

Abstract
A systems pharmacology approach was undertaken to define and identify the proteins/genes significantly associated with clinical incidence and severity of drug-induced peripheral neuropathy (DIPN). Pharmacological networks of 234 DIPN drugs, their known targets (both intended and unintended), and the intermediator proteins/genes interacting with these drugs via their known targets were examined. A permutation test identified 230 DIPN-associated intermediators that were enriched with apoptosis and stress response genes. Neuropathy incidence and severity were curated from drug labels and literature and were used to build a predictive model of DIPN using a regression tree algorithm, based on the drug targets and their intermediators. DIPN drugs whose targets interacted with both v-myc avian myelocytomatosis viral oncogene homolog (MYC) and proliferating cell nuclear antigen-associated factor (PAF15) were associated with a neuropathy incidence of 38.1%, whereas drugs interacting only with MYC had an incidence of 2.9%. These results warrant further investigation in order to develop a predictive tool for the DIPN potential of a new drug.
Drug-induced peripheral neuropathy (DIPN) is a common but unintended effect of many lifesaving drugs and often leads to dose reduction or regimen modification that may compromise clinical outcomes.1 Various chemotherapeutic agents and anti-infective drugs are known to cause peripheral neuropathy (PN) during treatment; however, other drugs can also induce PN following prolonged use. For example, the statins, which are widely prescribed for lowering total cholesterol, have been reported to cause PN with an incidence of 1 in 14,000 person-years of treatment.2
DIPN can be broadly divided into three groups based on pathophysiology: (i) axonal degeneration, (ii) segmental demyelination, and (iii) neuron soma damage. Chemotherapeutic agents not only act on their intended therapeutic targets but can also affect various cellular compartments of neurons and their surrounding cells (i.e., glial cells and macrophages), resulting in neurotoxicity and functional changes. For example, bortezomib, paclitaxel, platinum, and vincristine all can cause mitochondrial toxicity, with bortezomib also acting on endoplasmic reticulum, microtubules, and myelin sheath of the dorsal root ganglia.3
Genome-wide association studies have improved our understanding of the association between genes and differential clinical manifestations of DIPN. For example, genome-wide association studies have implicated regulatory factor X 2 (RFX2),4 ephrin type A receptor (EPHA4),5,6 and frizzled family receptor 3 (FZD3)6 in treatment-related PN of taxanes. However, it is generally not well understood how a wide spectrum of drug classes can cause treatment-related PN or how diverse these DIPN drugs are or whether there are any common biological factors connected to these particular compounds. Neither are the mechanisms of DIPN characterized across the drugs approved for treating different diseases.
To define the pharmacological space of DIPN drugs and to enable prediction and assessment of the likelihood of a new drug in causing DIPN, we adopted a hybrid approach of bioinformatics, systems pharmacology, and statistical regression tree analysis to link the clinical incidence and severity that are associated with individual DIPN drugs to their known and extended pharmacological targets. In this reported study, we mined the Drugs@FDA database, supplemented the findings by mining SIDER and NCBI DailyMed, curated the clinical incidence and severity of DIPN reported in published summaries of clinical trials, and then conducted statistical regression tree analyses to associate proteins in the extended pharmacological network with the clinical phenotypes of incidence.
Results
Compilation and curation of the drug list
To identify the DIPN drugs, we referred to the terms listed in Common Terminology Criteria for Adverse Events (v4.03: 14 June 2010) published by the National Cancer Institute of the National Institutes of Health.7 PN, peripheral sensory neuropathy, and peripheral motor neuropathy were used as search terms. Medical Dictionary for Regulatory Activities (MedDRA; http://www.meddra.com/) terms of hyperesthesia, hypoesthesia, and paresthesia were also referenced. A total of 3,200 current drug label PDF files were downloaded from the Drugs@FDA database8 and were converted to text files using Xpdf (http://www.foolabs.com/xpdf/). Among a total of 532 drug labels containing neuropathy terms, manual curation confirmed 214 nonredundant DIPN drugs. Similarly, two additional resources, the NCBI DailyMed data9 and the SIDER210 were surveyed for the aforementioned terms to include 20 additional drugs. A final list of 234 drugs was obtained (Supplementary Table S1), 183 (78%) of which were common in two or more resources (Figure 1a).
Figure 1.

DIPN drugs and the indication-based categories. Neuropathy-causing drugs were compiled from three resources (Drugs@FDA, DailyMed, and SIDER). (a) This Venn diagram illustrates the overlap among the identified drugs. (b) One representative indication category was assigned to each DIPN drug based on the indication information from the DrugBank database and Drugs@FDA. Those categories consisting of only one or two drugs were merged into the “miscellaneous” category, accounting for 14% of all DIPN drugs. DIPN, drug-induced peripheral neuropathy.
Indication-based drug categorization
DIPN drugs were categorized based on the indication for which each drug was approved by referencing DrugBank and Drugs@FDA. DrugBank included records for 224 out of the 234 drugs, with an average of three classes per drug. Referencing the US Food and Drug Administration (FDA)–approved labels allow the adoption of one indication for each drug. This categorization, though different from pharmacological target-based classification, was chosen to show the relation of DIPN to disease-specific treatment. The five largest categories were antineoplastic (n = 64; 27%), anti-infective (n = 38; 16%), anti-inflammatory (n = 16; 7%), anti-HIV (n = 16; 7%), and antidepressive (n = 10; 5%) (Figure 1b). Those categories with only one or two drugs were merged into the “Miscellaneous” category (n = 34; 14%).
Construction of DIPN pharmacological networks
The known intended and unintended targets of DIPN drugs are defined hereafter as targets and were collected from DrugBank and Therapeutic Target Database (TTD; http://bidd.nus.edu.sg/group/cjttd/). Among the 234 DIPN drugs, 204 had at least one target, whereas 30 anti-infective agents only had nonhuman targets in the databases. We then generated a pharmacological network (Figure 2a) using the remaining 174 DIPN drugs and their 280 human targets, referred to as the base network. This network contained multiple subnetworks (Figure 2b), identified by the Fast Greedy community structure analysis algorithm11 based solely on the network topology, and the 10 largest subnetworks are indicated by nongray node colors. The largest subnetwork, highlighted in yellow (Figure 2b), includes 23 drugs (mostly antineoplastic) and 62 targets. The second largest subnetwork consisted of 24 drugs and 35 targets (Figure 2c) and included the drugs that are mainly used for treating neurological disorders such as Parkinson disease, bipolar disorder, unipolar depression and seizures, and the drugs that shared serotonin receptors as common therapeutic targets.
Figure 2.

Base pharmacological network. (a) The base pharmacological network including 174 drug-induced peripheral neuropathy drugs and 280 human drug targets. The layout of the network was generated using edge-embedded spring layout available in Cytoscape, then was manually adjusted to make sure that all node names are visible. Node color represents the community structures identified by the Greedy algorithm implemented in GLay, where gray is used as a default color and nongray colors were used for the 10 biggest clusters. (b) The biggest subnetwork with 23 drugs and 62 targets, in yellow. Drugs were mostly from the antineoplastic class. (c) The second biggest subnetwork with 24 drugs and 35 targets. Drugs were mostly from antidepressive, antiparkinson, and anticonvulsant agents. Shape of notes represent types of entity: circle for drug target and rectangle for drug. The size of node corresponds to the number of interacting drugs.
Extension of DIPN pharmacological networks
The base network was extended by referencing the database of BioGRID (http://thebiogrid.org/)12 to add intermediators that had either genetic or physical protein–protein interactions with the targets. A total of 2,807 intermediators had interactions with any targets of at least one drug. For each intermediator, drug-degree was defined as the number of interacting drugs that the intermediator indirectly interacted through their known targets. With drug-degree as the cutoff, we generated 20 different extended networks, denoted as Min01 through Min20. The Min01 network included all 2,807 intermediators, whereas the Min20 network included 12 intermediators with at least 20 interacting drugs (Supplementary Table S2). The number of intermediators substantially decreased as the minimum drug-degree threshold increased. As an example, the Min05 extended network including 274 intermediators, ~10% of all intermediators, is depicted in Supplementary Figure S1. Compared with the base pharmacological network, the drugs and their targets in this extended network are highly interconnected via intermediators.
Gene set enrichment analysis on drug targets and intermediators
To identify significantly enriched biological functions among the targets and intermediators, gene set enrichment analysis was performed using local implementation of the Database for Annotation, Visualization and Integrated Discovery, v6.7 (DAVID; http://david.abcc.ncifcrf.gov/).13,14 Using Benjamini–Hochberg (BH)–adjusted P < 0.05 as the cutoff, we identified 698 biological functions in Gene Ontology (GO; http://geneontology.org/) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) terms in the drug targets and an average of 388 functional terms in the 20 sets of intermediators. Figure 3 is a heat-map of top 10 most significant biological functions in each set, in which the color gradient represents the statistical significance, log transformed by −log10 (BH-corrected P value). The targets were highly enriched with functions such as cell surface receptors, voltage-gated ion channels, and regulation of synaptic transmission, whereas the intermediators were highly enriched with functions related to transcription of mRNA, cell cycle, apoptosis, epidermal growth factor receptor signaling pathway, and other functions.
Figure 3.

Functional enrichment of drug targets and intermediators. A heat-map was generated using top 10 most enriched biological functions in each gene set (the human target set and 20 sets of intermediators at different level). Benjamini–Hochberg (BH)–corrected P values of the top 10 most significant functional terms are represented in a heat-map with −log10 (BH-corrected P value) as color gradient; the darker the red, the more significant. Any blank cell indicates that the corresponding biological function in the gene set is not statistically significant (BH-corrected P value ≥ 0.05).
Identification of significant DIPN-associated intermediators
To identify the intermediators significantly associated with DIPN, we generated 1,000 random drug sets from DrugBank and constructed extended networks for each random network. The drug-degree of each intermediator from the DIPN extended networks were compared with those from the extended networks of the 1,000 random networks using a Z-test. Among the total 2,807 intermediators, 230 (Supplementary Table S3) met our significance criteria: having at least five interacting drugs via their targets, a BH-corrected P value < 0.05, and a minimum of 1.5-fold enrichment of drug-degree in the DIPN networks compared with the average drug-degrees of the random networks. The 20 significant intermediators with the highest number of drug-degrees are listed in Table 1, which includes three transcription factors and nine transcription cofactors according to MatBase (Genomatix, Munich, Germany; http://www.genomatix.de/). Tumor protein 53 (TP53) was the most highly connected significant intermediator, interacting with 49 DIPN drugs in the Min01 network, which is 1.7 times more than the average 37.1 drugs in the 1,000 random networks.
Table 1. Top 20 highly connected significant intermediators in DIPN.
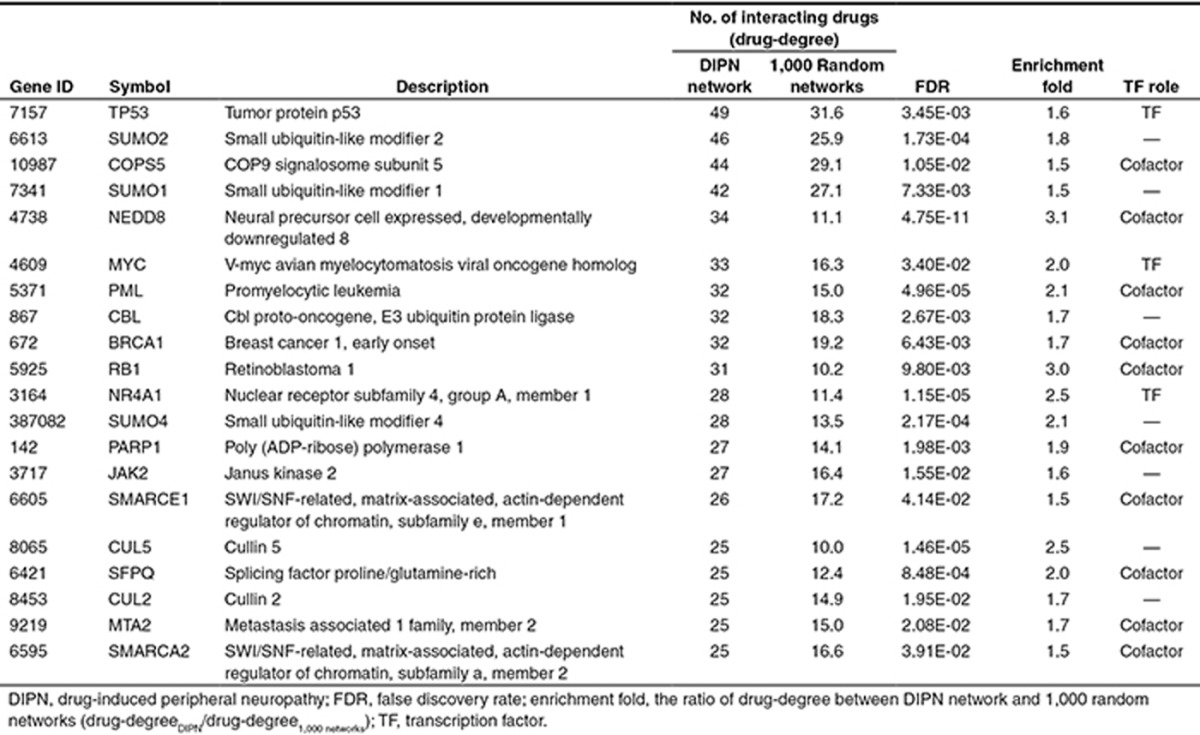
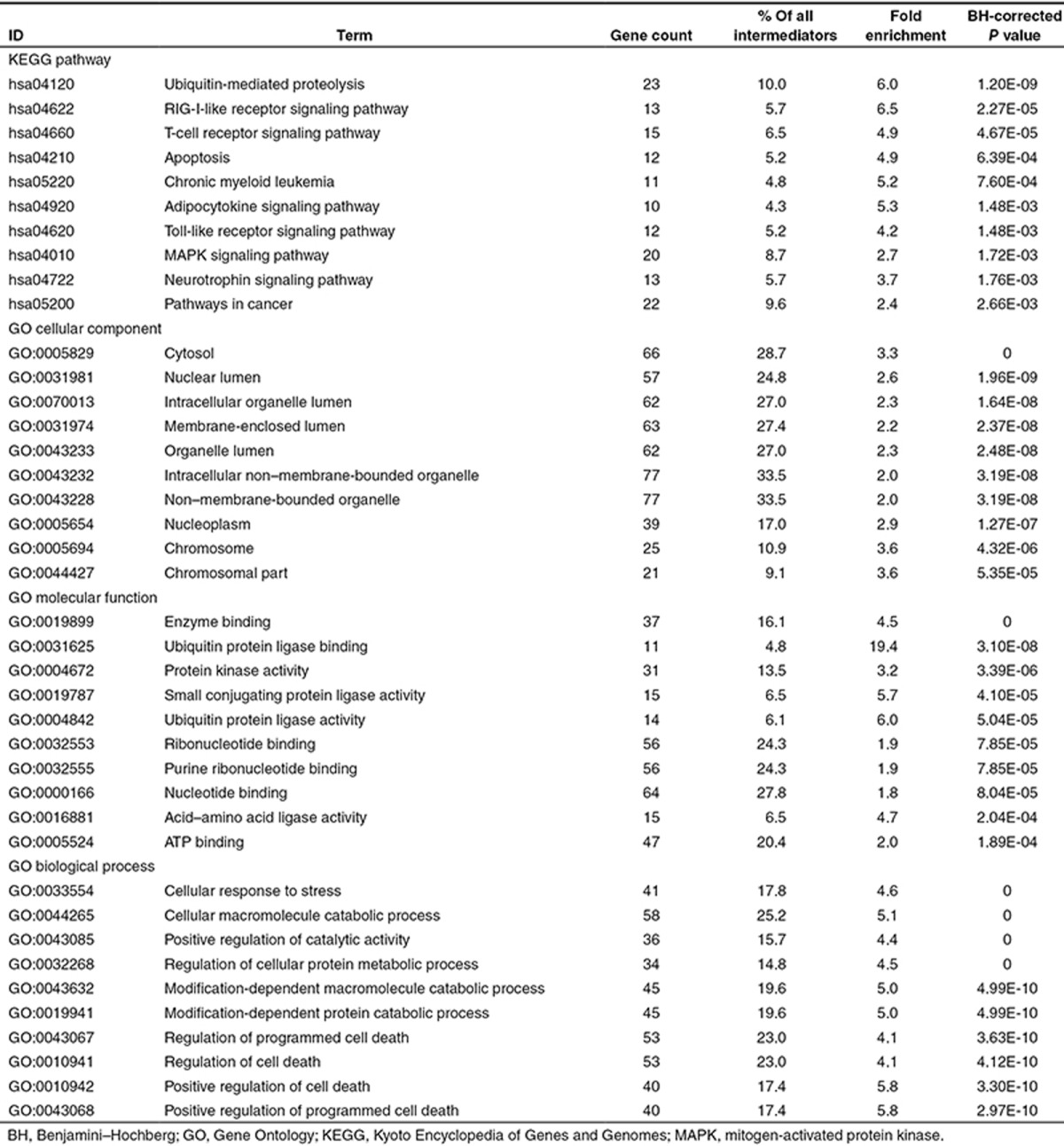
These significant intermediators were further examined for common features using gene set enrichment analysis. The 10 most significantly enriched biological functions in KEGG pathway and three GO categories are listed in Table 2. The ubiquitin-mediated proteolysis pathway was the most significantly enriched pathway, comprising 10% of the significant intermediators. Apoptosis-related terms were also highly enriched.
Table 2. Top 10 most significantly overrepresented biological functions in KEGG pathway and GO terms.
Comparison of known targets and intermediators with the genes implicated in neuropathy
We compiled 72 human genes whose mutations result in neurological disorders with neuropathy,15 and 36 genes for which single-nucleotide polymorphisms or differential expression was reportedly to be associated with chemotherapy-related neuropathy.4,6,16,17,18,19,20 When these two lists of genes were compared with the known targets and significant intermediators, little overlap was observed (Supplementary Table S4), suggesting that the DIPN pharmacological networks were not directly linked to the genes that are predisposed to neuropathy.
Integration of clinical information with pharmacological network
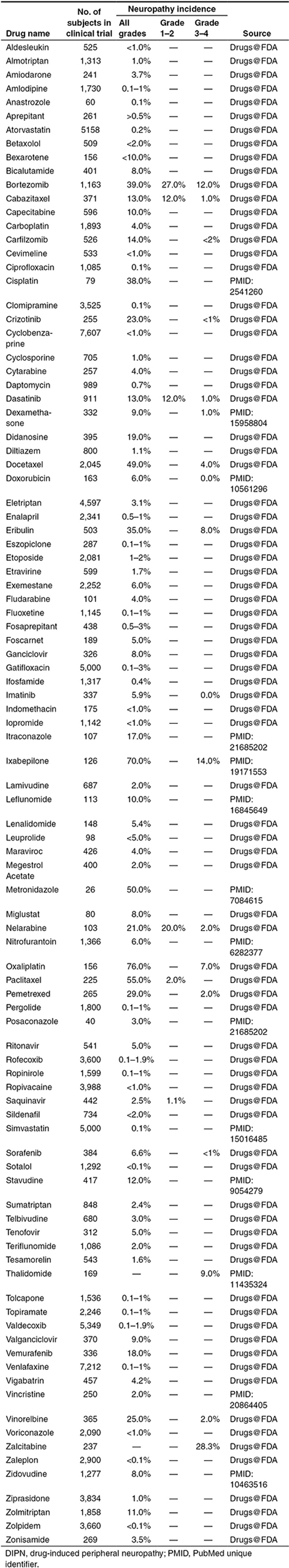
We extracted the clinical data (neuropathy incidence, grade, and the size of clinical trials) for 90 DIPN drugs from drug labels and published new drug application reviews in Drugs@FDA, and 43 drugs from published clinical studies, resulting in a nonredundant set of 97 drugs. Over 30 drugs included data from multiple clinical trials. The trial with the highest incidence was chosen first followed by highest severity if multiple trials of similar sizes were found for a drug. The majority of these 97 drugs had only the incidence of all grades (grade 0–4 combined), whereas only 20 drugs had a detailed incidence profile of grade 1–2 and/or grade 3–4 (Table 3).
Table 3. Incidence of DIPN.
Regression tree analysis was applied to the 97 drugs, of which treatment-related neuropathy incidences were reported, with their neuropathy incidence as the dependent variable. Multiple sets of factors, including intermediators from the pharmacological network (minimum level of 5), known molecular targets, and the drug categories and substructures obtained from DrugBank, were used as explanatory variables. Regression tree models for neuropathy incidence were obtained for all grades. A model of potentially important intermediators is shown in Figure 4a. Among the 17 drugs interacting with v-myc avian myelocytomatosis viral oncogene homolog (MYC), 8 drugs interacting also with PAF15 had an average neuropathy incidence of 38.1%, whereas 9 drugs not interacting with proliferating cell nuclear antigen-associated factor (PAF15) had an average incidence of 2.9% (Figure 4b; see Supplementary Table S5 for additional clinical and demographic data). Figure 4c illustrates a subnetwork centered on MYC, proliferating cell nuclear antigen-associated factor (PAF15), and all of their interacting drugs in the base DIPN pharmacological network. The subnetwork in the middle consists of 14 drugs and 11 drug targets interacting with both MYC and PAF15. Among these 14 drugs, 6 drugs highlighted in yellow had no known neuropathy incidence available from clinical trial data.
Figure 4.

The best working regression model. (a) The collected clinical frequencies of the drug-induced peripheral neuropathy drugs were used in a regression tree analysis modeling using known targets and intermediators at level 5. The numbers in the yellow circles are the numbers of drugs and the numbers beneath the circles are the average incidence of these drugs. (b) The clinical information of the 17 DIPN drugs interacting with MYC alone (n = 9) or with both MYC and PAF15 (n = 8). Y and N indicate whether the corresponding drug interacts with MYC or PAF15. (c) This network includes only those drugs interacting with MYC or PAF15. The drugs within the gray boundary in the middle interact with both MYC and PAF15. The yellow nodes were not included in the regression tree modeling because we were not able to collect appropriate neuropathy incidence data from drug labels or published literature. The drugs interacting with either MYC or PAF15 are colored differently depending on the availability of the neuropathy incidence: cyan and purple for with and without available incidence, respectively.
We also performed linear regression analyses to determine the effect of drug targets and intermediators on neuropathy incidence. Using univariate analysis, three targets and 18 intermediators were identified with false discovery rate < 0.05 as the cutoff (Supplementary Table S6). Based on this result, we performed three multivariate analyses: (i) using all 21 significant factors; (ii) using 12 factors, each with minimum three incidence data points; and (iii) using 7 factors, each with minimum five incidence data points. One drug target (tubulin beta-3 chain (TUBB3)) and three intermediators (protein phosphatase 1 alpha catalytic subunit (PPP1CA), proteasome 26s subunit ATPase 2 (PSMC2), and F-box protein 25 (FBXO25)) were found to be significant.
Discussion
To better understand the underlying mechanisms of DIPN and to facilitate future development of drugs with a minimal potential of inducing DIPN, we conducted a pharmacological network-based analysis. We first compiled the list of DIPN drugs and defined the pharmacological space of DIPN as broadly as possible. A substantial number of drugs with PN identified as an adverse reaction from the postmarketing surveillance were also included, as long as such information is available in their FDA-approved labels. The incidence for these drugs is typically less than 0.1% (Table 3). The DIPN causality for those identified from postmarketing surveillance cannot be established, nor can those from epidemiology studies. On the contrary, the identification of PN as a treatment-related problem in clinical trials is considered to have been judged by physicians in charge of the trials. In light of our goal to project an inclusive pharmacological network, it is not considered crucial whether the causality is established for each of the 234 drugs. Furthermore, the pharmacological networks in the current study do not distinguish severity and incidence. As a result, the DIPN list covered the drugs with a wide range of pharmacological characteristics. Because the 234 drugs were approved for treating various diseases (Figure 1), their pharmacological and extended pharmacological networks could conceivably highlight, at least, part of the network connecting those diseases. On the other hand, these drugs do share a common characteristic of clinical DIPN, and one could argue that these pharmacological networks would biologically reflect the DIPN network and, therefore, would be useful for understanding DIPN.
As shown in Table 3, clinical DIPN incidence varied from drug to drug. It should be noted that the underlying diseases could have affected the DIPN incidence; for instance, multiple myeloma itself can cause PN.21 In the present study, the impact of disease was, however, not taken into account due to the lack of a comprehensive understanding of how individual diseases at different stages had affected the incidence and severity of PN in individual clinical trials. When trials were conducted for a chemotherapeutic agent as compared with a “standard-of-care” treatment, we only adopted those from the trials that had a head-to-head comparison between two single-agent regimens. When multiple trial results for a drug for either the same indication or different indications were available, we adopted a subjective criterion of adopting the result from the trial with the highest incidence first followed by highest severity if multiple trials of similar sizes were found for a drug. As a result, we may have inflated the severity and/or incidence of DIPN for some drugs. Our goal was not to be exclusive by imposing strict rules but to employ consistent rules for adopting the published results.
To identify the pharmacological network with a broad coverage of biological pathways for future development of a useful predictive tool, we extended the base pharmacological network to include intermediators that interacted with the known targets of DIPN drugs. As shown in Figure 3, the most enriched biological functions of these intermediators, very similar across different sets of intermediators, included “transcription cofactor binding,” “cell cycle,” and “regulation of apoptosis.” We speculated that this was likely due to the fact that 27% of the DIPN drugs were antineoplastic agents; however, the significant intermediators, with the antineoplastic or anti-infective drugs excluded, were still highly enriched with these functions (Supplementary Figure S2). It was concluded that these enriched functions were not limited to antineoplastic or anti-infective agents. There seemed to be a distinct pattern in the enriched biological functions between the drug–target group and the various groups of intermediators (Figure 3). Drug targets are mainly receptors or specific kinases, whereas intermediators are cellular proteins that interact with drug targets. It seems reasonable that the spectrum of biological functions covered by intermediators would be different, including regulation and transcription.
Though all the 97 drugs listed in Table 3 were used for our regression tree analyses, many classes of drugs were pruned off the regression tree by the Generalized, Unbiased, Interaction Detection and Estimation tree algorithm due to low predictive power. As a result, higher clinical DIPN incidences of antineoplastic drugs as compared with other classes of drugs could have led to the dominance of these drugs identified in regression tree modeling. Our regression tree modeling of neuropathy incidence suggests that MYC and PAF15 might play an important role in DIPN (Figure 4). Interacting with either MYC or PAF15 alone was associated with relatively low neuropathy incidence of 2.94% or 2.90%, respectively, whereas interaction with both genes had a high rate of 38.14%. The drugs linked to MYC were dominated by floxacins, and then by tyrosine kinase inhibitors, whereas those linked to PAF15 seemed to include drugs from diverse categories. Those drugs linked to both MYC and PAF15 were mostly nucleoside analogues or metabolic inhibitors, followed by taxanes. When we examined the 10 drugs with the highest neuropathy incidences, 4 (paclitaxel, docetaxel, bortezomib, and vinorelbine) were connected to both MYC and PAF15 (Table 3 and Figure 4c), suggesting that these two genes might contribute to DIPN in some drugs. The remaining six drugs with top 10 incidences included four antineoplastic drugs (oxaliplatin, ixabepilone, eribulin, and pemetrexed), one anti-infective drug (metronidazole), and one anti-HIV drug (stavudine). In the pharmacological networks, four of these drugs (metronidazole, oxaliplatin, eribulin, and stavudine) did not have any known human targets; thus, unknown off-targets may play a role. It should be noted that our computational exercise is limited by what pharmacological targets and intermediators are available in the databases. Eribulin with the eighth highest rate (36%), for example, is known to directly bind tubulin, thus disrupting microtubule structures,22,23 which was not properly annotated in neither DrugBank nor TTD.
To demonstrate the usefulness of our theoretical exercises, we further explored the pharmacological networks focusing two antineoplastic agents. Both bortezomib and sorafenib are widely used anticancer drugs, where the former is a proteasome inhibitor and the latter is a tyrosine kinase inhibitor. They both interact with MYC through their targets: RAF1 (v-raf-1 murine leukemia viral oncogene homolog 1) for sorafenib and PSMB5 (proteasome subunit beta type-5) or PSMD2 (26S proteasome non-ATPase regulatory subunit 2) for bortezomib (Figure 4c). Interestingly, they had different incidences of DIPN: 39% for bortezomib and 6.6% for sorafenib. In the extended subnetwork centered on bortezomib and sorafenib, we observed that only the targets of bortezomib interacted with tubulins (Supplementary Figure S3). This finding is in line with other studies that related perturbation of tubulin or microtubules in neural cell lines24 and in rat dorsal root ganglion neurons25 to the neurotoxicity of bortezomib. In fact, five drugs (ixabepilone, paclitaxel, docetaxel, bortezomib, and vinorelbine) belonging to the top 10 list were connected to tubulins (Table 3 and Supplementary Figure S4). These results, though not new, are supported by other studies demonstrating that dysregulated microtubule polymerization by microtubule-targeting agents such as taxanes are strongly related to DIPN.24,25,26 Furthermore, these findings confirmed the quality of the databases and knowledge bases that were used for this study. The three intermediators (PPP1CA, PSMC2, and FBXO25) identified in the multivariate linear regression analysis are preliminary but worthy of further investigation.
The current study demonstrates the usefulness of leveraging the knowledge of the FDA-approved drugs and accumulated knowledge generated from pharmacological as well as medical research for constructing the extended pharmacological network of DIPN. Such extended pharmacological network can potentially be useful for predicting the neurotoxicity potential of new drugs. Based on the modeling result, the usefulness of targeting the genes regulated by MYC and PAF15 could be worthy of further investigation for the discovery of potential drugs for alleviating the symptoms of DIPN. Our next step will be to refine our network analysis by incorporating high-throughput omics data such as gene expression profiles of DIPN drugs in neuronal cells. Our ultimate goal is to develop our theoretical analysis into a predictive tool of DIPN with an acceptable specificity and sensitivity, based on the pharmacological networks and multiplex measurements including transcriptomic profiling. Such tool will be valuable for minimizing the severity and incidence of DIPN.
Methods
Compilation and curation of DIPN drugs
Text-mining of the drug labels and online resources. DIPN drugs were compiled and curated. A text-mining approach was first used to automatically retrieve and screen the latest drug labels, available as PDF files, from the Drugs@FDA database (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/) on 8 November 2012. The PDF files were converted to text files using Xpdf (http://www.foolabs.com/xpdf/). The flanking texts surrounding the identified neuropathy terms in the drug labels were manually examined for their accuracy, and a nonredundant set of DIPN drugs was compiled. Two other drug resources, DailyMed (http://dailymed.nlm.nih.gov/) and SIDER2 (http://sideeffects.embl.de/), were to identify additional DIPN drugs. The drugs collected only from DailyMed or SIDER2 were subjected to confirmation for inclusion by referencing the literature (PubMed).
Drug name normalization, integration, and manual curation. The drug names collected were normalized to be consistent and comparable. The official names in the DrugBank database were used as the representative names, if available. Drugs without a DrugBank record were processed by a Perl script to automatically identify the core ingredient name. For example, “sumatriptan succinate” was normalized to “sumatriptan.” The lists of drugs collected above were normalized and combined and then thoroughly reviewed by a pharmacology expert to include only the single active ingredients with evidence of DIPN.
Pharmacological network analysis
Construction of pharmacological network. A base pharmacological network consisting of DIPN drugs and their known intended and unintended targets was constructed by referencing the DrugBank version 3.0 (downloaded on 21 October 2012; http://www.drugbank.ca/)27 and Therapeutic Target Database version 4.3.02 released on 25 August 2011 (TTD; http://bidd.nus.edu.sg/group/cjttd/).28 This base network was extended by adding the proteins or genes, denoted as intermediators, that have protein–protein interactions or genetic interactions with the known targets of DIPN drugs. The protein–protein interactions and genetic interactions were derived from the BioGRID release 3.2.97 (http://thebiogrid.org/).12 Drug-degree was defined for each intermediator as the number of drugs indirectly interacting via the known targets. Twenty extended networks were generated using the criterion of drug-degrees ranging from 1 to 20 as the threshold for intermediators to be included in a network.
Functional enrichment of the pharmacological networks. To identify the enriched biological functions of the known targets of DIPN drugs and their intermediators, gene set enrichment analysis was employed using a locally implemented version of DAVID.13,14 GO terms and KEGG pathway terms were used as biological functions. Terms with a BH-corrected P < 0.05 were deemed significant. A heat-map with −log10 (BH-corrected P value) as a color index was generated for the top 10 most significantly enriched terms of the sets of known targets of DIPN drugs and their intermediators for visualization of the different contents of enriched biological functions across different gene sets.
Identification of significant DIPN-associated intermediators. One thousand random networks, of the same number of drugs as in DIPN, were generated from the 6,825 drugs in DrugBank. Extended networks using the same with a range of drug-degrees (1–20) were also generated for each random network. A Z-test was used to statistically evaluate the difference in drug-degrees of intermediators between the DIPN networks and the random networks. Intermediators meeting the following three criteria were deemed as DIPN-specific significant intermediators: (i) BH-corrected P value < 0.05 in the Min01 network, (ii) minimum drug-degree of 5, and (iii) the drug-degree in DIPN network is at least 1.5 times higher than that in random networks. These DIPN-specific intermediators were subjected to further functional enrichment analysis.
Integration of clinical information with pharmacological networks
Clinical information from drug labels and literature. The data of PN incidence, severity, and trial size from clinical trials were collected from drug labels, published new drug application reviews in Drugs@FDA, and published literature in PubMed. Following automatic text-mining, manual curation extracted the clinical phenotypes of incidence and severity of DIPN for individual DIPN drugs from Drugs@FDA. PubMed was also surveyed to compile relevant clinical information of DIPN drugs. Over 21 million abstracts in PubMed were surveyed using a text-mining script to identify publications containing any of DIPN drugs and keywords, “clinical trial” and “neuropathy.” Then, manual curation was performed to extract relevant clinical information. In the case of drugs with multiple clinical trial data available, the data from the source with the largest trial size first, and the highest incidence followed by the highest severity if the trial sizes were similar, were included for further analyses.
Modeling using regression tree algorithm. The collected neuropathy incidence data were used to build a statistical model with the Generalized, Unbiased, Interaction Detection and Estimation algorithm, a multipurpose machine-learning algorithm for constructing regression trees.29,30 Generalized, Unbiased, Interaction Detection and Estimation algorithm uses recursive partitioning to select the splits of the tree nodes. After an initial large tree is constructed, the algorithm uses 10-fold cross-validation to prune the nodes and estimate prediction accuracy. Only those models with estimated prediction accuracy within half a standard error of the minimum are reported.
Univariate and multivariate linear regression analysis. Linear regression analyses were performed to examine the effect of drug targets and intermediators on neuropathy incidence. Significant factors, defined at false discovery rate < 0.05 in univariate regression, were further examined in a multivariate regression analysis.
Author contributions
J.H. wrote the manuscript. J.P.F.B. and E.L.F. designed the research. J.H., J.P.F.B., A.Y.G., and W.-Y.L. performed the research. J.H., A.Y.G., and W.-Y.L. analyzed the data.
Conflict of Interest.
The authors declared no conflict of interest.
Study Highlights

Acknowledgments
The authors thank Dr. Darrell Abernethy at the US Food and Drug Administration for his support and comments on the manuscript. The authors also thank Dr. Phillipe O'Brien at the University of Michigan for his editorial support. This work was supported by the US Food and Drug Administration, the Program for Neurology Research and Discovery at the University of Michigan, the A. Alfred Taubman Medical Research Institute, and a Juvenile Diabetes Research Foundation Postdoctoral Fellowship (to J.H.). W.-Y.L.'s work was partially supported by the US Army Research Office grant W911NF-09-1-0205 and National Science Foundation grant DMS-1305725.
The views expressed in this article are those of the authors and do not necessarily reflect the official views of the US Food and Drug Administration.
Supplementary Material
References
- Paice J.A. Clinical challenges: chemotherapy-induced peripheral neuropathy. Semin. Oncol. Nurs. 2009;25:S8–S19. doi: 10.1016/j.soncn.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Gaist D., García Rodríguez L.A., Huerta C., Hallas J., Sindrup S.H. Are users of lipid-lowering drugs at increased risk of peripheral neuropathy. Eur. J. Clin. Pharmacol. 2001;56:931–933. doi: 10.1007/s002280000248. [DOI] [PubMed] [Google Scholar]
- Ferrier J., Pereira V., Busserolles J., Authier N., Balayssac D. Emerging trends in understanding chemotherapy-induced peripheral neuropathy. Curr. Pain Headache Rep. 2013;17:364. doi: 10.1007/s11916-013-0364-5. [DOI] [PubMed] [Google Scholar]
- Wheeler H.E., et al. Cancer and Leukemia Group B Integration of cell line and clinical trial genome-wide analyses supports a polygenic architecture of Paclitaxel-induced sensory peripheral neuropathy. Clin. Cancer Res. 2013;19:491–499. doi: 10.1158/1078-0432.CCR-12-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leandro-García L.J., et al. Genome-wide association study identifies ephrin type A receptors implicated in paclitaxel induced peripheral sensory neuropathy. J. Med. Genet. 2013;50:599–605. doi: 10.1136/jmedgenet-2012-101466. [DOI] [PubMed] [Google Scholar]
- Baldwin R.M., et al. A genome-wide association study identifies novel loci for paclitaxel-induced sensory peripheral neuropathy in CALGB 40101. Clin. Cancer Res. 2012;18:5099–5109. doi: 10.1158/1078-0432.CCR-12-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Cancer Institute. Common Terminology Criteria for Adverse Events v4.03 (CTCAE). < http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf >
- Drugs@FDA. < http://www.accessdata.fda.gov/scripts/cder/drugsatfda/ >. Accessed 25 November 2012.
- The Gene Ontology. < http://www.geneontology.org/ >.
- Kuhn M., Campillos M., Letunic I., Jensen L.J., Bork P. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol. 2010;6:343. doi: 10.1038/msb.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su G., Kuchinsky A., Morris J.H., States D.J., Meng F. GLay: community structure analysis of biological networks. Bioinformatics. 2010;26:3135–3137. doi: 10.1093/bioinformatics/btq596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A., et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 2013;41:D816–D823. doi: 10.1093/nar/gks1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang d.a.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH GeneReviews book . < http://www.ncbi.nlm.nih.gov/books/NBK1116/ >. Accessed 25 November 2012.
- Leandro-García L.J., et al. Genome-wide association study identifies ephrin type A receptors implicated in paclitaxel induced peripheral sensory neuropathy. J. Med. Genet. 2013;50:599–605. doi: 10.1136/jmedgenet-2012-101466. [DOI] [PubMed] [Google Scholar]
- Oguri T., et al. Genetic polymorphisms associated with oxaliplatin-induced peripheral neurotoxicity in Japanese patients with colorectal cancer. Int. J. Clin. Pharmacol. Ther. 2013;51:475–481. doi: 10.5414/CP201851. [DOI] [PubMed] [Google Scholar]
- Leandro-García L.J., et al. Regulatory polymorphisms in β-tubulin IIa are associated with paclitaxel-induced peripheral neuropathy. Clin. Cancer Res. 2012;18:4441–4448. doi: 10.1158/1078-0432.CCR-12-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H.H., et al. Polymorphic markers associated with severe oxaliplatin-induced, chronic peripheral neuropathy in colon cancer patients. Cancer. 2012;118:2828–2836. doi: 10.1002/cncr.26614. [DOI] [PubMed] [Google Scholar]
- Broyl A., et al. Mechanisms of peripheral neuropathy associated with bortezomib and vincristine in patients with newly diagnosed multiple myeloma: a prospective analysis of data from the HOVON-65/GMMG-HD4 trial. Lancet Oncol. 2010;11:1057–1065. doi: 10.1016/S1470-2045(10)70206-0. [DOI] [PubMed] [Google Scholar]
- Richardson P.G., et al. Management of treatment-emergent peripheral neuropathy in multiple myeloma. Leukemia. 2012;26:595–608. doi: 10.1038/leu.2011.346. [DOI] [PubMed] [Google Scholar]
- LaPointe N.E., Morfini G., Brady S.T., Feinstein S.C., Wilson L., Jordan M.A. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology. 2013;37:231–239. doi: 10.1016/j.neuro.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.A., et al. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry. 2010;49:1331–1337. doi: 10.1021/bi901810u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poruchynsky M.S., Sackett D.L., Robey R.W., Ward Y., Annunziata C., Fojo T. Proteasome inhibitors increase tubulin polymerization and stabilization in tissue culture cells: a possible mechanism contributing to peripheral neuropathy and cellular toxicity following proteasome inhibition. Cell Cycle. 2008;7:940–949. doi: 10.4161/cc.7.7.5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff N.P., et al. Bortezomib alters microtubule polymerization and axonal transport in rat dorsal root ganglion neurons. Neurotoxicology. 2013;39:124–131. doi: 10.1016/j.neuro.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson K., Ocean A.J. Peripheral neuropathy with microtubule-targeting agents: occurrence and management approach. Clin. Breast Cancer. 2011;11:73–81. doi: 10.1016/j.clbc.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Knox C., et al. DrugBank 3.0: a comprehensive resource for ‘omics' research on drugs. Nucleic Acids Res. 2011;39:D1035–D1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F., et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012;40:D1128–D1136. doi: 10.1093/nar/gkr797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh W.-Y. Regression trees with unbiased variable selection and interaction detection. Stat. Sinica. 2002;12:361–386. [Google Scholar]
- Loh W.-Y. Probability Approximations and Beyond. Springer; 2012. Variable selection for classification and regression in large p, small n problems. pp. 135–159. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.