:6]-reversing loop. We have reported folding times (at 300K) as short as 0.4 μs with the longest folding timescale for species that have comparable or higher fold stability on the order of 18 μs (18). The fold population (χF) for stHP1 in H2O is 0.72 at 320K; the native hairpin of Pin1 is much less stable (Tm ≪ 0 °C). Thus, it is now clear that the native WW hairpin, stHP1wt, even though it is viewed as the fold-nucleating site (9,26), is not a stable hairpin removed from the protein context. This suggests that a significant portion of the pin1 WW domain folding barrier may represent entropy loss upon formation of this nucleating hairpin.
Beta-loop-beta Constructs
While circular permutation of stHP1 reduces its fold stability the permuted hairpin still has far greater stability than stHP1wt, suggesting that this β-strand alignment should still provide enthalpic stabilization to a circular permutant fold with wild-type core and secondary structural features. However, in our WW domain circular permutants the DRGSGK loop of cpHP1, which has some intrinsic propensity to form a [4:6]-hairpin turn, would be replaced by a long “loop”. What effect would this have on fold stability and folding rates? Beta-loop-beta (blb) constructs with long flexible loops, RWITVTI-(GGGGKK)n-I RVWE (n = 3 and 4, blb30 and blb36), were prepared to examine this issue. These species have loop lengths comparable to those present in model C (Fig.1) of our permuted WW domain design. The NMR and CD data indicated β sheet formation, at 280K χF = 0.78 (n = 3) and 0.46 (n = 4) (Fig. S4), with the expected EtF Trp/Trp pair in the capping unit. The extreme broadening of the ring-current shifted hydrogens of the edge indoles in each EtF interaction, indicated very slow folding rates: n = 3 (1/kF ≈ 248 μs at 280 K, 125 μs at 295 K) and for n = 4 (1/kF ≈ 250 μs at 295 K). On this basis, the worst-case scenario for our WW domain circular permutant was a surmountable loop-search which should produce a modestly stable folded state.
Circular permutation of Pin1 WW
Given our cautious optimism, we decided to incorporate the hairpin-stabilizing features of stHP1 and made a few other single site mutations expected to stabilize both the standard-topology and circularly-permuted structures.(27) The M10W / V17W double-mutation that introduced the end-capping Trp/Trp interaction and all other mutation sites are shown below. At this point, we proceeded directly to the synthesis of the full-length circular permutant of the WW domain (WW cp34).
The translocated segment including the β1 strand (blue highlighting) is shown at the end of WW cp34 in Scheme 1. The circular permutation was carried out via an excision/insertion strategy, whereby four residues from turn 1 were moved to become part of the new loop connecting the former N and C termini. The resulting construct proved surprisingly stable. We observed a far-upfield Hε3 proton of the “edge” indole in the C-terminal β-capping Trp residue and a very strong exciton couplet in the CD (Fig. 3). The CD melt indicated a 2-state transition with Tm of 63 °C (∼8 °C higher than the wild-type Pin1 WW domain). An NMR shift comparison of chemical shift deviations from random coil norms (CSDs) (Fig. S6A) with wild-type Pin1 indicated that the same fold formed.
Scheme 1. Pin1-WW Circular Permutation and Truncation.

Figure 3.
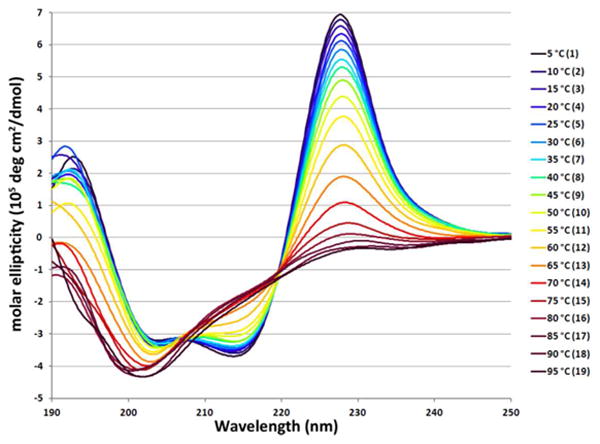
The temperature dependence of the circular dichroism spectrum of WW cp34. The intense Trp/Trp exciton couplet maxima at ∼228 nm adds to the less intense W/Y exciton couplet present in wild-type WW domain.
The NMR spectra of WW cp34 suggested that there were minor alternative geometries within the loop in the folded state ensemble (see supporting information, Fig. S5). We were able to reduce the loop length by 5 residues and retain fold formation, with only a single folded-state species evident. This truncated version (WW cp29, Tm = 53 °C) was very similar to wild-type Pin1 (WW st34wt) in fold stability. The NMR structure ensemble obtained based exclusively on the observed NOEs was a very close match to the crystal structure reported (28) for wild-type Pin1 (Fig. 4). Points of significant backbone deviation are limited to the region corresponding to the connecting loop of WW cp29 / the N and C terminal tails of the wild-type. Packing interactions disrupted in this manner include the docking of the L2 and P3 sidechains against hydrophobic core residues W6, Y19, P32 (wild-type numbering). The hydrophobic cores both above and below the sheet are otherwise packed in a very similar fashion, with nearly-identical sidechain rotamers. See figure S9 (supporting information) for details, and structural overlays that establish the high degree of similarity in both the three stranded sheet and the hydrophobic core.
Figure 4.
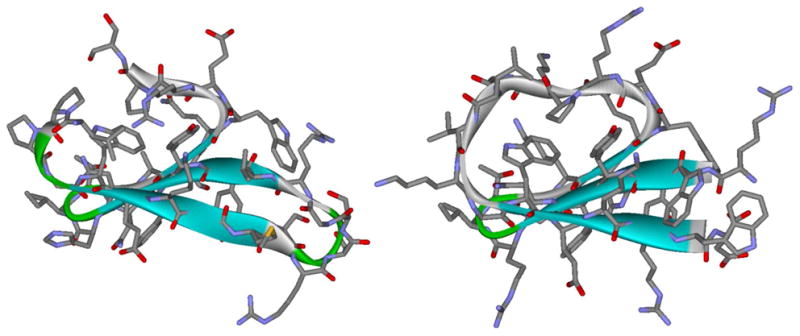
Comparison of the crystal structure of wild-type Pin1 WW domain (left, from PDB ID # 3tcz)(28) and NMR-derived structure of WW domain circular permutant WW cp29 (PDB ID # 2mdu). The EtF Trp/Trp interaction that takes the place of turn-1 is evident in the permutant structure which also has the same packing of a Trp, Tyr and Pro in the hydrophobic core. The backbone RMSD between the two structures (not counting excised loops) was 1.1 Angstroms; see Supporting Information Fig. S9 for structural overlays and more detailed comparisons.
In order to have a comparison point for ascertaining the effect of the circular permutation upon fold stability, we prepared a standard-topology Pin1 analog with all of the mutations included in our circular permutant: the WW st34 sequence shown in Scheme 1. As expected, these mutations effected a significant increase in fold stability (Tm = 94 °C). The spectra recorded for WW st34 also provided an even better match to the NMR structuring shifts observed for our circular permutants (see Fig. S6B).
Truncating the standard-topolgy WW sequence
At this point, we faced an unusual problem: we sought to compare the relative folding rates of the two structurally-identical but sequentially-disparate constructs (WW st and WW cp), but one of them (WW st34) was too stable for our dynamics method: dynamics by NMR relaxation requires the presence of a significant population of the unfolded state at the temperature of the NMR linewidth experiments.(18,21) We did not want to resort to denaturant (e.g., urea) or co-solvent addition for the standard topology construct. Since residues at the very ends of the wild-type WW domain (+KLPPG…SG-) provide some fold stability enhancement, but are not required for folding, we explored the effects of truncation as a way of lowering thermal stability of WW st34 while preserving its core structure. The truncation included deletion of the leucine residue in the N-terminal segment. This leucine is a hydrophobic core feature in WT-Pin1 (a 7.2 kJ/mol destabilization has been reported (26) for a L→A mutation.(29) These truncations resulted in a sequence with the same 29-residue length and more nearly comparable fold stability. (See Supporting Information for details.) A CD melt comparison for the hyperstable construct, its truncated version, and the 29-resdue circular permutant appears as Fig. 5. Thus, the stage was thus set for an exploration of the effects of circular permutation on folding dynamics.
Figure 5.
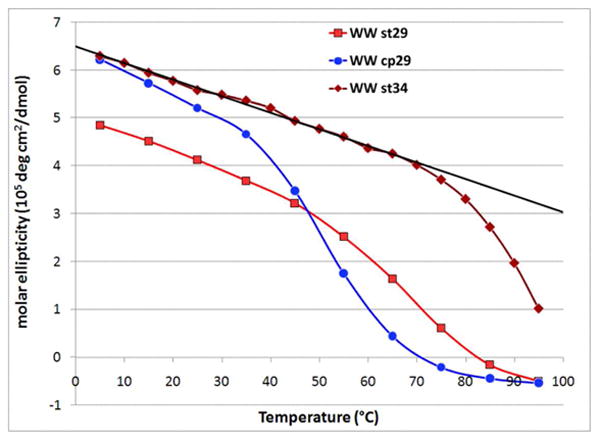
Molar ellipticity at the exciton couplet maximum (228 nm) versus temperature for three WW sequence mutants. The steeper pre-melting slope for the circular permutant likely reflects greater Trp χ angle variation with warming when the EtF Trp/Trp unit involves the chain terminal residues rather than being at a sequence-internal, turn-flanking position.
A contact plot of NOEs (gray) and H-bonding interactions (blue) shows that the key contacts of the WW domain are nearly identical in both the standard-topology and circularly permuted constructs. Though effectively identical contacts are formed, the contact order (sequence-distance) for most of these contacts is grossly altered; see figure 6.
Figure 6.
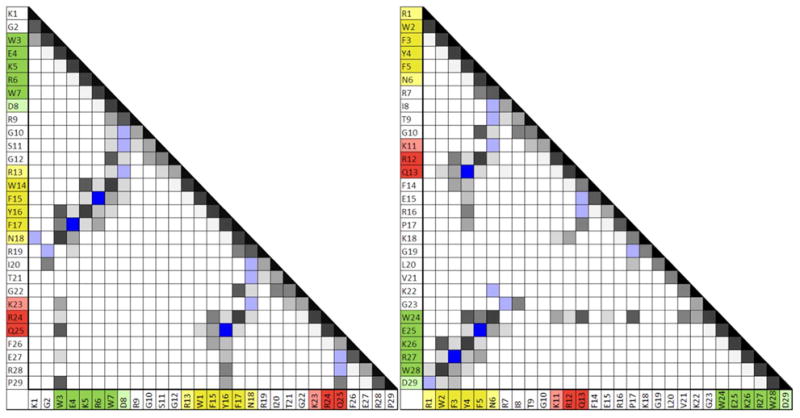
Contact plots for the 29 residue standard-topology WW-domain mutant (left) and its circular permutant (right). Contacts with more and/or more intense NOEs are shown in darker shades of gray. H-bonding interactions are shown in blue. (These are evidenced by chemical shift deviations as well as NOEs. Deeper blue = two cross-strand backbone H-bonds are present.) The three strands of the core WW domain structure are color-coded: strand 1 is green, strand 2 is yellow, and strand 3 is red; the 1, 2, 3 numbering is based on the standard topology (see figure 1). Note the greater prevalence of much longer range interactions in the circular permutant.
Circular Permutation Effects on Folding Dynamics
Using our truncated constructs (WW st29 and cp29), folding dynamics data could be obtained over a range of temperatures at different probe sites. A total of 9 NMR lineshape probes, all of which displayed similar (<15% difference) CSDs to those of the hyperstable WW st34 construct, were used (full length WW st34 numbering throughout): 3 indole sites displaying upfield ring shifts (W10Hζ3, Hδ1 and Hε3), 2 protons shielded by the central Trp6 sidechain (N21Hβ3, P32Hγ2) and 4 downfield β-strand Hα protons (W17, Y19, F20 & R25). In the case of WW cp29, data for 6 – 9 probes were available at each point (ΔT = 5 °C) in the 310 – 325 °C span. The coverage was nearly as good for WW st29 and a larger temperature range could be probed. The probes (shown in red) are well-dispersed in both sequences: st29 ( KGWEKRW–DRGSGR–WFYFNRITGKRQFERP) and cp29 ( RWFYFNRITGKRQFERPKGLVKG–WEKRWD). Their locations within the folded motif are shown for WW cp29 in the upper panel of Fig. 7.
Figure 7.
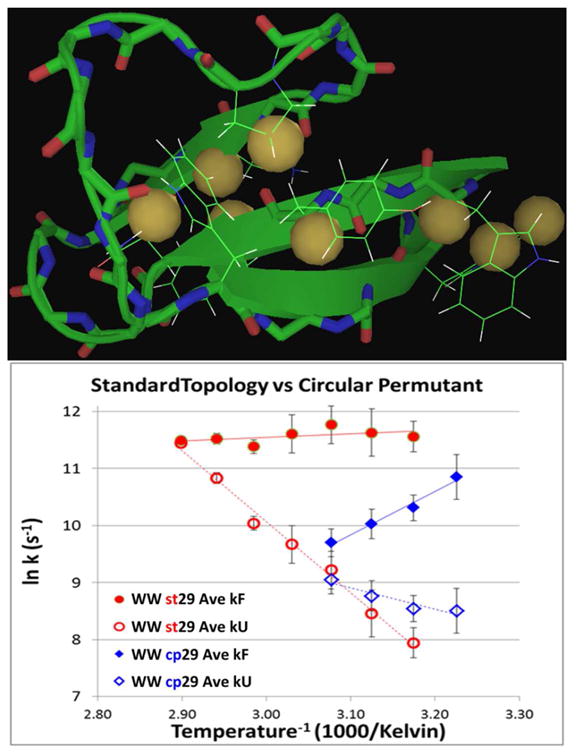
Upper panel: locations of the probes in the WW domain fold (illustrated on the NMR structure of the circular permutant). Lower panel: Arrhenius plots for folding and unfolding rates of the standard- topology WW domain (st29) and its circular permutant (cp29). The points are data averaged over all available probes at each temperature. The probe identities and individual ln k values appear in the SI (Fig. S8).
In both the case of WW st29 and its circular permutant cp29, the Arrhenius plots for both folding and unfolding for all probes are well-clustered and nearly linear. The plots in Fig. 7 (lower panel) are rates averaged over all available probes at each temperature. WW st29 with 1/kF = 8.9 μs at 320K (ln kF = 11.63 ± 0.21, over 9 sequence-dispersed probes) folds more than 8 times as fast as WW st34wt [W29F] (9) at this temperature and is amongst the very fastest folding WW domains even though it does not have a turn sequence optimized for faster folding (7,18). The folding rates calculated for the less-stable circularly permuted construct were slower; an expected consequence of abolishing the primary nucleation site and increasing the contact order between key stabilizing interactions.
The flat folding rate vs. temperature plots observed for WW st29 suggest a very low enthalpic barrier for this standard-topology WW domain. Small barriers and negative slopes for the folding Arrhenius plots have also been observed in T-jump studies of WW domain folding dynamics (9, 30). The circular permutant, with its presumably more distributed nucleation (e.g., not exclusively at the classic N-terminal hairpin nucleation site) appears to fold via a more diffuse, even-less-barrier-mediated mechanism.
An Arrhenius plot comparison for WW cp29 and cpHP1, the hairpin with the same strands and β-capping motif, RWFYFN–RITGKRQFERPKGLVKG–WEKRWD versus RWFYFN–DRGSGR–WEKRWD, appears in Figure 8.
Figure 8.
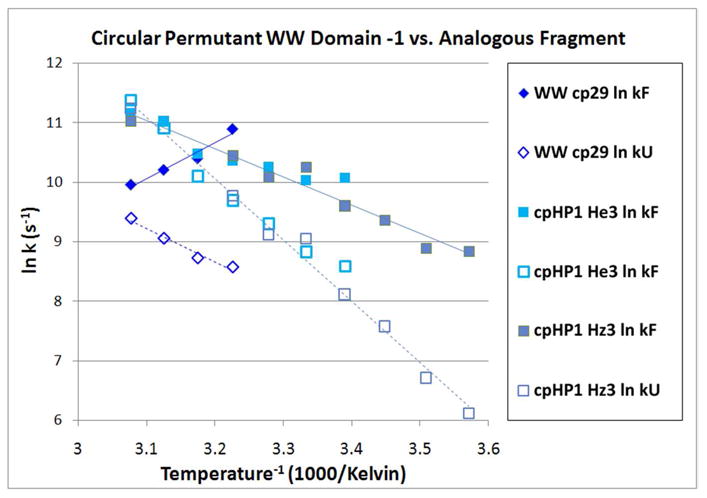
Inverse log plot folding & unfolding rates plots for WW cp29 and hairpin fragment cpHP1. In the case of the hairpin, rate data from two ring-current shifted sites (W17 Hε3 and Hζ3) are shown: the lines represent a single linear fit to all of the data.
In WW cpHP1, the entire central section of WW cp29 is excised and replaced with a 6-residue hairpin turn; the resulting hairpin has a melting temperature that is only ∼9 °C lower than full-length domain. This large loop size reduction, however, does not enhance the folding rate significantly; in fact, at colder temperatures the hairpin folding rates are slower than that of WW cp29. Some comparisons of folding rates and Arrhenius plot slopes, including the 16-loop construct and turn replacement analogs of stHP1 and cpHP1, are collected in Table 1. For both hairpins, replacing the loop region with a better turn can result in as much as a 10-fold increase in the folding rate.
Table 1. Fold Stability and Arrhenius Plot Dynamics Data Comparisons a.
| Fold Stability | Arrhenius plot dynamics parameters | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Slope (J/mol) | 300 K | 310 K | 320 K | ||||||
| ΔGU 310K | folding | unfolding | 1/kF (μs) | 1/kU (μs) | 1/kF (μs) | 1/kU (μs) | 1/kF (μs) | 1/kU (μs) | |
| WW st29 | -10.8 | -161 | 1277 | 8.1 b | 8.9 | 230.3 | |||
| stHP1 DRGSGR-turn | -6.5 | 899 | 1574 | 63.1 | 1742 | 27.7 | 445 | 12.6 | 95.5 |
| DPATGR-turn | -8.7 | 363 | 742 | 2.4 | 69.8 | 2.0 | 40.0 | ||
| WW cp29 | -6.0 | -663 | 671 | 11 b | 21 | 213 | 49.3 | 172.7 | |
| cpHP1 DRGSGK-turn | -1.75 | 531 c | 1188 | 39.3 | 132 | 30.1 | 59.6 | 16.2 | 18.4 |
| NPATGK-turn | -5.4 | 329 | 857 | 6.4 | 76.8 | 4.5 | 36.5 | 3.7 | 17.1 |
| IpGK-turn | -7.0 | 889 | 1551 | 7.1 | 213 | 3.2 | 55.2 | 1.4 | 12.9 |
| BLB30 (n=3) | +0.62 | 372 | 865 | 112 | 162 | 92 | 72 | ||
The rate data at 320K provides the most complete comparison over all constructs; the rates shown are the averages over all available probes. Arrhenius inverse-log plot slopes are an average over all probes for each construct. In the case of the unfolding rates, all species display strict linearity within experimental error. There is some curvature suggested by the folding rate data for the DPATGR turn mutant of stHP1 and the NPATGK turn mutant of cpHP1.
Extrapolated folding rates based on the linear fit equation for ln k vs 1/T.
This is the clearest case of a curved ln k vs 1/T plot. The slopes at 300 and 320 K are: 80 and 450 J/mol, respectively.
Discussion
WW domains have been viewed as borderline downhill folding systems (7, 30). Previous studies of Pin1 dynamics have been based on a single probe, the fluorescence associated primarily with the buried N-terminal Trp. Multiple probes provide a better criterion for strict 2-state versus downhill folding. A total of nine probes, well-dispersed along the sequences (Fig. 7) were used in our studies of the optimized WW domain and its circular permutant; thus, our data supports a strict 2-state folding scenario. While it is possible that the fold-stabilizing mutations that we have inserted are essential for this observation, the analogies to other Pin1 WW domain systems displayed by our constructs and their constituent hairpins (vide infra) lead us to view downhill folding for Pin1 WW domain species as unlikely. We begin our discussion with the hairpin species.
The hairpin fragments corresponding to our Pin1 constructs and the circular permutant are, for hairpins that display comparable fold stability, the slowest folding species reported to date (18, 31). At temperatures below 320K, both hairpins appear to fold more slowly than the analogous sequences that provide the complete WW fold (Table 1). This raises a number of questions.
The Pin1 WW fold is not the only case in which the formation of one hairpin has been established to be the fold-nucleating event for a protein. Another well-studied system is the B1 domain of protein G. The GB1 folding motif has a four-stranded p-sheet made up of two hairpins. Of these, the C-terminal hairpin, GB1p, has been shown to be the fold-nucleating site (32). GB1p has a large enough fold population (χF ≈ 0.22 at 298K) to allow dynamics studies (21, 33). The folding rate of GB1p (1/kF = 20 μs), with a DDATKT turn flanked by a cross-strand Y/F interaction, is in the same time range as similar aryl-flanked [4:6]-hairpins (18), but at least a factor of 30 faster than the fastest folding rate reported (34) for intact GB1. A ΦF value of 0.96 has been reported for a destabilizing DDATKT to ADATKT mutation in the intact GB1 protein (32). In the case of GB1p, we have established that a DDATKT to NPATGK turn mutation increases both fold stability and the folding rate, the latter by a factor of 5.1. The ΦF value for this turn mutation can be estimated as 0.92 from the data in the most recent report (18).
Our data for the nucleating N-terminal hairpin of Pin1 WW implies much less intrinsic hairpin stability than was the case for nucleating hairpin of GB1. However, the mutations introduced in the turn and the flanking W/W interaction in WW st29 and its fragment hairpin (stHP1) provide a basis for comparison. A stabilizing turn mutation, DRGSGR to DPATGR, in hairpin stHP1 results in accelerated folding (ΦF = 1.9 at 320K, ΦF > 2.5 at 310K). Phi values significantly greater than unity have also been noted for mutations in this loop in wild-type WW Pin1.(26, 35) These analogies imply a similar requirement for N-terminal hairpin formation and are viewed as validating WW st29 as a model for WW domain fold-nucleation. The same stabilizing RGS → PAT mutation within the [4:6]-turn of the circularly permuted hairpin series displays somewhat smaller ΦF values: 1.05 at 320K, and 1.42 at 300K. We attribute this to movement of the most important stabilizing interaction (the EtF W/W cluster) from the turn-flanking position to the remote chain termini. The increase in intrinsic turn formation associated with the turn locus mutation does not remove the need for a further loop search in cpHP1: even with an optimal turn, achieving the folding transition state still requires the Trp residues at the strand ends to find each other. The stabilizing mutations within the [4:6]-hairpin turns decreased the barrier to folding in both peptides examined. Comparison between the full-length domains and their analogous hairpin fragments provide further insights. The hairpins display the large increase in both folding and unfolding rates on warming previously observed for other hairpins (e.g. 18, 21) while the complete 29-residue WW-fold-forming sequences display faster folding at lower temperatures.
In the case of the stHP1 to WW st29 comparison, we are examining a direct excision of the presumably fold-nucleating hairpin. Thus it came as a surprise to observe slower folding for this hairpin vs. its full-length parent at temperature below 325K (see Table 1). This observation in and of itself would be enough to suggest efficient, multi-pathway folding mechanisms for WW domain formation. While the fragment (stHP1) is slow folding for its structural class, its parent WW domain (WW st29) is at the head of the pack (1/kF = 9 μs versus 43 μs @Tmax for WT-Pin1 (9): there is only one WW domain (the GTT mutant of Fip35, at 4.2 μs) (36) that folds faster.
With the structural characterization of WW cp29 given herein, we have shown that local fold-nucleating elements can be replaced by highly stabilizing long-range interactions with modest impact on overall fold stability and folding rate. Specifically, the strategy of replacing an essential turn with a non-covalent Trp/Trp β-cap was sufficiently stability-preserving to afford a robust circularly permuted WW domain. The folding rate of the circular permutant is faster than the wild-type Pin1 WW domain and only three to six times slower (depending on temperature) than the equivalent standard-topology mutant. This is remarkable when one considers the orders-of-magnitude differences between the slowest and fastest folding turn-replaced WW domain mutants investigated by the Kelly group. (7, 8, 26) Curiously, removing the nucleating turn altogether is on par with a modestly destabilizing turn mutation; thus, it would appear, for folding dynamics, that a poor “nucleating” turn is worse than no turn at all.
As was the case with WW st29, the circular permutant folds more rapidly than its closest hairpin equivalent at the lower temperatures examined. In this case, the hairpin represents the replacement of the central 17 residues of sequence with a 6-residue turn: this does not improve folding rate acceleration at temperatures below 315K. The comparison of WW cp29 ( RWFYFN–RITGKRQF–ERPKGLVKG–WEKRWD) and a β-capped loop model (blb30, RWITVTI–GGGGKKGG–GGKKGGGG–KKIRVWE) (Fig. 9) also illustrates the consequences of a structure-forming versus unstructured chain between key tertiary structural elements: the two halves of the β-capping motif situated at the extreme chain termini. The sequence-distance separating the two halves of the beta cap is nearly identical for blb30 and WW st29. Yet, despite the presence of some strand residues with a lower β-propensity, the WW domain circular permutant folds significantly faster than its analogous long-loop counterpart. Presumably, this is because the intervening WW domain sequence encodes components of WW domain structure which can form prior to association of the terminal β strands and the development of the key Trp/Trp capping interaction. In contrast, most of the intervening residues of the long-Gly/Lys-loop constructs are always disordered, providing no pre-folding boost to search times. Put another way, the WW circular permutant contains fold-nucleating regions with low contact-order, while the long-loop construct has no possibilities for persistent low-contact-order structure which could speed the loop search.
Figure 9.
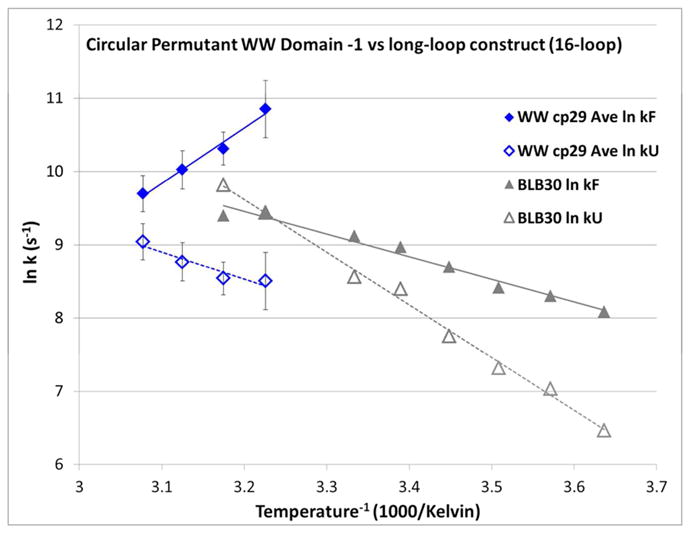
Arrhenius plots of folding and unfolding rates for WW cp29 versus blb30: RWITVTI-(GGGGKK)3-IRVWE. Rates for WW cp29 are given as the average of all probes, with standard errors shown.
In addition to overall slower folding rates, the long-loop construct has a negative slope for the folding Arrhenius plot: folding rates increase upon warming, as is routinely observed for small peptide folds including hairpins. The inverted temperature versus folding rate dependence of WW cp29 may be related to the increasing importance of high-chain-friction molten-globule states, which would not be a feature of the (GGGGKK)n loop constructs. It appears that a construct must be “protein-like” (possess multiple potential nucleating or trapping sites) to have an inverse folding rate / temperature dependence, and that this factor can outweigh loop-search considerations. Thus we can reaffirm that folding processes for proteins (more so than for smaller structured fragments) are robust: alternative folding pathways activate when one pathway is blocked or conditions change. This is what would be predicted for a protein folding landscape that can be modeled as a less-than-perfectly smooth folding funnel.
We would not expect the wild-type WW domain to survive circular permutation without our crucial design element: the β-cap. This view is supported by model studies and β-cap disruption of a blb analog: an even less severe alteration in the β-cap of a blb analog with a (GGGGKK)2 loop resulted in a ΔΔGU of −8 kJ/mol(17).
Further evidence of the importance of Trp/Trp interactions in stabilizing circularly permuted folds comes from a study of the Trp-cage. It had been established that introducing a Trp/Trp interaction in the Trp-cage, via a P→W mutation, results in ∼3 kJ/mol fold-stabilization. (37) This interaction affords greater stability to a circularly permuted Trp-cage (2.7 kJ/mol) vs. its standard topology analog (2.0 kJ/mol). (13) The Trp residues are only separated by five residues in either case, but in the circularly permuted Trp-cage the introduced tryptophan is near the C-terminus and thus better able to reinforce the long-range hydrophobic contacts between the chain ends.
Disruption of structure-stapling hydrophobic interactions at chain termini can be highly deleterious to folding. The beta cap is a crucial termini-stabilizing interaction with a ΔG contribution in excess of 8 kJ/mol. On this basis, we expect that a WW domain circular permutant lacking the chain terminal β-cap will populate unfolded states to such an extent that it will be subject to aggregate formation via its exposed hydrophobic sites.
Conclusions
We have demonstrated that circular permutation is viable for a system as small and compact as a WW domain, and that the scission to yield new termini can bisect (and thus sideline) the fold-nucleating site. We view this success as validating not only our circular permutation strategy, but also the viability of our β cap as a replacement for fold-crucial features in β-sheet proteins. It also provides a clear example of multi-path folding where contact order dictates folding path and the nature of a folding transition state.
Our observation of nearly-identical rates over multiple probes confirms the two-state nature of WW domain folding trajectories. Yet this work also illustrates the complex nature of the domain's transition state via comparisons with hairpin fragments and related beta-loop-beta constructs. The fact that independently stable hairpins comprising the supposed fold-nucleating site of a larger β-sheet motif, at some temperatures, fold slower than their parent domains demands that the WW domain fold-nucleation event be more complex than simple hairpin formation followed by downhill accretion of tertiary, high contact order, structural features. This work will be foundational in our ongoing efforts to design WW domains with tunable kinetic traps. These will include WW domain folds, where the native state must fold in competition with a mutually exclusive beta hairpin alternatively stabilized by Trp/Trp interactions or particularly favorable turn sequences.
NMR lineshape analysis of circular permutants and stable structure fragments versus their parent protein domains has yielded new insights into the folding mechanism of the WW domain: a key model system of biological importance. This strategy has been successful for the Trp-cage miniprotein (13) and is currently being explored for the Villin Headpiece domain, so far with promising results.
Supplementary Material
Acknowledgments
This work was supported in large part by National Institutes of Health (NIH) grant GM099889 (N.H.A., P.I.). Support for the loop model studies came from an NSF grant (CHE-1152218).
Footnotes
Notes: The authors declare no competing financial interest.
Supporting Information: A table of all peptides used in this study with their melting temperatures and justifications, figures comparing peptide chemical shift deviations, raw CD melts, example NMR spectra used for peak-shape rates derivations, example NMR spectra illustrating spurious peaks in WW cp34, plots of 100%-folded chemical shift values vs. temperature, a plot of rates data for all probes of WW st29 and cp29, statistics & tables for the NMR structure of WW cp29, and an overlay (with discussion) of WW cp29 structure with that of wild-type WW Pin1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Macias MJ, Wiesner S, Sudol M. FEBS Lett. 2002;513:30. doi: 10.1016/s0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- 2.Otte L, Wiedemann U, Schlegel B, Pires JR, Beyermann M, Schmieder P, Krause G, Volkmer-Engert R, Schneider-Mergener J, Oschkinat H. Prot Sci. 2003;12:491. doi: 10.1110/ps.0233203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macias MJ, Gervais V, Civera C, Oschkinat H. Nat Struct Mol Biol. 2000;7:375. doi: 10.1038/75144. [DOI] [PubMed] [Google Scholar]
- 4.Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R. Nature. 2005;437:579. doi: 10.1038/nature03990. [DOI] [PubMed] [Google Scholar]
- 5.Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Science. 2010;330:341. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 6.Jäger M, Zhang Y, Bieschke J, Nguyen H, Dendle M, Bowman ME, Noel JP, Gruebele M, Kelly JW. Proc Natl Acad Sci USA. 2006;103:10648. doi: 10.1073/pnas.0600511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen H, Jäger M, Kelly JW, Gruebele M. J Phys Chem B. 2005;109:15182. doi: 10.1021/jp052373y. [DOI] [PubMed] [Google Scholar]
- 8.Liu F, Du D, Fuller AA, Davoren JE, Wipf P, Kelly JW, Gruebele M. Proc Natl Acad Sci USA. 2008;105:2369. doi: 10.1073/pnas.0711908105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuller AA, Du D, Liu F, Davoren JE, Bhabha G, Kroon G, Case DA, Dyson HJ, Powers ET, Wipf P, Gruebele M, Kelly JW. Proc Natl Acad Sci USA. 2009;106:11067. doi: 10.1073/pnas.0813012106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sibanda BL, Thornton JM. Nature. 1985;316:170. doi: 10.1038/316170a0. [DOI] [PubMed] [Google Scholar]
- 11.Lindberg MO, Haglund E, Hubner IA, Shakhnovich EI, Oliveberg M. Proc Natl Acad Sci USA. 2006;103:4083. doi: 10.1073/pnas.0508863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lo WC, Lee CC, Lee CY, Lyu PC. Nuc Acid Res. 2009;37:D328. doi: 10.1093/nar/gkn679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrne A, Kier BL, Williams DV, Scian M, Andersen NH. RSC Adv. 2013;3:19824. doi: 10.1039/C3RA43674H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camerero JA, Pavel J, Muir TW. Angew Chem Int Ed. 1998;37:347. doi: 10.1002/(SICI)1521-3773(19980216)37:3<347::AID-ANIE347>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 15.Deechongkit S, Kelly JW. J Am Chem Soc. 2002;124:4980. doi: 10.1021/ja0123608. [DOI] [PubMed] [Google Scholar]
- 16.Jäger M, Dendle M, Fuller AA, Kelly JW. Prot Sci. 2007;16:2306. doi: 10.1110/ps.072904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kier BL, Shu I, Eidenschink LA, Andersen NH. Proc Natl Acad Sci USA. 2010;107:10466. doi: 10.1073/pnas.0913534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scian M, Shu I, Olsen KA, Hassam K, Andersen NH. Biochemistry. 2013;52:2556. doi: 10.1021/bi400146c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eidenschink LA, Kier BL, Huggins KNL, Andersen NH. Prot Struct Funct Bioinf. 2009;75:308. doi: 10.1002/prot.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kier BL, Andersen NH. J Am Chem Soc. 2008;130:14675. doi: 10.1021/ja804656h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsen KA, Fesinmeyer RM, Stewart JM, Andersen NH. Proc Natl Acad Sci USA. 2005;102:15483. doi: 10.1073/pnas.0504392102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersen NH, Olsen KA, Fesinmeyer RM, Tan X, Hudson FM, Eidenschink LA, Farazi SR. J Am Chem Soc. 2006;128:6101. doi: 10.1021/ja054971w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fesinmeyer RM, Hudson FM, Andersen NH. J Am Chem Soc. 2004;126:7238. doi: 10.1021/ja0379520. [DOI] [PubMed] [Google Scholar]
- 24.Williams DV, Byrne A, Stewart JM, Andersen NH. Biochemistry. 2011;50:1143. doi: 10.1021/bi101555y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The melting temperatures reported are from the temperature dependence of the CD spectra (Fig. S2B). The proton sites with large CSDs, all of which melt in consort, provide comparable data. NMR melts carried out in D2O medium, which was used for the dynamics studies, report Tm's that are 6 – 9 °C higher; this behavior, indicating greater hairpin fold stability in D2O, has also been observed for other hairpins with cross-strand aryl/aryl interactions (18, 21).
- 26.Jäger M, Nguyen H, Crane JC, Kelly JW, Gruebele M. J Mol Biol. 2001;311:373. doi: 10.1006/jmbi.2001.4873. [DOI] [PubMed] [Google Scholar]
- 27.Other mutations, besides those in stHP1, were included in the WW st34 sequence; these are listed here together with the basis for their inclusion, in wild-type numbering: A26K, S27R for increased beta propensity and solubility, H22R for solubility and simpler pH dependence, W29F to reduce chromophore redundancy (a common mutation in WW systems), N25G to increase fold-stability, G34D for solubility and to accommodate for the loss of carboxylate residues typically found in the linker region between WW domains and their partner domains in complete proteins.
- 28.Zhang Y, Daum S, Wildemann D, Zhou XZ, Verdecia MA, Bowman ME, Lucke C, Hunter T, Lu KP, Fischer G, Noel JP. ACS Chem Biol. 2007;2:320. doi: 10.1021/cb7000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The contact between the L2 sidechain and residues W6 and Y19 in the wild-type hydrophobic core is absent in WW cp29 since the connecting loop of WW cp29 cannot adopt the same orientation as the very N and C terminal tails of wild-type Pin1. This interaction is not present in some NMR and crystal structures, presumably due to flexibility (e.g., PDB ID 1zr7). Since it is also absent WW st34 - the leucine sites do not display significant CSDs and the NOEs to hydrophobic core residues are much weaker than would be predicted based on an analogous structure, we viewed its deletion from WW st29 as unlikely to change any folding preferences or pathways.
- 30.Liu F, Du D, Fuller AA, Davoren JE, Wipf P, Kelly JW, Gruebele M. Proc Natl Acad Sci USA. 2008;105:2369. doi: 10.1073/pnas.0711908105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du D, Zhu Y, Huang CY, Gai F. Proc Natl Acad Sci USA. 2004;101:15915. doi: 10.1073/pnas.0405904101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCallister EL, Alm E, Baker D. Nat Struct Biol. 2000;7:669. doi: 10.1038/77971. [DOI] [PubMed] [Google Scholar]
- 33.Muñoz V, Thompson PA, Hofrichter J, Eaton WA. Nature. 1997;390:196. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 34.Park SH, Shastry MC, Roder H. Nat Struct Biol. 1999;6:943. doi: 10.1038/13311. [DOI] [PubMed] [Google Scholar]
- 35.Petrovich M, Jonsson AL, Ferguson N, Daggett V, Fersht AR. J Mol Biol. 2006;360:865. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 36.Piana S, Sarkar K, Lindorff-Larsen K, Guo M, Gruebele M, Shaw DE. J Mol Biol. 2011;405:43. doi: 10.1016/j.jmb.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 37.Bunagan MR, Yang X, Saven JG, Gai F. J Phys Chem B. 2006;110:3759. doi: 10.1021/jp055288z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.