

Abstract
Brugada syndrome is a rare cardiac arrhythmia characterized by electrocardiographic right bundle branch block and persistent ST-segment elevation in the right precordial leads. It is associated with ventricular fibrillation and a high risk for sudden cardiac death, predominantly in younger males with structurally normal hearts. Patients can remain asymptomatic, and electrocardiographic patterns can occur both spontaneously or after pharmacological induction. So far, several pathogenic genes have been identified as associated with the disease, but SCN5A is the most prevalent one. Two consensus reports to define the diagnostic criteria, risk stratification, and management of patients have been published in the last few years. This brief review focuses on the recent clinical diagnosis, genetic basis, and advances in pharmacological treatment of Brugada syndrome.
Keywords: Brugada syndrome, arrhythmias, sudden cardiac death

Clinical Characteristics
Brugada syndrome (BrS) was described 20 years ago as a new clinical entity characterized by the presence of a typical electrocardiographic (ECG) pattern (right bundle branch block and persistent ST-segment elevation in right precordial leads) and associated with a high risk of sudden cardiac death (SCD).1 Currently, it is believed to be responsible for 12% of SCD cases and 20% of SCD in patients with structurally normal hearts.2 Patients may suffer syncope or SCD secondary to polymorphic ventricular tachycardia (PVT)/ventricular fibrillation (VF). However, the majority of patients remain completely asymptomatic. Some of the arrhythmias may occur after large meals, during rest, or while sleeping, believed to be due to high vagal tone.3 The symptoms usually appear around 40 years of age; however, there are reports of patients affected from ages 1 to 84. Males are more often symptomatic than females, probably from the influence of hormones and gender distribution of ion channels across the heart. There is little information regarding the pediatric population, but studies performed in children have failed to identify a male predominance, perhaps due to low levels of testosterone in children of both genders.4 The prevalence of the disease is difficult to estimate because the pattern is not always recognized or because it may transiently normalize. Nevertheless, global prevalence varies from 5 to 20 in every 10,000, and it is considered endemic in Southeast Asian countries, where the prevalence is higher.5
Diagnosis
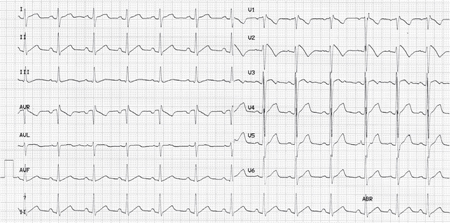
The diagnosis of BrS may be hampered because of incomplete penetrance and dynamic ECG manifestations.6 Originally, three repolarization patterns were described: a) Type-1 ECG pattern, in which a coved ST-segment elevation ≥ 2 mm is followed by a negative T-wave, with little or no isoelectric separation, with this feature being present in > 1 right precordial lead (from V1 to V3); b) Type-2 ECG pattern, also characterized by a ST-segment elevation but followed by a positive or biphasic T-wave that results in a saddle-back configuration; c) Type-3 ECG pattern, a right precordial ST-segment elevation ≤ 1 mm either with a coved-type or a saddle-back morphology.7 In 2012, Bayés de Luna et al. reported two specific ECG patterns considered descriptive of BrS.8 However, so far, only the ECG type 1 pattern is the sine qua non BrS diagnosis: J-point elevation of > 2 mm with a coved (downward convex) ST segment (Figure 1).9 Both type 2 and 3 are not considered diagnostic. The ECG type 1 pattern may be spontaneously evident or induced by a provocative drug challenge test using intravenous Class 1A or 1C antiarrhythmic drugs. Flecainide, ajmaline, procainamide, disopyramide, propafenone, and pilsicainide have been used to unmask BrS, but ajmaline and flecainide are the drugs of choice at present.3 The diagnosis of BrS is based on clinical and electrocardiographic features. So, BrS is definitely diagnosed when the patient also presents at least one of the following criteria10:
Figure 1.
Electrocardiogram showing Brugada syndrome pattern type I
A. Family history: SCD in a family member < 45 years or ECG type 1 in relatives
B. Arrhythmia-related symptoms: syncope, seizures, or nocturnal agonal respiration
C. Ventricular arrhythmias: PVT or VF
Treatment
Currently, the only proven effective strategy for preventing SCD in BrS patients is the use of an implantable cardioverter-defibrillator (ICD).11 Several pharmacological treatments are presently being used, especially quinidine and phosphodiesterase III inhibitors, but further studies have to be performed to clarify their benefit in BrS patients.3 In addition, radiofrequency ablation of ventricular ectopy was postulated as a therapeutical approach in BrS patients. Thus, in 2011, Nademanee et al. published the first study showing prevention of VF in BrS patients by catheter ablation over the anterior right ventricular (RV) outflow tract epicardium.12
Molecular Mechanism
The characteristic right precordial ST-segment elevation in the ECG is not well understood. Currently there are two mechanisms that may explain the ECG alteration; neither mechanism has been conclusively confirmed, nor are they mutually exclusive.13 The first hypothesis, repolarization, focuses on the presence of transmural voltage gradients due to heterogeneity in action potential duration between the RV epicardium and endocardium (disequilibrium between INa and Ito). This generates transmural dispersion of repolarization and causes the ST-segment elevation.14 The second hypothesis, depolarization, involves preferential conduction slowing in the RV outflow tract, leading to ST-segment elevation in the right precordial leads.15 Regional differences in conduction velocity in the RV epicardium would be aggravated by INa reduction and trigger the occurrence of epicardial reentrant excitation waves. Additionally, in 2009, Boukens et al. suggested that the embryological development of the right ventricle could explain the electrophysiological heterogeneity in the ventricular myocardium, including the RV outflow tract, which could provide the arrhythmogenic substrate.16
Genetics
Brugada syndrome is a disease with an autosomal dominant pattern of transmission. Incomplete penetrance is frequent in families, and the disease can be sporadic in up to 60% of patients.17 In 1998, the first pathogenic mutation in the SCN5A gene was identified.18 This gene encodes the alpha subunit of the cardiac sodium channel (Nav1.5). Since then, more than 350 pathogenic mutations in several genes have been published (SCN5A, GPD1L, SCN1B, SCN2B, SCN3B, RANGRF, SLMAP, KCNE3, KCNJ8, HCN4, KCNE5, KCND3, CACNA1C, CACNB2B, CACNA2D1, and TRPM4) (Table 1).19 These genes encode subunits of cardiac sodium, potassium, and calcium channels as well as genes involved in the trafficking or regulation of these channels. Despite the high number of gene mutations, only about 35% of BrS patients have been determined to have a genetic cause. Of them, nearly 30% carry a pathogenic mutation in the SCN5A gene.20 All other genes together are responsible for about 5% of all BrS cases. Therefore, 65% of cases do not have a genetic origin.
Table 1.
Genes associated with Brugada syndrome.
| CHANNEL | GENE | PROTEIN |
| SODIUM | SCN5A GPD1-L SCN1B SCN3B |
Nav1.5 glycerol-3-P-DH-1 Navβ1 Navβ3 |
| SCN2B RANGRF SLMAP |
Navβ2 RAN-G-release factor (or MOG1) sarcolemma associated protein |
|
| POTASSIUM |
KCNE3 KCNJ8 KCN4 KCNE5 KCND3 |
MiRP2 Kv6.1 Kir6.1 hyperpolarization cyclic nucleotide-gated 4 K voltage-gated subfamily E member 1 like Kv4.3 Kir4.3 |
| CALCIUM |
CACNA1C CACNB2B CACNA2D1 TRPM4 |
Cav1.2 voltage-dependent β-2 voltage-dependent α2/δ1 transient receptor potential M4 |
Several factors could explain the high number of BrS patients without genetic alteration after genetic screening. For example, copy number variations have already been reported in SCN5A.21 In addition, pathogenic mutations associated with BrS could be localized in unknown genes, or the disease could be related to epigenetic factors, mainly DNA methylation, post-translational modifications, and RNA mechanisms.22, 23 All these factors could also explain, at least in part, incomplete penetrance and variable expressivity characteristics in BrS families.24
Phenotype Modulators
Several modulating factors that play a key role in the ECG dynamic nature have been published,24 with bradycardia and vagal tone thought to contribute to ST-segment elevation and arrhythmia initiation. This fact explains the greater ST-segment elevation documented in vagal situations, such as arrhythmias and SCD at night. The role of hormones is also debated, in that a regression of the typical ECG features has been observed in castrated men, and the levels of testosterone seem to be higher in male BrS patients. In addition, temperature is also a main modulator in BrS. Febrile states may unmask certain BrS patients and temporarily increase the risk of arrhythmias. It seems that fever would be a particularly important trigger factor among the pediatric population despite that limited data exists thus far of BrS in children.3, 5
Risk Stratification
It is well accepted that the etiology of BrS is multifactorial, involving genetic, environmental, and hormonal components that contribute to its phenotype manifestation. In addition, some clinical features have been identified as high-risk markers in BrS. It is established that symptomatic patients with recurrent syncope, agonal respiration during sleep, or unknown seizures are at risk of sudden death and need ICD. However, a debate is still ongoing on the value of risk stratification parameters, such as electrophysiological inducibility, in asymptomatic patients.6 Some will argue that it has no value, while others will claim that the electrophysiology study (EPS) enables the identification of a subgroup of asymptomatic patients at higher risk who will benefit from ICD implantation.
Other modulating factors also have been investigated. For example, genetic studies have reported that compound pathogenic mutations in BrS patients cause more severe phenotype25 and that common polymorphisms may modulate the effect caused by pathogenic mutations.26 In addition, it recently has been published that pathogenic mutations in combination with common single nucleotide polymorphisms could increase the risk of arrhythmias in BrS patients,27 though at present genetics are not useful in risk stratification. At this time, genetic screening is only recommended as a diagnostic tool.28
Conclusion
The amount of information about BrS has been exponentially increasing since it was first described two decades ago despite the fact that many questions and controversies remain. In summary, patients should only be diagnosed if a type 1 pattern is present in the ECG. This is not an ECG that can be ignored, no matter the age of the patient or the context in which the ECG was obtained. Second, since the ECG is an indicator of a possible familial disease, family members should be investigated in case they might be at risk of SCD. Third, BrS-type ECG patterns that are induced by acute fever or drugs is a medical emergency, and patients should remain monitored. Finally, despite the controversy over the value of the EPS for risk stratification, the inducibility during the EPS is a clear indicator that the heart may be more excitable and that the patient may be at higher risk of SCD. At present, without any other tool available to identify the few asymptomatic patients at risk, the use of EPS can be lifesaving.
Funding Statement
Funding/Support: The authors have no funding disclosures.
Footnotes
Conflict of Interest Disclosure: All authors have completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported.
References
- 1.Brugada P, Brugada J. Right bundle branch block persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992 Nov;20(6):1391–6.. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 2.Juang JM, Huang SK. Brugada syndrome—an under-recognized electrical disease in patients with sudden cardiac death. Cardiology. 2004;101(4):157–69.. doi: 10.1159/000076693. [DOI] [PubMed] [Google Scholar]
- 3.Berne P, Brugada J. Brugada syndrome 2012. Circ J. 2012;76(7):1563–71.. doi: 10.1253/circj.cj-12-0717. [DOI] [PubMed] [Google Scholar]
- 4.Benito B, Brugada J, Brugada R, Brugada P. Brugada syndrome. Rev Esp Cardiol. 2009 Nov;62(11):1297–315.. doi: 10.1016/s1885-5857(09)73357-2. [DOI] [PubMed] [Google Scholar]
- 5.Jellins J, Milanovic M, Taitz DJ, Wan SH, Yam PW. Brugada syndrome. Hong Kong Med J. 2013 Apr;19(2):159–67.. [PubMed] [Google Scholar]
- 6.Veerakul G, Nademanee K. Brugada syndrome: Two decades of progress. Circ J. 2012;76(12):2713–22.. doi: 10.1253/circj.cj-12-1352. [DOI] [PubMed] [Google Scholar]
- 7.Brugada syndrome. Benito B Brugada R, Brugada J, Brugada P. In: Brugada R, Brugada J, Brugada P, eds. Clinical approach to sudden cardiac death syndromes. London, UK: Springer-Verlag; 2010. [Google Scholar]
- 8.Bayés de Luna A, Brugada J, Baranchuk A, Borggrefe M, Breithardt G, Goldwasser D, et al. Current electrocardiographic criteria for diagnosis of Brugada pattern: a consensus report. J Electrocardiol. 2012 Sep;45(5):433–42.. doi: 10.1016/j.jelectrocard.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002 Nov 5;106(19):2514–9.. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 10.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: report of the second consensus conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005 Feb 8;111(5):659–70.. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 11.Benito B, Brugada R, Brugada J, Brugada P. Brugada syndrome. Prog Cardiovasc Dis. 2008 Jul-Aug;51(1):1–22.. doi: 10.1016/j.pcad.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Nademanee K, Veerakul G, Chandanamattha P, Chaothawee L, Ariyachaipanich A, Jirasirirojanakorn K, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011 Mar 29;123(12):1270–79.. doi: 10.1161/CIRCULATIONAHA.110.972612. [DOI] [PubMed] [Google Scholar]
- 13.Wilde AA, Postema PG, Di Diego JM, Viskin S, Morita H, Fish JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol. 2010 Oct;49(4):543–53.. doi: 10.1016/j.yjmcc.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999 Oct 12;100(15):1660–6.. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 15.Tukkie R, Sogaard P, Vleugels J, de Groot IK, Wilde AA, Tan HL. Delay in right ventricular activation contributes to Brugada syndrome. Circulation. 2004 Mar 16;109(10):1272–7.. doi: 10.1161/01.CIR.0000118467.53182.D1. [DOI] [PubMed] [Google Scholar]
- 16.Boukens BJ, Christoffels VM, Coronel R, Moorman AF. Developmental basis for electrophysiological heterogeneity in the ventricular and outflow tract myocardium as a substrate for life-threatening ventricular arrhythmias. Circ Res. 2009 Jan 2;104(1):19–31.. doi: 10.1161/CIRCRESAHA.108.188698. [DOI] [PubMed] [Google Scholar]
- 17.Campuzano O, Brugada R, Iglesias A. Genetics of Brugada syndrome. Curr Opin Cardiol. 2010 May;25(3):210–5.. doi: 10.1097/HCO.0b013e32833846ee. [DOI] [PubMed] [Google Scholar]
- 18.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998 Mar 19;392(6673):293–6.. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen MW, Holst AG, Olesen SP, Olesen MS. The genetic component of Brugada syndrome. Front Physiol. 2013 Jul 15;4 doi: 10.3389/fphys.2013.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010 Jan;7(1):33–46.. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eastaugh LJ, James PA, Phelan DG, Davis AM. Brugada syndrome caused by a large deletion in SCN5A only detected by multiplex ligation-dependent probe amplification. J Cardiovasc Electrophysiol. 2011 Sep;22(9):1073–6.. doi: 10.1111/j.1540-8167.2010.02003.x. [DOI] [PubMed] [Google Scholar]
- 22.Webster AL, Yan MS, Marsden PA. Epigenetics and cardiovascular disease. Can J Cardiol. 2013 Jan;29(1):46–57.. doi: 10.1016/j.cjca.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 23.Kim GH, Ryan JJ, Archer SL. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid Redox Signal. 2013 May 20;18(15):1920–36.. doi: 10.1089/ars.2012.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Translational research : the journal of laboratory and clinical medicine. 2013 Jan;161(1):1–14.. doi: 10.1016/j.trsl.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cordeiro JM, Barajas-Martinez H, Hong K, Burashnikov E, Pfeiffer R, Orsino AM, et al. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation. 2006 Nov 7;114(19):2026–33.. doi: 10.1161/CIRCULATIONAHA.106.627489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lizotte E, Junttila MJ, Dube MP, Hong K, Benito B, DE Zutter M, et al. Genetic modulation of Brugada syndrome by a common polymorphism. J Cardiovasc Electrophysiol. 2009 Oct;20(10):1137–41.. doi: 10.1111/j.1540-8167.2009.01508.x. [DOI] [PubMed] [Google Scholar]
- 27.Sommariva E, Pappone C, Martinelli Boneschi F, Di Resta C, Rosaria Carbone M, Salvi E, et al. Genetics can contribute to the prognosis of Brugada syndrome: a pilot model for risk stratification. Eur J Hum Genet. 2013 Sep;21(9):911–7.. doi: 10.1038/ejhg.2012.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011 Aug;8(8):1308–39.. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]