

Abstract
Prenatal exposure of the developing brain to various environmental challenges increases susceptibility to late-onset of neuropsychiatric dysfunction; still the underlying mechanisms remain obscure. Here we show that exposure of embryos to a variety of environmental factors such as alcohol, methylmercury and maternal seizure activates HSF1 in cerebral cortical cells. Furthermore, Hsf1 deficiency in the mouse cortex exposed in utero to subthreshold levels of these challenges causes structural abnormalities and increases seizure susceptibility after birth. In addition, we found that human neural progenitor cells differentiated from induced pluripotent stem cells derived from schizophrenia patients show higher variability in the levels of HSF1 activation induced by environmental challenges compared to controls. We propose that HSF1 plays a crucial role in the response of brain cells to prenatal environmental insults and may be a key component in the pathogenesis of late–onset neuropsychiatric disorders.
Introduction
It is generally accepted that disturbance of genetic programs and prenatal exposure to harmful environmental factors increase the susceptibility of the brain to neurological and psychiatric disorders such as epilepsy, autism and schizophrenia (SZ) (Ben-Ari, 2008; Thompson et al., 2009). Since new types of detrimental environmental agents are continuously added to the list and individual differences in susceptibility become evident, increased attention is being paid to the possible interaction between prenatal environmental and genetic factors in fetal development.
It has become apparent that functional and anatomical anomalies in the brain provoked by diverse prenatal environmental challenges include very similar phenotypes (van Loo and Martens, 2007; Edwards et al., 2003; Edwards, 2006). The cerebral cortex, which develops through coordinated developmental events such as neurogenesis, migration and maturation (Rakic, 1988; Ayala et al., 2007), is particularly vulnerable to disturbances in the prenatal environment. For example, rodent and non-human primate models have shown that prenatal exposure to alcohol (e.g., Mooney et al., 2004), hypoxia (De Riu et al., 1995), cocaine (e.g. Lidow, 2003), methylazoxymethanol (Chevassus-au-Louis et al., 1999b), X-irradiation (Algan and Rakic, 1997) and methylmercury (MeHg) (e.g., Kakita et al., 2001) can cause similar structural abnormalities in the cortex; e.g., heterotopias and reduced cortical volume and thickness, in conjunction with decreases in the seizure threshold of offspring (Norman et al., 2009; Chevassus-au-Louis et al., 1999a,b; Oghlakian et al., 2009; Szasz et al., 1999; Russo et al., 2008; De Feo et al., 1995). This finding is consistent with human clinical cases; in addition to genetic mutations (Dixon-Salazar and Gleeson, 2010; Walsh and Engle, 2010), there is a clear correlation between the pathogenesis of epilepsy and exposure to alcohol, methylmercury (MeHg), maternal epileptic seizure (with or without antiepileptic drugs), stroke and infection (Chevassus-au-Louis et al., 1999a).
A main drawback of these animal studies is that, even taking species differences in metabolic rates of substrates into consideration, exposures were made at much higher levels than are clinically relevant for humans to yield specific phenotypes to examine. In fact, although phenotypic manifestation of environmental impacts generally correlates with the magnitude of exposed environmental challenges, the penetrance is relatively small. On the other hand, even exposure to subthreshold levels can lead to devastating consequences (Miller et al., 2006). These lines of evidence imply the presence of shared mechanisms in the developing brain that increase tolerance against the shared impact of different environmental challenges and suggest that defects in these mechanisms in certain individuals may increase the chance of phenotypic manifestation, i.e., onset of disease, which occurs later in life, even in or post adolescence.
Heat shock proteins (HSPs) are considered essential molecular chaperones that respond to diverse external challenges in organisms ranging from bacteria to humans (Lindquist, 1986). Transcription of HSPs is primarily regulated by the transcription factor, heat shock factor 1 (HSF1) (Wu, 1995; Morimoto, 1998), which plays a dominant role in stress responses, whereas other HSFs function in normal cellular processes (Christians and Benjamin, 2006; Akerfelt et al., 2010). The HSF1 response to cellular stresses is deficient in mature neurons in the adult brain (Morimoto, 2008) but rather strong in the embryonic brain. However, nothing is known about its role in response to stresses during brain development.
In the present study, we show that HSF1-HSP signaling is strongly activated in the embryonic cerebral cortex upon exposure to alcohol, maternal seizure and MeHg, even at subthreshold levels that do not exert obvious cortical anomalies alone, and this activation contributes critically to reduce the risk of cortical malformation and postnatal seizure susceptibility. We also provide evidence of unique abnormalities in the activation of the HSF1-HSP signaling in neural progenitor cells (NPCs) derived from inducible pluripotent stem cells (iPSCs) from subjects with SZ. These results demonstrate a novel role for HSF1 in increasing fetal cortical tolerance to harsh prenatal environment, thereby decreasing the prevalence and severity of neuropsychiatric diseases, and they suggest the possible contribution of prenatal HSF1 malresponse to the pathogenesis of several neuropsychiatric disorders.
Results
Subthreshold environmental challenges activate HSF1 in the embryonic cerebral cortex
Among the major prenatal risk factors that are known to increase susceptibility to neurological and psychiatric disorders, we selected three factors for study: ethanol (ETOH), methylmercury (MeHg) and PTZ-induced maternal seizure. We found that prenatal exposure to these factors commonly activates HSF1 signaling in the embryonic cerebral cortex (Fig.1). It is important to note that all three factors were employed at subthreshold levels, which barely induce aberrant structural abnormalities such as heterotopias in the cerebral cortex (Hashimoto-Torii et al., 2011). Pregnant CD-1 mice received intraperitoneal injections of ETOH [25% in phosphate buffered saline [PBS], 2.0 g/kg weight every 24 hours at embryonic days [E] 14-16], MeHg (5 mg/kg weight at E14) or PTZ (40 mg/kg weight at E14) (Fig1A). The blood alcohol concentration (BAC) returned to normal 24 hours after each injection of ETOH in pregnant mice (Hashimoto-Torii et al., 2011). In contrast to the rapid metabolism of ETOH, the level of inorganic mercury remained stable in the embryonic brain for 2 days subsequent to injection (Lewandowski et al., 2002). The average behavioral seizure scale of the PTZ-induced maternal seizure is shown in Figure S1A. Using chromatin immunoprecipitation, we found a robust increase in Hsf1 binding to the Hsp70 promoter in the cerebral cortex 3 hours after the injection of EtOH, MeHG or PTZ at E14 compared with control injections of PBS (Fig.1B, S1B). The effect of prenatal ETOH exposure, in particular, was also addressed using an in vitro human model, in which human fetal cortical slices (gestational week 15 to 18) were cultured with 235 mg/dl ETOH for 24 hours (Hashimoto-Torii et al., 2011). Consistent with the mouse model, HSF1 binding to the Hsp70 promoter was increased in the slices exposed to ETOH, compared with control slices cultured with medium containing PBS (Fig.1B, Fig.S1C). Taken together with our observation that prenatal exposure to these challenges leads to increases of HSP70 mRNA 3 hours after injection compared with PBS in the mouse cortex (ETOH: Hashimoto-Torii et al., 2011; PTZ: fold-change 10.99+/-4.08 (SEM) p=0.0369; MeHg: fold-change 12.67+/-4.799 (SEM) p=0.0498), these results substantiate activation of HSP70 transcription by HSF1.
Fig.1. Environmental challenges activate HSF1 signaling in the embryonic cerebral cortex.
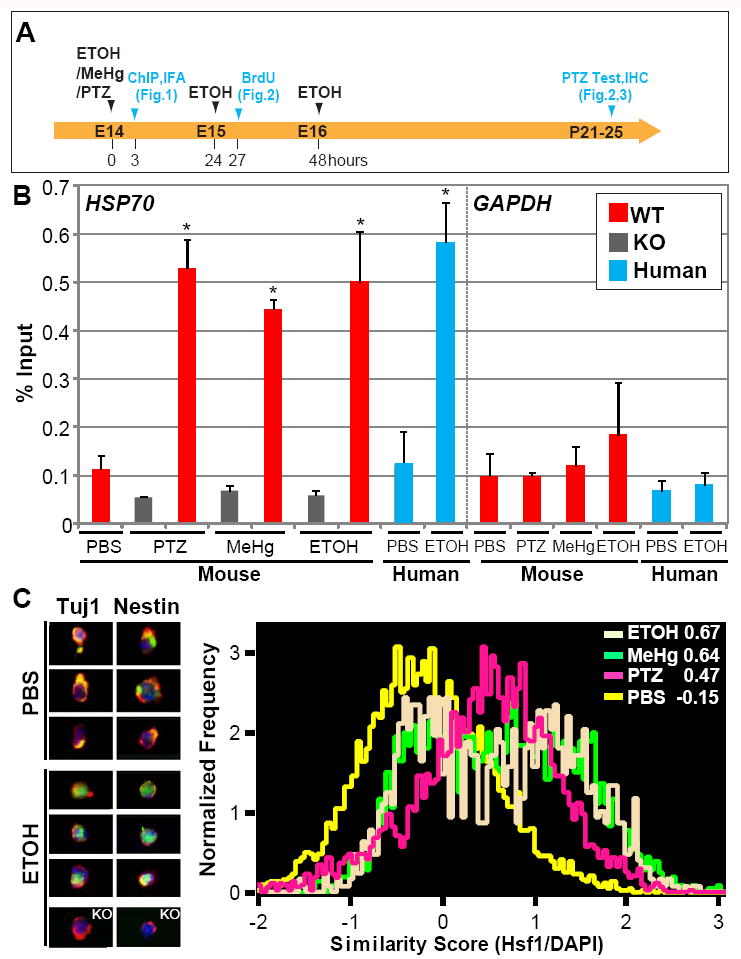
(A) Experimental scheme for Figs.1-3. (B) Chromatin immunoprecipitation analysis showing that the association of HSF1 with the HSP70 promoter is increased in embryonic mouse and human cortices after exposure to the indicated substrates. Occupancy of Hsp70 and Gapdh promoters was analyzed 3 hours after the challenge exposure. Quantification of the band intensities compared with input is shown. “WT” and “KO” indicate wild-type and Hsf1 KO mouse samples, respectively. *p < 0.005 by Student’s t-test compared with controls (n = 4 samples per condition, experiments were repeated at least two times for each sample). (C) Subcellular localization analysis of Hsf1 shows nuclear translocation of Hsf1 by prenatal exposure to indicated challenges. Left panels show representative flow cytometry images of co-immunostaining for Hsf1 (green) and Tuj1 or Nestin (red) with DAPI nuclear counter-stain (blue) in the cortical cells dissected from control (PBS)- and ETOH-exposed embryos. The cells from Hsf1 KO mouse embryonic cortices did not show Hsf1 labeling, confirming the specificity of labeling by the Hsf1 antibody (images at the bottom). Subcellular localization was analyzed using Imaging flow cytometry (right). Numbers at the right top corner are similarity scores indicating the extent to which the fluorescent signals of Hsf1 labeling coincided with nuclear DAPI staining in each cell. Analysis for each condition included more than 100,000 cells obtained from the embryos from multiple litters. See also Figure S1.
We further examined nuclear translocation of Hsf1 protein in cortical cells dissociated from mouse embryos 3 hours after injection of the three substances. Using imaging flow cytometric analysis (IFA), we observed fluorescent signals indicating that Hsf1 labeling coincided with nuclear DAPI staining, as evidenced by the similarity score (Fig.1A,C). We found higher rates of Hsf1 nuclear translocation in Tuj1+ young neurons and Nestin+ NPCs (Fig.1C) than in MAP2+ mature neurons (Similarity Scores of PBS, ETOH, MeHg and maternal epilepsy in MAP2+ cells were -0.22, 0.19, 0.15, 0.11 respectively.). These observations suggest cell type specificity in the Hsf1 response to prenatal challenges.
Hsf1 deletion plus subthreshold prenatal exposure to challenges lead to leptomeningeal heterotopia and reduced cortical size
To address the role of activated Hsf1 in response to external challenges, we next examined the consequence of prenatal exposure to ETOH, MeHg and PTZ-induced maternal seizure in the absence of Hsf1 using Hsf1 knockout (KO) mice (Fig.2). Hsf1 heterozygote pairs were crossed (Fig.2A). In the case of ETOH, we first established that there were no differences in the BAC profiles between wild-type and Hsf1 heterozygous pregnant mice (Fig.S2A). Hsf1 KO mice typically show defects in oocyte meiosis and the development of preimplantation embryos but no obvious phenotypic aberrations in their embryonic brains (reviewed in Christians and Benjamin, 2006). When Hsf1 KO mice were challenged either by ETOH, MeHg or maternal epilepsy, occupancy of the Hsp70 promoter by Hsf1 was abolished in the cortex (Fig.1B). However, this observation on heterozygote was comparable to wild-type mice (PTZ 0.585+/- 0.0929; MeHg 0.508+/-0.0574; ETOH 0.647+/-0.0618, p=n.s by Student’s t-test). Hsf1 KO pups were born from untreated dams at a lower frequency than would be expected from Mendelian inheritance. Dams exposed to PBS bore KO pups at a similar frequency, but dams exposed to ETOH, MeHg or maternal epilepsy bore KO pups at a lower frequency with normal male to female ratios. The body weight of Hsf1 KO mice was the same as wild-type and heterozygous mice at birth but became lower than wild-type and heterozygous mice by postnatal days (P) 10 (14% average reduction at P25). However, weight loss was not influenced further by the various types of prenatal exposure (data not shown). In contrast, when examined at P25, the volume and weight of the cerebral cortex were decreased by the combination of the Hsf1 deletion and exposure to ETOH, MeHg or maternal epilepsy (Fig.2B,C).
Fig.2. Cortical dysplasia induced by prenatal challenges in Hsf1 KO mice.
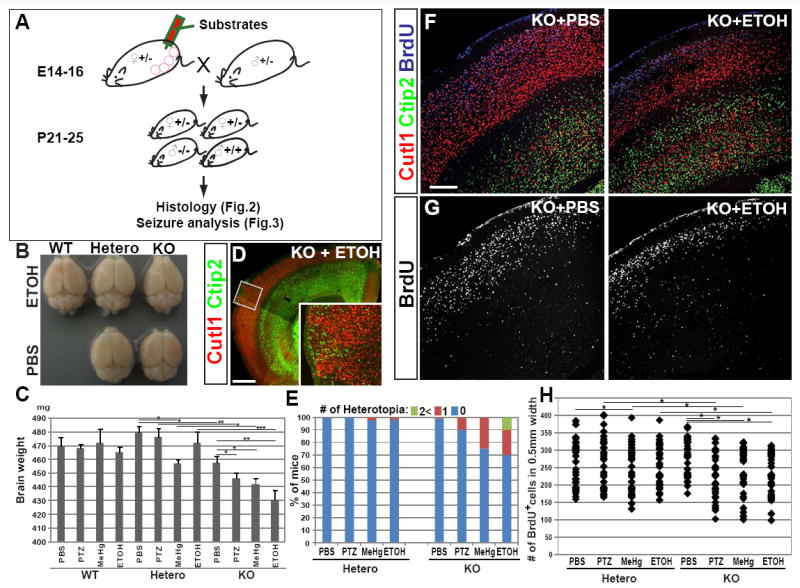
(A) Scheme for substrate administration and experiments. (B) An image of whole brains of P25 mice with indicated Hsf1 genotypes and substrate exposure, showing a slightly smaller cerebral cortex in the Hsf1 KO mouse prenatally exposed to ETOH. (C) Weight of the brains dissected at P25 from the mice with indicated Hsf1 genotypes and prenatal treatments. *p < 0.05; **p < 0.001; ***p < 0.0001 by Student’s t-test. (D) Immunohistochemistry for Cutl1 and Ctip2 in a cortical section of a P25 Hsf1 KO mouse prenatally exposed to ETOH, showing the formation of heterotopia. The inset shows the higher magnification view of leptomeningeal heterotopia seen in the boxed area. Bar = 0.5 mm. (E) Percentages of mice with one or multiple leptomeningeal heterotopias in the group of each genotype and prenatal exposure. (F,G) Immunohistochemistry for Cutl1, Ctip2 and BrdU (labeled at E15) in cortical sections of P25 mice with indicated genotypes and prenatal exposures. Bar = 0.2 mm. (H) The dot plot shows the number of BrdU+ cells in 0.5 mm wide cortical regions. BrdU+ cells were counted in the sections represented in G (n = 6 from more than 3 dams per condition). *p < 0.05 by Mann-Whitney U test. See also Figure S2.
Numerous structural abnormalities were identified in the cerebral cortex of KO and heterozygous mice exposed to subthreshold prenatal challenges. Notably, a higher incidence of leptomeningeal heterotopia formation was observed in Hsf1 KO mice that were prenatally exposed to any of the three factors, compared to the controls (Hsf1 KO mice with PBS exposure, or Hsf1 heterozygotes exposed to the factors) (Fig.2D,E,S2D). Multiple heterotopic nodules were also observed in the Hsf1 KO mice exposed to ETOH (Fig.2E). These heterotopias were most pronounced in the frontal cortex. In addition, we found that the thickness of the cortical plate (CP) was slightly reduced in the Hsf1 KO mice exposed to each of the factors prenatally (ETOH: Fig.2F,G, MeHg and PTZ: data not shown). This reduction was not observed in Hsf1 KO mice exposed to PBS, although mild hydrocephaly developed, as reported previously (Santos and Saraiva 2004; Homma et al., 2007). Within the CP, the thickness of Cutl1+ upper cortical layers (II-IV) was reduced more than that of CTIP2+ lower layers (V and VI) (Fig.2F). The reduction of upper layer thickness was evident at birth, before the emergence of hydrocephaly in Hsf1 KO mice (Fig.S2B).
Because upper layer neurons are generated during the exposure time to our, subthreshold environmental challenges, we hypothesized that the process of neurogenesis was a likely target of the environmental challenges. To examine this possibility, we performed birth date labeling with Bromodeoxyuridine (BrdU) at E15 in Hsf1 mice exposed to challenges or PBS and we counted the labeled cells at P25. The results showed that the number of cells produced in Hsf1 KO cortex upon prenatal exposure to each factor was decreased versus PBS-exposed Hsf1 KO or challenge-exposed heterozygous cortices (Fig.2G,H). In contrast, the number of cells labeled by BrdU at E12, when lower layer neurons are generated but before challenge exposure, was not affected (Fig.S2C). This finding is consistent with moderate effects on the thickness of lower layers. Altogether, these results suggest that the smaller brain is likely caused, at least in part, by temporal reduction of neuronal production after the exposure in Hsf1 KO mice. Although some neurons extended their migration into layer 1, where they formed leptomeningeal heterotopias (Fig.S2D), gross neuronal migration in the cortex was unaffected (Fig.S2E). The ratio of excitatory to inhibitory neurons and the ratios of interneuron subtypes were not affected (Fig.S2F,G). Cross-fostering between PBS- and challenge-exposed Hsf1 KO litters did not influence cortical phenotypes in each litter (Fig.S2H), indicating that the phenotypes depend neither on the effects of the challenge to the dams nor on maternal behavior.
Prenatal exposure to subthreshold challenges upon deletion of Hsf1 increases susceptibility to epilepsy
The correlation between prenatal exposure to environmental risk factors and the pathogenesis of epilepsy associated with malformation of the brain, especially heterotopia in the cortex, has been reported both in humans and rodent models (reviewed in Chevassus-au-Louis et al., 1999). Accordingly, we investigated whether the increase in seizure susceptibility upon prenatal exposure to challenges is affected by the loss of Hsf1. A series of subconvulsive doses of PTZ (5 mg/kg) was administered intraperitoneally every 10 min to mice at P21-25 until each mouse had a generalized seizure. Seizure severity was quantified with Racine’s canonical behavioral seizure scale, and the dose required to produce a generalized tonic-clonic seizure was compared. As shown in Figure 3A, tonic-clonic seizures occurred at significantly lower doses of PTZ in Hsf1 KO offspring with a prenatal history of exposure to any of the 3 challenges versus doses required to elicit seizures in Hsf1 KO offspring prenatally exposed to PBS or their heterozygous littermates exposed to the challenges (Fig.3A). In another set of experiments in which the behavioral seizure score was examined with cumulative dosing of PTZ (15, 20 and 25mg/kg weight), we found that Hsf1 KO mice with prenatal challenge exposure exhibited overall higher seizure scores than their heterozygous littermates or the KO mice exposed to PBS (Fig.3B). These results indicate that Hsf1 plays a role in curtailing the increase of seizure susceptibility elicited by prenatal exposure to external challenges.
Fig.3. Hsf1 KO mice with a prenatal history of challenge exposure show increased susceptibility to PTZ-induced seizures at the juvenile stage.
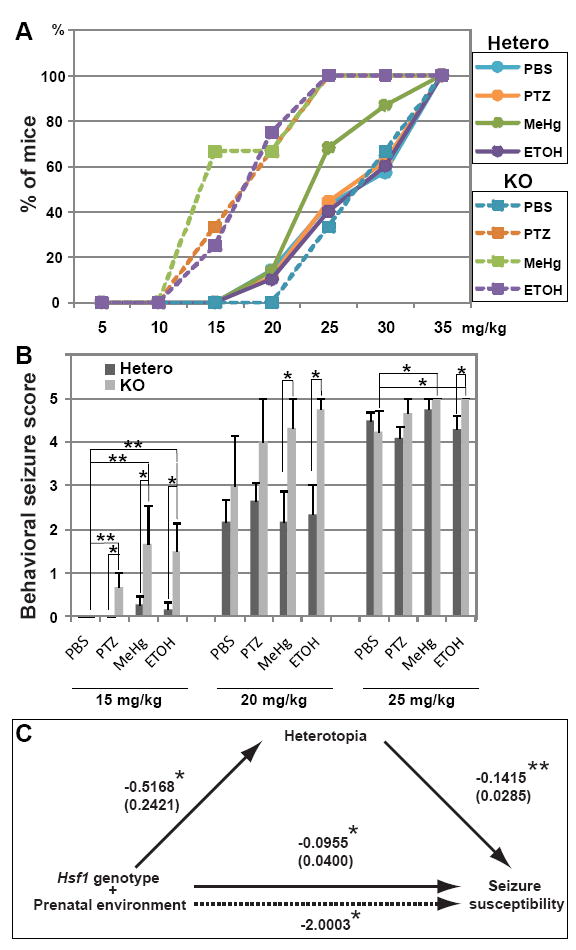
(A) Dose-response cumulative curves for induction of tonic-clonic seizures demonstrate greater PTZ sensitivity of Hsf1 KO mice prenatally exposed to challenges compared to heterozygous littermates and KO mice exposed to control treatment. p < 0.001 by the Kolmogorov-Smirnov test between Hsf1 KO mice prenatally exposed to indicated substrates (except PBS) and heterozygotes with the same exposure or Hsf1 KO mice exposed to PBS. The comparison between heterozygotes prenatally exposed to challenges and those exposed to PBS shows no significant difference, except that a weak effect of MeHg is detected under our experimental condition (p = 0.0082 between MeHg- and PBS-exposed heterozygotes). n ≥ 4 offspring born from multiple dams for each experimental condition. (B) Seizure score values with increasing cumulative doses of PTZ. Hsf1 KO mice prenatally exposed to challenges showed significantly greater behavioral seizure score values at 15 to 25 mg/kg compared with their heterozygous littermates. *p < 0.05; **p < 0.001 by the Mann-Whitney U test. n ≥ 4 offspring born from multiple dams for each experimental condition. (C) Mediational regression analysis of Hsf1 genotype, prenatal environment, heterotopia formation and seizure susceptibility. The values on continuous lines denote the standardized regression coefficient (Beta). Standard errors are indicated in parentheses. The value on a dotted line, which represents the path when heterotopia formation is treated as a mediator, is the Sobel statistic value that indicates the strength of the indirect effect. *p < 0.05; **p < 0.0001. Both the direct effect and indirect effect (through heterotopia formation) of both Hsf1 genotype and prenatal environment on seizure susceptibility are significant, indicating partial mediation of Hsf1 genotype and prenatal environment on seizure susceptibility.
Next, we performed meditational regression analysis (Baron and Kenny, 1986) to test the model that heterotopia formation is a mediator of the effect of Hsf1 gene dosage and prenatal environment on seizure susceptibility. The analysis indicated partial mediation by heterotopia formation on seizure susceptibility, with an effect ratio of 0.73 (Fig.3C). This result suggests a correlation between the heterotopias and increased seizure susceptibility, but identifying a definitive role that heterotopias may play in promoting epileptiform activity still awaits electrophysiological studies.
Subthreshold challenge exposure increases apoptotic cell death of NPCs in Hsf1 KO embryos
Having found that the loss of Hsf1 causes the emergence of cortical dysplasia upon prenatal exposure to various agents or treatments, we sought to determine possible cellular mechanisms. Given the well-known role that Hsf1 plays as a cellular protective factor against stress and our results showing reduced thickness of CP in prenatally challenged Hsf1 KO mice at P0, we first tested whether the survival of cortical cells in response to environmental challenges was affected in the Hsf1 KO embryonic cortex. The degree of cell death was determined by counting apoptotic cells at E16 by TUNEL staining after exposure to substrates at E14-16 (Fig.4,S3A). Compared with heterozygous embryos exposed to ETOH and KO embryos exposed to PBS, the number of TUNEL+ cells was significantly increased in the entire cortex of KO embryos exposed to ETOH (Fig.4A). The increase was particularly evident in the ventricular zone (VZ) and subventricular zone (SVZ) (Fig.4A,C). Apoptosis was similarly increased in the KO cortex exposed to MeHg or PTZ-induced maternal seizure (Fig.4C). However, prenatal challenges did not induce apoptosis after birth (Fig.S3E). Interestingly, the NPCs in the hippocampal primordium appeared to be more vulnerable to maternal epilepsy than to the other agents (Fig.S3B, n=8). Taken together, these results suggest that NPCs and likely also their immediate progeny, immature neurons, depend upon Hsf1 for survival in a deleterious prenatal environment.
Fig.4. Increase of apoptotic cell death in the cortex of Hsf1 KO embryos exposed to challenges.
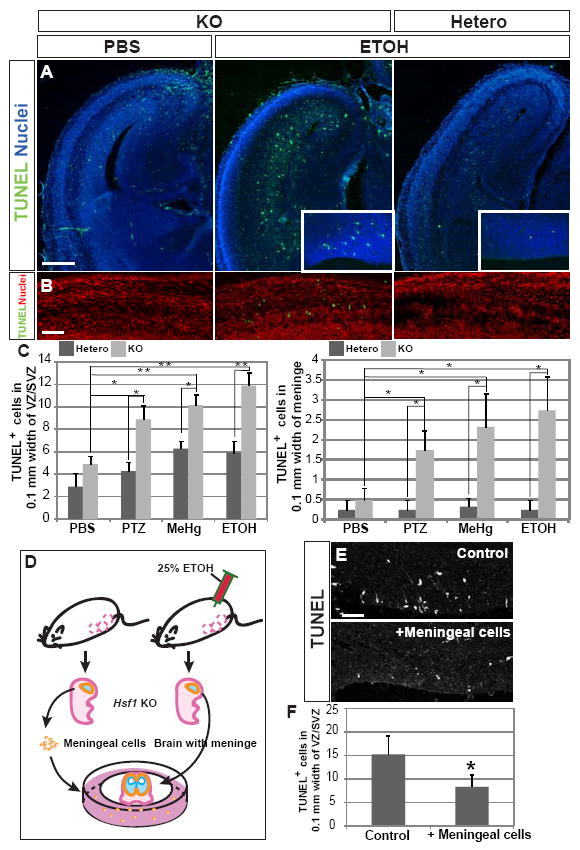
(A) TUNEL staining at E16 in cortical slices from embryos with indicated genotypes and prenatal exposure. An increase in apoptosis in the cortex of Hsf1 KO embryos exposed to ETOH is evident, particularly in the VZ/SVZ (insets). Bar = 0.5 mm. (B) TUNEL staining in meninges of the E16 Hsf1 KO embryos exposed to PBS or ETOH. ETOH-exposed Hsf1 KO embryos show increased apoptosis. Bar = 50 μm. (C) Quantification of TUNEL+ cells in the VZ/SVZ (left) and in the meninge in the cortices of Hsf1 mice exposed to indicated substrates. *p < 0.05; **p < 0.0001 by Student’s t-test. n = 8 (left) or 4 (right) embryos from more than 3 dams for each experimental condition. (D) Scheme for the preparation of in vitro slice culture with meningeal cells (see the details in Experimental Procedures). Slices of cerebral cortex and surrounding tissues were prepared from the Hsf1 KO embryos exposed to ETOH at E14 and 15, 3 hours after the last exposure to ETOH at E15. Fig.S3F demonstrates the increase in apoptosis in the meningeal cells at this time. The slices were cultured on filter inserts in the plates on which meningeal cells collected from other Hsf1 KO embryos (without challenge exposure) were plated. (E) Compared with control cultures (without meningeal cells, top), the culture with meningeal cells from embryos unexposed to challenge (bottom) showed significantly decrease in TUNEL+ cells in the VZ/SVZ. Bar = 50 μm. (F) Quantification of TUNEL+ cells in the VZ/SVZ. *p = 0.027 compared with the control culture (without meningeal cells) by Student’s t-test. n = 4 samples from 2 independent experiments per condition. See also Figure S3.
Meningeal cells of the Hsf1 KO embryos are vulnerable to prenatal subthreshold ETOH exposure
In Figure 2, we showed evidence of leptomeningeal heterotopias in the cortex of Hsf1 KO mice exposed prenatally to the substrates. Because pial basement membrane disruption is thought to be a main cause of leptomeningeal heterotopias (Siegenthaler and Pleasure, 2011; Barros et al., 2011), we examined the effects of prenatal challenge exposure at the pial region using the ETOH model, which showed the most pronounced effects on heterotopia formation (Fig.2). In wild-type mice, nuclear translocation of Hsf1 in meningeal cells was promoted in the cerebral cortex of ETOH-exposed embryos observed 3 hours after the exposure at E14 (Fig.S3C). In Hsf1 KO mice, but not in heterozygotes, we observed an increase in apoptotic meningeal cells by ETOH but not by PBS at E16 (Fig.4B,C). In addition, the KO cortex occasionally showed cortical regions with disrupted integrity of basal membrane and fewer RC2+ radial glial endfeet (Fig.S3D), an observation that often precedes heterotopia formation. Since the number of apoptotic cells in these particular regions was similar to that in surrounding regions with normal integrity of basement membrane, basal membrane disruption might be due to mechanisms independent of apoptosis in meningeal cells, e.g., impaired attachment between pial cells and extracellular matrix. Previous studies have shown that meningeal cells provide neurotrophic factors to NPCs through their endfeet attached to the basal membrane and that the lack of meningeal cells increases apoptotic cell death of NPCs in the VZ (Radakovits et al.,2009). To address whether meningeal cells contribute to progression of apoptotic cell death of NPCs in the ETOH-exposed Hsf1 KO cortex, cortical slices of Hsf1 KO embryos were prepared with meninges at E15, 3 hours after the second injection of ETOH, and cultured either with or without meningeal cells purified from other, unchallenged E15 Hsf1 KO embryos (Fig.4D). We observed a significant reduction in apoptotic cells in the culture supplemented with unchallenged meningeal cells (Fig.4E,F). The rescue of cell death was also observed to a similar extent with the meningeal cells purified from wild-type embryos (data not shown). These results suggest that the death of meningeal cells contributes to apoptosis in neural progenitors in the ETOHexposed Hsf1 KO cortex.
Subthreshold challenge exposure affects cell cycling of NPCs in Hsf1 KO embryos
Entering growth arrest is a pathway alternative to apoptotic death when cells are exposed to harsh conditions (Klein and Ackerman, 2003). We therefore measured the proliferation rate of NPCs at E15 by obtaining BrdU labeling indices in the embryonic cortex after exposure to substrates (Fig.S3A). Having found a significant reduction of proliferation rate in Hsf1 KO embryos exposed to challenges compared with heterozygous littermate or KO embryos exposed to PBS (Fig.5A,B), we further examined cell cycling of NPCs with several molecular markers following subthreshold ETOH challenge. A decrease in Ki67+ (a marker for active mitotic cells in G2, S and M phases) cells and, conversely, increases in MCM2+/Ki67- (MCM2 is a marker for quiescent progenitors in G1 phase, Stoeber et al., 2001) cells and MCM2-/Ki67- postmitotic cells were evident in the VZ/SVZ of the ETOH-exposed Hsf1 KO cortex at E16 (Fig.5C-E). Consistently, the number of PH3+ (a marker for cells in M phase) cells was reduced, but the neuronal marker Tuj1 increased, revealing elevated and ectopic neurogenesis in the VZ/SVZ of Hsf1 KO cortex exposed to ETOH (Fig.S4A,B). The increase in cell cycle exit was also confirmed in these cortices by counting the number of Ki67- cells labeled with BrdU 1 day before fixation (Fig.S3A, S4C). These results suggest that Hsf1 activation is indispensable for the NPCs to escape from being arrested at the G1 phase or exiting the cell cycle (G0) prematurely upon exposure to challenge.
Fig.5. Hsf1 maintains cell cycling of NPCs upon exposure to environmental challenges.
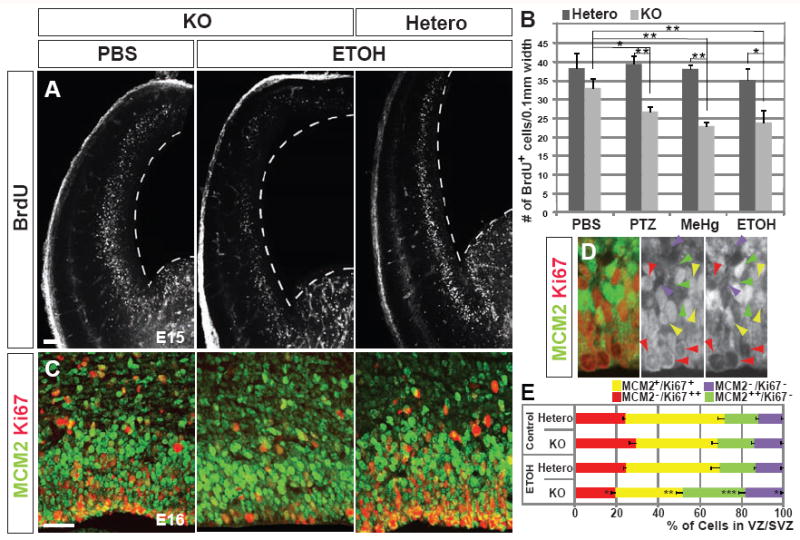
(A) Pulse-labeling with BrdU for 30 minutes at E15 in the cortex of PBS- or ETOH-exposed embryos with indicated genotypes. The number of BrdU+ progenitors is decreased in the Hsf1 KO cortex exposed to ETOH. Broken lines indicate the VZ surface. Bar = 0.25 mm. (B) Quantification of BrdU+ cells in the cortices of Hsf1 mice exposed to indicated substrates. *p < 0.05; **p < 0.005 by Student’s t-test. n = 8 embryos from multiple dams for each experimental condition. (C, D) Immunostaining for MCM2 and Ki67 in PBS- and ETOH-exposed E16 embryonic cortices with indicated genotypes. The numbers of MCM2++/Ki67- quiescent NPCs and MCM2-/Ki67- post-mitotic cells are increased, whereas MCM2-/Ki67++ active NPCs and MCM2+/Ki67+ NPCs, which are likely in transitional states at M/G1(0) or G1(0)/s, are decreased in the KO cortex exposed to ETOH. Bar = 0.1 mm. (D) Higher magnification view shows immunostaining in the PBS-exposed heterozygote cortical slice. Green, red, yellow and purple arrowheads indicate the MCM2++/Ki67-, MCM2-/Ki67+, MCM2+/Ki67+, and MCM2-/Ki67- cells, respectively. (E) The percentages of cells with indicated labeling of MCM2 and Ki67 in the VZ/SVZ. *p = 0.05; **p = 0.02; ***p =0.01, respectively, compared with heterozygotes exposed to ETOH or KO embryos exposed to PBS by the Student’s t-test. n = 4 embryos from multiple dams for each experimental condition. See also Figure S4.
RNAi-mediated Hsf1 knockdown in cortical cells revealed cell-autonomous function of Hsf1
Aberrant placental development in Hsf1 KO embryos (Xiao et al., 1999) or indirect effects from other tissues may contribute to the damage to cortical cells in challenge-exposed embryonic cortex. To assess a direct role for Hsf1 in the cortical cells using the ETOH mouse model, we used an RNA interference (RNAi)-mediated approach to knockdown Hsf1 in the cortical cells. Screening of small hairpin RNA (shRNA) clones with N2A mouse neuroblastoma cells identified clones C2 and S4 to be the most effective in suppressing the endogeneous expression of Hsf1 (56 and 62% reduction, respectively)(Fig.S4D). Mouse embryos were electroporated in utero with either of the two shRNAs at E14, and ETOH was administered 6 hours after complete recovery of the dams from anesthesia (Fig.S3A). ETOH was administered once a day until E15 (Fig.S4F,G,I,J) or E16 (Fig.S4E,H). Consistent with our previous results with Hsf1 KO mice, the percentage of TUNEL+ apoptotic cells was increased (Fig.S4E,H) and the proliferation rate of NPCs was reduced (Fig.S4F,I) by the combination of ETOH exposure and Hsf1 knockdown. An increase in NPCs in the quiescent state or exiting the cell cycle was also confirmed using MCM2, Ki67 and BrdU immunohistochemistry (data not shown). These results were further confirmed by Fucci-G1/0 Orange reporter labeling, which detects G1 and G0 phase cells by expression of monomeric Kusabira-Orange2 (Sakaue-Sawano et al., 2008)(Fig.S4G,J). All the effects of Hsf1 knockdown were rescued by co-expressing full-length human HSF1 (Fig.S4H-J). These results suggest that the cortical phenotypes induced by challenge exposure in Hsf1 KO mice critically depend upon the loss of Hsf1 intrinsic to the cortical cells.
HSP70 mediates the function of HSF1 upon prenatal exposure to ETOH
HSP70 is known as a major transcriptional target of HSF1 (reviewed in Wu, 1995; Morimoto, 1998), and its transcription is strongly induced by prenatal exposure to challenges (ETOH: Hashimoto-Torii et al., 2011, PTZ, MeHg: data not shown). To test whether introduction of HSP70 mitigates the adverse effects of challenge exposure in the Hsf1 KO cortex, an HSP70 expression plasmid was electroporated into the Hsf1 KO cortex at E14. We again used the ETOH model for the analysis. ETOH was injected 6 hours after electroporation and every 24 hours afterward until E15 or E16 (Fig.S3A). Overexpression of HSP70 reduced the number of apoptotic cells in response to ETOH exposure to that comparable with heterozygotes (Fig.6A,B,G). However, HSP70 had no effect on the cell cycling defects as assessed by BrdU labeling and Fucci-G1/0 Orange indices (Fig.6C-F,H,I), although full-length HSF1 was able to mitigate both, consistent with shRNA studies in Fig.S4. We detected no effect of prenatal ETOH exposure on apoptotic cell death and cell cycling in Hsp70 KO mice (data not shown), likely because of the compensation by other HSPs. Overexpression of HSP70 in wild-type cortex did not show any overt effects on cortical development (data not shown). Altogether, our results suggest that HSP70 partially mediates HSF1 functions under environmental challenges, i.e., the inhibition of apoptotic cell death but has no effect on cell cycling.
Fig.6. HSP70 mitigates ETOH exposure-induced cell death in the cortex of Hsf1 KO mice.
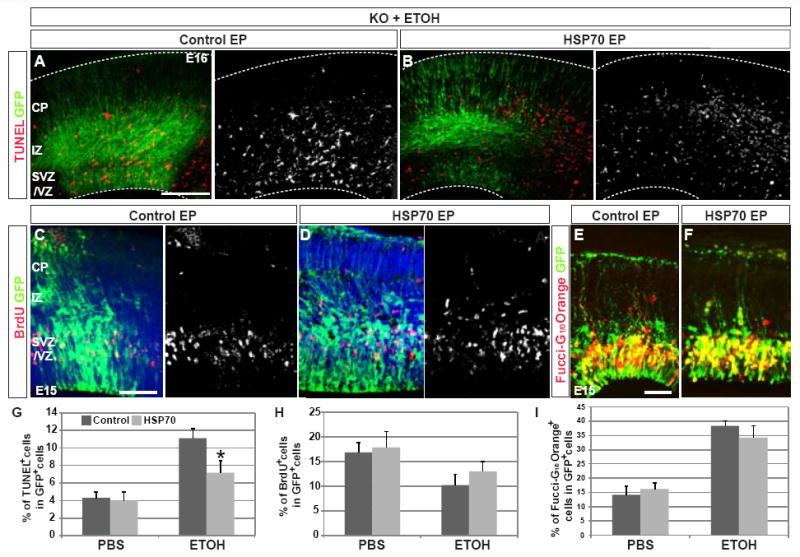
(A,B,G) Increased apoptosis in the cortex of Hsf1 KO embryos by prenatal ETOH exposure is mitigated by HSP70 overexpression. The number of TUNEL+ cells is reduced in the electroporated domain labeled by GFP compared to electroporation with control vector (A,B). Bar = 0.2 mm. The reduction is statistically significant (G). *p = 0.04 compared with control electroporation plus ETOH exposure (immediate left column) by Student’s t-test. n = 4 from 4 dams for each experimental condition. (C,D,H) HSP70 overexpression does not improve the decreased proliferation of progenitors in Hsf1 KO embryos exposed to ETOH. Immunohistochemistry for BrdU and GFP in control- (C) and HSP70-electroporated (D) cortices; the quantification (H) shows no effect of HSP70 overexperssion. p = 0.23, comparison between control and HSP70 overepxression with ETOH exposure (right two columns) by Student’s t-test (n > 4 each). Bar = 0.1 mm. (E,F,I) HSP70 overexpression does not affect the increase in G1 and G0 cells in Hsf1 KO embryos exposed to ETOH. Immunohistochemistry for Fucci-G1/0 Orange and GFP in control- (E) and HSP70-electroporated (F) cortices; the quantification (I) shows no effect of HSP70 overexpression. p = 0.44, comparison between control and HSP70 overexpression in ETOH exposure (right two columns) by Student’s t-test (n > 4 each). Bar = 0.1 mm.
Increased cell-to-cell variation in the HSF1 activation level among NPCs differentiated from human iPSCs derived from subjects diagnosed with SZ
Lastly, we examined the possibility that HSF1 activation is altered in patients with neuropsychiatric disorders that are thought to be linked to exposure to harsh prenatal environmental conditions. We used iPSCs derived from patients with SZ (Brennand et al., 2011) as a model because a number of prenatal environmental risk factors have been reported to potentially cause or contribute to SZ (Sullivan 2005) and because abnormalities in HSF1-Hsp70 signaling have been demonstrated in SZ (see Discussion). Two independent NPC lines from four patients with SZ were employed in the experiments. As our studies using MeHg and ETOH indicated that NPCs are more susceptible to environmental disturbances than neurons (Fig.4), we applied the same subthreshold environmental challenges [MeHg at 0.02 mM, ETOH at 50 mM or PBS (equal volume)] to iPSC NPCs for 3 hours (Fig.7A). We measured the copy number of HSP70 and GAPDH in individual cells and found that all cell lines showed a robust increase in HSP70 expression in response to those challenges (Fig.7D). Notably, although the mean of the HSP70 expression change showed no differences between control and SZ NPCs, we found that cell-to-cell variability in HSP70 expression was significantly larger in the SZ NPCs (Fig.7B-D). We did not observe any difference in cell-to-cell variability in GAPDH measurements (Fig.7D). In addition, Pearson’s correlation analysis showed no significant correlation (p=0.22) between the expression levels of GAPDH and HSP70, suggesting that cell-to-cell variability was specific to HSP70. The increase of expression variability in SZ NPCs was also not due to the increase or decrease in the mean expression value of each cell line. We performed measurements of single cell equivalents (Fig.7D); after lysis of individual cells, the lyses of 10 cells were pooled and a 1/10th aliquot was used as single cell equivalent. This procedure provided very small variability among the samples (Fig.7D). Thus, our results demonstrated that HSF1-mediated stress response is abnormal in a subpopulation of SZ NPCs at the single cell level.
Fig.7. Cell-to-cell variability of HSP70 mRNA levels in response to environmental challenges is increased in SZ NPCs.
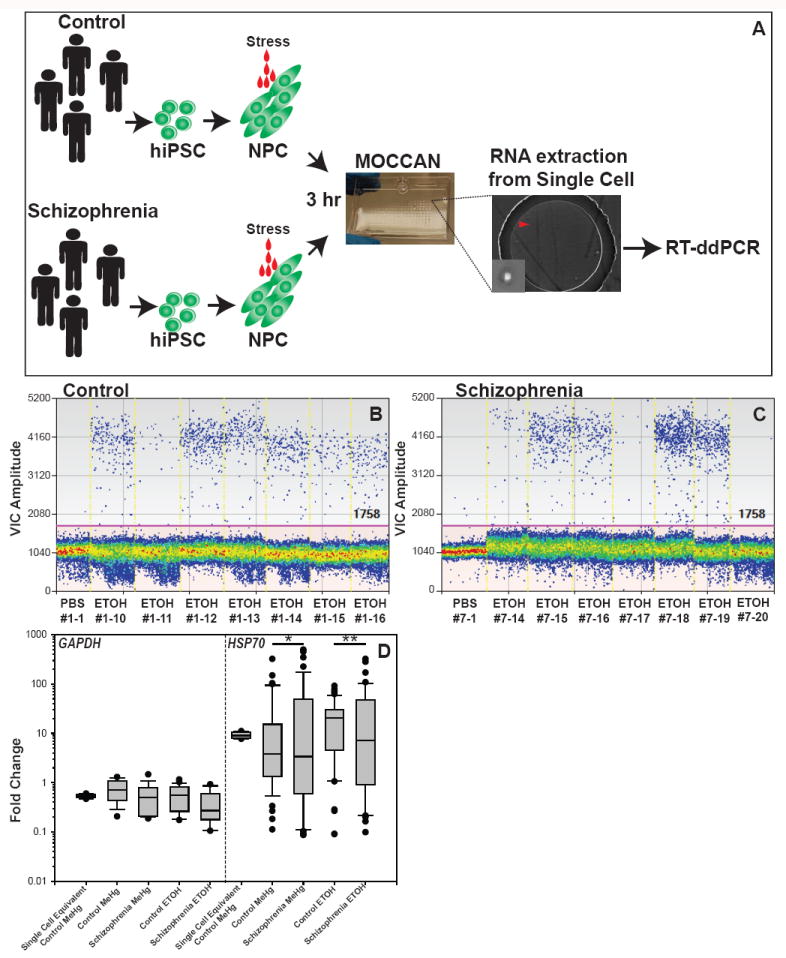
(A) Schematic representation of the experiment. (B,C) Representative results of single cell ddPCR of human HSP70 (VIC label) in control (B) and SZ NPCs (C). The clone number of each cell and substrates applied are shown under the graph. The red line shows cut-off of positive and negative droplets. (D) Graph shows fold change of GAPDH and HSP70 expression compared with PBS exposure. Significantly increased variability was observed in schizophrenic NPCs as compared with the control (*p<0.0001, **p=0.002 by Levene’s test, n>50). Significant differences in the comparison of means were not observed in all sets of comparisons (p>0.05 by Welch’s t-test). The single cell equivalents were made by using 1/10 of a pool of 10 lysed cells for the template of reverse transcription (10 biological replicates were used).
Discussion
It is not understood how diverse types of environmental factors lead to similar brain anomalies and commonly increase susceptibility to neuropsychiatric disorders by interacting with genetic factors. In this study, we demonstrated that HSF1 is activated by prenatal exposure to subthreshold levels of different types of environmental challenge and that impaired HSF1 responses against these subthreshold challenges uncovers common cryptic cortical anomalies such as formation of leptomeningeal heterotopias, reduction of brain volume, cortical thickness, and increased seizure susceptibility.
In addition to the three environmental challenges that are the focus of the present study, exposure to other environmental challenges listed as prenatal risk factors for neurological and psychiatric disorders, such as virus infection, hypoxia, inflammation (all well-known factors in the epidemiology of SZ), irradiation and heat are also factors that increase transcription of HSF1 target genes (Hsps) in many biological contexts (e.g. Melkonyan et al., 1995) This body of knowledge supports the possibility that HSF1-mediated mechanisms may be shared by many types of neuropsychiatric disorders thought to be linked to destructive environmental factors.
Recent studies have revealed that leptomeningeal heterotopias are caused by disruption of basal lamina that delineates meningeal invagination (reviewed in Siegenthaler and Pleasure, 2011). Therefore, the increased incidence of leptomeningeal heterotopias that we observed in prenatally exposed Hsf1 KO mice is likely associated with lesions in meninges as well. Lesions in other non-neural tissues such as placenta may also be involved in the generation of cortical phenotypes (Hsiao and Patterson, 2011).
We found that Hsp70 harbors Hsf1’s function in protecting cells from apoptotic cell death in prenatal exposure to challenges but not in protecting NPCs from being arrested in a quiescent phase or from exiting the cell cycle prematurely. Recent genome-wide high-throughput analysis using ChIP-on-chip has identified numerous potential direct target genes of HSF1 (Hahn et al., 2004). These lines of evidence support the model that HSF1 exerts its pleiotropic functions through a variety of downstream pathways. Understanding the divergent downstream pathways is essential for the development of interventions to prevent or moderate prenatal environment-induced mental illness. An alternative hypothesis for the cell cycle arrest and premature exit of progenitors is that mechanisms compensating for the loss of Hsf1 may modify cell cycling to minimize the impact of de novo apoptosis caused by environmental perturbation.
The observed aberrant HSF1 activation is likely due to unidentified genetic mutations in the schizophrenic patients. Importantly, the iPSC NPC lines from patients with SZ used for the study represent different genetic backgrounds and include different sets of gene mutations (Brennand et al., 2011). Thus, aberrant regulation of HSF1 signaling may underlie diverse genetic mutations, thereby contributing to NPC vulnerability to various environmental risk factors believed to contribute to SZ. Consistent with this hypothesis, the disturbance of this signaling cascade has been reported; human genetic studies have reported HSP70 polymorphism in SZ as well as in bipolar disorder among individuals in the Korean population (Kim et al., 2001; Pae et al., 2005). Furthermore, the production of antibodies against the HSPs, including HSP70, has been reported in never-medicated SZ patients as well as in cases of autism and bipolar disorder, and an increase in HSP70 expression has also been shown in the prefrontal cortex of SZ patients (Schwarz et al., 1999). A recent paper by Ottis et al. (2011) has also provided evidence for cell-invasive pathogenic aggregates of HSP70 with DISC1, which is one of the putative susceptible SZ genes linked to neural development (Jaaro-Peled et al., 2009). Cortical thinning, one of the structural abnormalities seen in Hsf1 deficiency plus challenge exposure, has a strong correlation with some of the attributes associated with human SZ (Kuperberg et al., 2003). We have not performed tests examining an association of behavioral traits in Hsf1 KO mice exposed to challenges. However, given that epilepsy, a common comorbidity alongside psychiatric disorders, was observed in these mice and that the observed structural anomalies are known to be associated with abnormalities of sociability, sensory hypersensitivity and anxiety, behavior is likely to be impacted. In addition, our results in the small cohort of SZ NPCs should be confirmed by using larger cohorts of SZ patients in the future.
The mechanism through which increased cellular variation in stress response contributes to SZ can only be speculated. Cell-to-cell variability at the transcription level has been well recognized in microbial cells, and, interestingly, is particularly evident in cellular stress-responsive genes (reviewed in Avery, 2006). In contrast to increased survival rates in populations of microbial cells cultivated in a harsh environment (reviewed in Avery, 2006), this rudimentary biological mechanism could cause adverse impacts on mammalian brain development, laying the groundwork for the eventual development of SZ and/or other human brain disorders. Interestingly, elevated cell-to-cell transcriptional variability in aged cardiomyocytes is a result of increased genome damage by oxidative injury (Bahar et al., 2006). The mechanisms producing higher variability of the HSF1 activation in SZ NPCs requires further investigation. Because cells and tissues derived from SZ patients exhibit increased oxidative DNA damage (Clay et al., 2011), another avenue of research may be to assess the state of oxidative DNA damage in association with cell-to-cell variability among SZ iPSCs. This work could uncover the utility of prenatal treatment with antioxidants in pregnancies with known genetic risk factors for SZ.
Experimental Procedures
Animals
Generation and genotyping of Hsf1 KO mice are described in Extended Experimental Procedures. Hsp70 null homozygotes (MMRRC) were crossed to obtain embryos. All in utero electroporation experiments were performed at E14 as described previously (Torii and Levitt, 2005). BrdU labeling for proliferation analysis was performed on the next day according to the previous study (Hashimoto-Torii et al., 2008). DNA constructs used for electroporation are listed in Extended Experimental Procedures. An empty vector was electroporated as the control. All animals were handled according to protocols approved by the Institutional Animal Care and Use Committee of the Yale University School of Medicine.
Substrate administration and the seizure susceptibility test
Details of experimental paradigms of substrate administration are described in Extended Experimental Procedures. Administration of ETOH to pregnant mice and cortical slice cultures of human fetuses were performed as described previously (Hashimoto-Torii et al., 2011). No differences in the BAC profile between wild-type and Hsf1 heterozygous pregnant mice were observed (Fig.S2A). For MeHg administration to pregnant mice, a single dose of methylmercury chloride solution (Sigma-Aldrich) at 5 mg/kg was given by intraperitoneal injection. To induce maternal seizure, PTZ (Sigma-Aldrich) was injected to pregnant mice at 40 mg/kg, and the behavior of the mice was observed for 30 minutes. To test the seizure susceptibility of the mice exposed to prenatal challenges, PTZ (5 mg/kg) was administered to P21-25 juvenile mice by intraperitoneal injection. Mice were observed every 10 minutes with increasing cumulative doses of PTZ until generalized seizures occurred. The behavioral seizure score was based on Racine’s behavioral seizure scale. In a separate series of experiments, each mouse received only a single injection at 15, 20 or 25 mg/kg and was euthanized immediately after exhibiting convulsions or at the end of the 30-minute observation period.
Mouse cortical slice culture with meningeal cells
For culturing cortical slices, we followed the procedure described by Siegenthaler et al. (2009). Briefly, pregnant HSF1 heterozygous dams were injected with ETOH at E14 and E15. Embryonic brains with intact meninges were collected 3 hours later and embedded in 3% SeaPlaque low-melt agarose (Cambrex). The brains were cut into 300-μm coronal sections using a vibratome (Leica). The slices were then cultured at 37°C, 5% CO2 for 24 hours in 3 ml Neurobasal medium (Invitrogen) supplemented with 2% B27 with vitamin A (Invitrogen), 2 mM L-glutamine and 100 μM penicillin/streptomycin on 0.40-μm filter inserts (Millipore) prior to supplementation with meningeal cells. Four days before the slice culture experiments, meningeal cultures were obtained by collecting the forebrain meninges from E15 Hsf1 KO or wild-type mice in DMEM medium containing TripLE Select (Invitrogen). Following dispersion with a glass pipette, the cells were pelleted by centrifugation, resuspended in DMEM with 10% fetal bovine serum, and then plated on collagen-coated, 6-well plates (Costar) at a density of 1.0 × 105 cells/well. The culture medium was changed to Neurobasal medium 2 days prior to the slice culture experiments.
HiPSC-derived NPCs
Five control and four SZ hiPSCs were reprogrammed from the fibroblasts of subjects. Three replicates per sample (= each cell) were analyzed. Generation and differentiation of the hiPSCs were described in Brennand et al. (2011). Briefly, the iPSCs were reprogrammed using inducible lentiviral system, and the hiPSCs were differentiacted via embryoid bodies and neural rosette formation. The pluripotency was confirmed using REX1, NANOG, CRUPTO, OCT4 and SOX2 in vitro. Three germ layers were similarly generated from every one of the hiPSC-lines. Normal katyotypes were also confirmed in every one of the control and SZ hiPSCs. The proliferation capability and neural patterning of NPCs were same among all control and SZ lines (Brennand et al., 2014).
MOCCAN and Single cell RT-ddPCR
Three hours after application of agents into NPC cultures in 24-well plates, the NPCs were scraped and dissociated into single cell suspensions. The cells were stored up to 7 days in 1 ml RNAlater at 4 degrees. Single cell separation was performed with MOCCAN (modified MOCCA for Neural cells) by principally following Lin et al. (2009). The droplets containing a single cell were screened under a DIC microscope. Cell lysis solution with DNase1 was applied directly into single-cell containing microfluid. Single cell lysis kit (Ambion) and Superscript Vilo cDNA synthesis kit (Invitrogen) were used. The Taqman gene expression assay (Invitrogen) was used with the Bio-Rad droplet digital PCR QX100.
Data analysis
All statistical data are presented as mean ± SD. Statistical tests were performed to assess the significance of the results from multiple experiments (at least n ≥ 4 for each experimental condition). For the meditational analysis, we used a SPSS macro with a resample procedure of 10,000 bootstrap samples (Preacher and Hayes, 2008). The variables used for the analysis were Hsf1 genotype, three types of challenge treatments, seizure susceptibility (PTZ concentrations that induce tonic-chronic convolution), and heterotopia formation (number of heterotopias in the cerebral cortex).
Details of other experimental procedures are available in Extended Experimental Procedures.
Supplementary Material
Highlights.
HSF1 is activated in the cortex by various types of prenatal environmental stresses.
Loss of HSF1 increases seizure susceptibility upon prenatal exposure to challenges.
Hsp70 partially mediates the pluripotent roles of HSF1.
Cell-to-cell variability of HSF1 activation is increased in schizophrenia iPSC-NPC.
Acknowledgments
We thank S. Rodriguez and M. Pappy for technical assistance and the Yale Statistical Clinic for technical advice. This work was supported by the Kavli Institute for Neuroscience at Yale (K.H-T., M.T.), R01-DA023999, R01NS014841 (P.R.), NARSAD (K.H-T., M.T., K.J.B.), CTSI-CN, K99/R00-AA018387 (K.H-T.), Institut de Recherche sur les Boissons (V.M, R.E.), R01 MH101454 and the New York Stem Cell Foundation (K.J.B.) and CIRM Grant RL1-00649-1, the G. Harold & Leila Y. Mathers Foundation, the JPB Foundation, the Leona M. and Harry B. Helmsley Charitable Trust, Annette Merle-Smith, and Robert and Mary Jane Engman (F.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algan O, Rakic P. Radiation-induced area- and lamina-specific deletion of neurons in the primate visual cortex. J Comp Neurol. 1997;381:335–352. doi: 10.1002/(sici)1096-9861(19970512)381:3<335::aid-cne6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Avery SV. Microbial cell individuality and the underlying sources of heterogeneity. Nat Rev Microbiology. 2006;4:577–587. doi: 10.1038/nrmicro1460. [DOI] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441:1011–1014. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research: Conceptual, strategic and statistical considerations. J Pers Soc Psychol. 1986;51:1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- Barros CS, Franco SJ, Muller U. Extracellular matrix: functions in the nervous system. Cold Spring Harb Perspect Biol. 2011;3:a005108. doi: 10.1101/cshperspect.a005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Neuro-archaeology: pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci. 2008;31:626–636. doi: 10.1016/j.tins.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboun-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;479:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol A, Ladran I, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry. 2014 doi: 10.1038/mp.2014.22. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevassus-au-Louis N, Baraban SC, Gaiarsa JL, Ben-Ari Y. Cortical malformations and epilepsy: new insights from animal models. Epilepsia. 1999;40:811–821. doi: 10.1111/j.1528-1157.1999.tb00786.x. [DOI] [PubMed] [Google Scholar]
- Chevassus-Au-Louis N, Jorquera I, Ben-Ari Y, Represa A. Abnormal connections in the malformed cortex of rats with prenatal treatment with methylazoxymethanol may support hyperexcitability. Dev Neurosci. 1999;21:385–392. doi: 10.1159/000017388. [DOI] [PubMed] [Google Scholar]
- Christians ES, Benjamin IJ. Heat shock response: lessons from mouse knockouts. Handb Exp Pharmacol. 2006:139–152. doi: 10.1007/3-540-29717-0_6. [DOI] [PubMed] [Google Scholar]
- Clay HB, Sillivan S, Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci. 2011;29:311–324. doi: 10.1016/j.ijdevneu.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Feo MR, Del Priore D, Mecarelli O. Prenatal cocaine: seizure susceptibility in rat offspring. Pharmacol Res. 1995;31:137–141. doi: 10.1016/1043-6618(95)80060-3. [DOI] [PubMed] [Google Scholar]
- De Riu PL, Mameli P, Becciu A, Simula ME, Mameli O. Effect of fetal hypoxia on seizure susceptibility in rats. Physiol Behav. 1995;57:315–318. doi: 10.1016/0031-9384(94)00270-f. [DOI] [PubMed] [Google Scholar]
- Dixon-Salazar TJ, Gleeson JG. Genetic regulation of human brain development: lessons from Mendelian diseases. Ann N Y Acad Sci. 2010;1214:156–167. doi: 10.1111/j.1749-6632.2010.05819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MJ. Review: Hyperthermia and fever during pregnancy. Birth Defects Res A Clin Mol Teratol. 2006;76:507–516. doi: 10.1002/bdra.20277. [DOI] [PubMed] [Google Scholar]
- Edwards MJ, Saunders RD, Shiota K. Effects of heat on embryos and foetuses. Int J Hyperthermia. 2003;19:295–324. doi: 10.1080/0265673021000039628. [DOI] [PubMed] [Google Scholar]
- Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Torii K, Kawasawa YI, Kuhn A, Rakic P. Combined transcriptome analysis of fetal human and mouse cerebral cortex exposed to alcohol. Proc Natl Acad Sci U S A. 2011;108:4212–4217. doi: 10.1073/pnas.1100903108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Torii K, Torii M, Sarkisian MR, Bartley CM, Shen J, Radtke F, Gridley T, Sestan N, Rakic P. Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron. 2008;60:273–284. doi: 10.1016/j.neuron.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25:604–615. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma S, Jin X, Wang G, Tu N, Min J, Yanasak N, Mivechi NF. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J Neurosci. 2007;27:7974–7986. doi: 10.1523/JNEUROSCI.0006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaro-Peled H, Hayashi-Takagi A, Seshadri S, Kamiya A, Brandon NJ, Sawa A. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 2009;32:485–495. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz DF, Taipale M, Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet. 2010;44:189–216. doi: 10.1146/annurev.genet.40.110405.090412. [DOI] [PubMed] [Google Scholar]
- Kakita A, Wakabayashi K, Su M, Piao YS, Takahashi H. Experimentally induced leptomeningeal glioneuronal heterotopia and underlying cortical dysplasia of the lateral limbic area in rats treated transplacentally with methylmercury. J Neuropathol Exp Neurol. 2001;60:768–777. doi: 10.1093/jnen/60.8.768. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Lee SJ, Toh KY, Lee CU, Lee C, Paik IH. Identification of antibodies to heat shock proteins 90 kDa and 70 kDa in patients with schizophrenia. Schizophr Res. 2001;52:127–135. doi: 10.1016/s0920-9964(00)00091-8. [DOI] [PubMed] [Google Scholar]
- Klein JA, Ackerman SL. Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest. 2003;111:785–793. doi: 10.1172/JCI18182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperberg GR, Broome MR, McGuire PK, David AS, Eddy M, Ozawa F, Goff D, West WC, William SC, van der Kouwe AJ, et al. Regionally localized thinning of the cerebral cortex in schizophrenia. Arch Gen Psychiatry. 2003;60:878–888. doi: 10.1001/archpsyc.60.9.878. [DOI] [PubMed] [Google Scholar]
- Lewandowski TA, Pierce CH, Pingree SD, Hong S, Faustman EM. Methylmercury distribution in the pregnant rat and embryo during early midbrain organogenesis. Teratology. 2002;66:235–241. doi: 10.1002/tera.10098. [DOI] [PubMed] [Google Scholar]
- Lidow MS. Consequences of prenatal cocaine exposure in nonhuman primates. Brain Res Dev Brain Res. 2003 Dec 30;147(1-2):23–36. doi: 10.1016/j.devbrainres.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Lin LI, Chao SH, Meldrum DR. Practical, microfabrication-free device for single-cell isolation. PLoS One. 2009;4:e6710. doi: 10.1371/journal.pone.0006710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist S. The heat-shock response. Annu Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Melkonyan HS, Ushakova TE, Umansky SR. Hsp70 gene expression in mouselung cells upon chronic gamma-irradiation. Int J Radiat Biol. 1995;68:277–280. doi: 10.1080/09553009514551201. [DOI] [PubMed] [Google Scholar]
- Miller MW. Brain Development: Normal Processes and the Effects of Alcohol and Nicotine. Oxford University Press; 2006. [Google Scholar]
- Mooney SM, Siegenthaler JA, Miller MW. Ethanol induces heterotopias in organotypic cultures of rat cerebral cortex. Cereb Cortex. 2004;14:1071–1080. doi: 10.1093/cercor/bhh066. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman AL, Crocker N, Mattson SN, Riley EP. Neuroimaging and fetal alcohol spectrum disorders. Dev Disabil Res Rev. 2009;15:209–217. doi: 10.1002/ddrr.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oghlakian RO, Tilelli CQ, Hiremath GK, Alexopoulos AV, Najm IM. Single injection of a low dose of pentylenetetrazole leads to epileptogenesis in an animal model of cortical dysplasia. Epilepsia. 2009;50:801–810. doi: 10.1111/j.1528-1167.2008.01815.x. [DOI] [PubMed] [Google Scholar]
- Ottis P, Bader V, Trossbach SV, Kretzchmar H, Leliveld SR, Korth C. Convergence of two independent mental disease genes on the protein level: recruitment of dysbindin to cell-invasive disrupted-in-schizophrenia 1 aggresomes. Biol Psychiatry. 2011;70:604–610. doi: 10.1016/j.biopsych.2011.03.027. [DOI] [PubMed] [Google Scholar]
- Pae CU, Kim TS, Kwon OJ, Artioli P, Serretti A, Lee CU, Lee SJ, Lee C, Paik IH, Kim JJ. Polymorphisms of heat shock protein 70 gene (HSPA1A, HSPA1B and HSPA1L) and schizophrenia. Neurosci Res. 2005;53:8–13. doi: 10.1016/j.neures.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Preacher KJ, Hayes AF. Asymptotic and resampling strategies for assessing and comparing indirect effects in multiple mediator models. Behav res Methods. 2008;40:879–891. doi: 10.3758/brm.40.3.879. [DOI] [PubMed] [Google Scholar]
- Radakovits R, Barros CS, Belvindrah R, Patton B, Muller U. Regulation of radial glial survival by signals from the meninges. J Neurosci. 2009;29:7694–7705. doi: 10.1523/JNEUROSCI.5537-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Defects of neuronal migration and the pathogenesis of cortical malformations. Prog Brain Res. 1988;73:15–37. doi: 10.1016/s0079-6123(08)60494-x. [DOI] [PubMed] [Google Scholar]
- Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- Russo E, Citraro R, De Fazio S, Torcasio G, De Sarro G, Di Paola ED. Effects of ethanol on the development of genetically determined epilepsies in rats. Int J Dev Neurosci. 2008;26:739–744. doi: 10.1016/j.ijdevneu.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Santos SD, Saraiva MJ. Enlarged ventricles, astrogliosis and neurodegenerationin heat shock factor 1 null mouse brain. Neuroscience. 2004;126:657–663. doi: 10.1016/j.neuroscience.2004.03.023. [DOI] [PubMed] [Google Scholar]
- Schwarz MJ, Riedel M, Gruber R, Ackenhell M, Muller R. Antibodies to heat shock proteins in schizophrenia patients: implications for the mechanism of the disease. Am J Psychiatry. 1999;156:1103–1104. doi: 10.1176/ajp.156.7.1103. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, Folias AE, Choe Y, May SR, Kume T, et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. doi: 10.1016/j.cell.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler JA, Pleasure SJ. We have got you ‘covered’: how the meninges control brain development. Curr Opin Genet Dev. 2011;21:249–255. doi: 10.1016/j.gde.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeber K, Tlsty TD, Happerfield L, Thomas GA, Romanov S, Bobrow L, Williams ED, Williams GH. DNA replication licensing and human cell proliferation. J Cell Sci. 2001;114:2027–2041. doi: 10.1242/jcs.114.11.2027. [DOI] [PubMed] [Google Scholar]
- Szasz A, Barna B, Szupera Z, De Visscher G, Galbacs Z, Kirsch-Volders M, Szente M. Chronic low-dose maternal exposure to methylmercury enhances epileptogenicity in developing rats. Int J Dev Neurosci. 1999;17:733–742. doi: 10.1016/s0736-5748(99)00041-6. [DOI] [PubMed] [Google Scholar]
- Tamasi L, Bohacs A, Tamasi V, Stenczer B, Prohaszka Z, Rigo J, Jr, Losonczy G, Molvarec A. Increased circulating heat shock protein 70 levels in pregnant asthmatics. Cell Stress Chaperones. 2010;15:295–300. doi: 10.1007/s12192-009-0143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BL, Levitt P, Stanwood GD. Prenatal exposure to drugs: effects on brain development and implications for policy and education. Nat Rev Neurosci. 2009;10:303–312. doi: 10.1038/nrn2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii M, Levitt P. Dissociation of corticothalamic and thalamocortical axon targeting by an EphA7-mediated mechanism. Neuron. 2005;48:563–575. doi: 10.1016/j.neuron.2005.09.021. [DOI] [PubMed] [Google Scholar]
- van Loo KM, Martens GJ. Genetic and environmental factors in complex neurodevelopmental disorders. Curr Genomics. 2007;8:429–444. doi: 10.2174/138920207783591717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CA, Engle EC. Allelic diversity in human developmental neurogenetics: insights into biology and disease. Neuron. 2010;68:245–253. doi: 10.1016/j.neuron.2010.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol. 1995;11:441–469. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999;18:5943–5952. doi: 10.1093/emboj/18.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.