

Abstract
RNA-guided Cas9 nucleases derived from clustered regularly interspaced short palindromic repeats (CRISPR)-Cas systems have dramatically transformed our ability to edit the genomes of diverse organisms. We believe tools and techniques based on Cas9, a single unifying factor capable of colocalizing RNA, DNA and protein, will grant unprecedented control over cellular organization, regulation and behavior. Here we describe the Cas9 targeting methodology, detail current and prospective engineering advances and suggest potential applications ranging from basic science to the clinic.
Bacteria and archaea have evolved adaptive immune defenses termed CRISPR-CRISPR–associated (Cas) systems that use short RNA to direct degradation of foreign nucleic acids1–3. Type II CRISPR-Cas systems have been engineered to effect robust RNA-guided genome modifications in multiple eukaryotic systems4–17, substantially improving the ease of genome editing and, more recently, genome regulation18–23. As an RNA-guided dsDNA-binding protein, the Cas9 effector nuclease is the first known example of a programmable unifying factor capable of colocalizing all three types of sequence-defined biological polymers, a capability with tremendous potential for engineering living systems. Here we review the Cas9 targeting methodology, outline key steps toward enhancing the efficacy, specificity and versatility of Cas9-mediated genome editing and regulation, and highlight its transformative potential for basic science, cellular engineering and therapeutics.
Engineering CRISPR-Cas systems
In bacteria and archaea, CRISPR-Cas systems provide immunity by incorporating fragments of invading phage and plasmid DNA into CRISPR loci and using the corresponding CRISPR RNAs (crRNAs) to guide the degradation of homologous sequences24. Each CRISPR locus encodes acquired ‘spacers’ that are separated by repeat sequences. Transcription of the locus yields a pre-crRNA, which is processed to yield crRNAs consisting of spacer-repeat fragments that guide effector nuclease complexes to cleave dsDNA sequences complementary to the spacer. Hence, CRISPR systems are readily retargeted by expressing or delivering appropriate crRNAs25–30.
The type II effector system3 (Fig. 1), the focus of this Perspective, is comprised of a long pre-crRNA transcribed from the spacer-repeat CRISPR locus, the multifunctional Cas9 protein and a trans-activating crRNA (tracrRNA) important for processing the pre-crRNA and formation of the Cas9 complex. Type II CRISPR interference is a multistep process31. First, tracrRNAs hybridize to repeat regions of the pre-crRNA. Second, endogenous RNase III cleaves the hybridized crRNA-tracrRNAs, and a second event removes the 5′ end of each spacer, yielding mature crRNAs that remain associated with both the tracrRNA and Cas9. Third, each mature complex locates a target dsDNA sequence and cuts both strands. Although the mechanisms underlying target search remain unknown (with no crystal structures determined to date), target recognition and subsequent cleavage by the crRNA-tracrRNA-Cas9 requires both sequence complementary between the spacer and the target ‘protospacer’ sequence as well as the presence of an appropriate protospacer-adjacent motif (PAM) sequence at the 3′ end of the proto-spacer sequence (Fig. 2a)32,33. The PAM is an essential targeting component that also serves as a self versus non-self recognition system to prevent the CRISPR locus itself from being targeted. Many type II systems have differing PAM requirements, which can limit their ease of targeting34. The most commonly engineered system thus far, that of Streptococcus pyogenes, requires a PAM with sequence NGG, where N is any nucleotide33. Type II systems may differ in the details of pre-crRNA production and crRNA-tracrRNA processing35.
Figure 1.

Functioning of the type II CRISPR-Cas systems in bacteria3. Phase 1: in the immunization phase, the CRISPR system stores the molecular signature of a previous infection by integrating fragments of invading phage or plasmid DNA into the CRISPR locus as ‘spacers’. Phase 2: in the immunity phase, the bacterium uses this stored information to defend against invading pathogens by transcribing the locus and processing the resulting transcript to produce CRISPR RNAs (crRNAs) that guide effector nucleases to locate and cleave nucleic acids complementary to the spacer. First, tracrRNAs hybridize to repeat regions of the pre-crRNA. Second, endogenous RNase III cleaves the hybridized crRNA-tracrRNA, and a second event removes the 5′ end of the spacer, yielding mature crRNAs that remain associated with the tracrRNA and Cas9. The complex cleaves complementary ‘protospacer’ sequences only if a PAM sequence is present.
Figure 2.

Cas9-sgRNA targeting complexes. (a) The basic S. pyogenes Cas9-sgRNA RNA-guided nuclease complex for eukaryotic genome engineering. Target recognition and cleavage require protospacer sequence complementary to the spacer and presence of the appropriate NGG PAM sequence at the 3′ of the protospacer. (b) Cas9 enables programmable localization of dsDNA, RNA and proteins. Proteins can be targeted to any dsDNA sequence by simply fusing them to Cas9nuclease-null, and additional RNA can be tethered to sgRNA termini without compromising Cas9 binding. Attaching RNA binding sites can in turn recruit RNA-binding proteins or directly recruit other RNAs via sequence hybridization. Finally, Cas9 can theoretically bring together any two dsDNA regions by employing sgRNA-Cas9 ‘staples’ that bind to the targeted loci and to one another. Consequently, Cas9 can in principle bring together any fusion proteins, any natural or fusion RNAs and/or any dsDNA locus to any other dsDNA sequence of interest. (c) The diverse potential applications of Cas9 range from targeted genome editing (via simplex and multiplex double-strand breaks and nicks) to targeted genome regulation (via tethering of epigenetic effector domains to either the Cas9 or sgRNA, and via competition with endogenous DNA binding factors) and possibly programmable genome reorganization and visualization. Cas9 might also be engineered to function as an RNA-guided recombinase, and via RNA tethers could serve as a scaffold for the assembly of multiprotein and nucleic acid complexes.
Implementing this system in a given organism requires appropriate reconstitution of the functional crRNA-tracrRNA-Cas9 functional unit. In bacteria, the system can be used as is28, but in the human setting this involves expression of a human-codon–optimized Cas9 protein with an appropriate nuclear localization signal, and the crRNA and tracrRNA expressed either individually or as a single chimera via a RNA polymerase III promoter5,11,15. Alternatively, in vitro– transcribed RNA can be delivered directly to the target cell types4,10. Expressing a chimeric crRNA-tracrRNA, also termed a short guide RNA (sgRNA), is the most common approach by virtue of enhanced simplicity and robust targeting, especially if the sgRNA is not truncated15. This general methodology has been used to edit genomes of numerous model eukaryotic organisms4–17.
Expanding Cas9 functionality
For all its demonstrated utility in facilitating genome editing, we suggest that the true versatility and potential of the Cas9 unifying factor is in its singular ability to bring together all three major classes of biopolymers. Proteins can be targeted to any dsDNA sequence by simply fusing them to a nuclease-null Cas9 (Cas9nuclease-null)18–23 and expressing a suitable sgRNA, whereas RNAs can be attached to sgRNA termini without compromising binding of Cas9 (ref. 21). Consequently, Cas9 can bring any fusion protein together with any fusion RNA at any dsDNA sequence by covalent attachment to Cas9nuclease-null or to sgRNAs, or by noncovalent binding to covalently attached molecules (Fig. 2b). Because so many biological elements are primarily regulated by effective concentration, a single unifying factor capable of mediating these interactions has extraordinary potential for use in investigating and engineering living systems (Fig. 2c).
For example, transcription is exquisitely dependent on the assembly of regulatory complexes and their interactions with chromatin. By targeting Cas9nuclease-null to important binding sites for putative transcription factors, it should be possible to obstruct the recruitment of these factors and thereby elucidate their role in transcription. Similarly, individual factors with unknown roles could be selectively recruited to almost any desired sequence by Cas9nuclease-null fusions or sgRNA tethers with only slightly less precision. Together, these capabilities may allow a reductionist, component-by-component approach to perturbing endogenous gene regulation.
Transcriptional activation
For engineering purposes, it is most useful to directly upregulate the transcription of endogenous genes to a desired level of activity. Experiments with zinc finger effectors and transcription activator–like (TAL) effectors demonstrated that multiple VP64 activator domains localized 5′ of the transcription start site yield synergistic effects36–39. We and others showed that Cas9-mediated localization functions similarly with both Cas9nuclease-null–VP64 (refs. 19–22) and sgRNA-tethered MS2-VP64 proteins21,40. As a caveat, the extent of activation can vary markedly among targeted genes and requires synergy between multiple Cas9-sgRNA activators for robust transcription, presumably owing to local chromatin structure, unique interactions with endogenous transcriptional machinery or the underlying Cas9 biochemistry. Elucidation of these effects as well as evaluation of additional Cas9 orthologs will be necessary to fine-tune our control over endogenous transcription.
The capability to upregulate any endogenous gene or combination of genes in trans has tremendous implications for our ability to investigate and control cellular behavior. In particular, multiplexed sgRNA libraries15 targeting every known gene could help pinpoint the factors responsible for important cellular processes such as differentiation.
Transcriptional repression
Recruitment of repressor domains by zinc finger effector or TAL effector proteins potently suppresses endogenous transcription. By using a similar architecture for Cas9nuclease-null–KRAB or related fusion proteins or sgRNA-based tethers, it should be possible to repress genes with equivalent efficacy and far greater ease of targeting. Indeed, a Cas9nuclease-null–KRAB fusion has been recently shown to induce modest repression using single guide RNAs19. Localizing additional repressors and optimizing the structure of the fusion protein could greatly increase the potency of this approach. Adding the ability to repress transcription to our tool-box will not only complement studies using transcriptional activation, but may also be useful for antiviral applications in eukaryotic cells. By preventing the transcription of invading viral genomes, Cas9 repressors could in principle render a transgenic organism immune to many DNA viruses targeted with sufficient sgRNAs, a notable advantage for both crops and domesticated animals.
Modulation of epigenetic marks
Although no attempts to engineer chromatin modifications at endogenous loci using Cas9 have been published, recruiting the appropriate effector domains should result in the desired effects. In principle, Cas9 can precisely recruit any of the major chromatin-remodeling complexes, including Swi-Snf, histone acetylases and deacetylases, methylases and demethylases, kinases and phosphatases, DNA methylases and demethylases, and others. If such approaches prove successful, these capabilities will simultaneously transform our ability to investigate the nature of epigenetic control and to engineer long-lasting expression ‘states’.
Modulation of genome architecture
Regulation may also be achieved via programmable alterations to genome architecture41,42. Cas9nuclease-null has the potential to bring together any two or more regions of the genome via multivalent sgRNAs that recruit Cas9 and also other sgRNAs. In one scheme, two sgRNAs with complementary 3′ regions target sequences in the regions to be stapled together. Each sgRNA binds to the other and to a copy of Cas9nuclease-null, which in turn binds to the specified dsDNA regions. Alternatively, a single sgRNA with multiple Cas9nuclease-null binding sites and spacers might have a similar role in bringing together two target dsDNA regions. In these scenarios, Cas9nuclease-null and sgRNAs would serve as ‘staples’ with the entire genome having the role of the ‘scaffold strand’ in a cellular-scale equivalent of DNA origami43. These genomic rearrangements might in turn be visualized by attaching fluorescent proteins to the Cas9nuclease-null–effector complex by direct fusion, sgRNA tethering or both.
Cas9-targeted recombinases
Despite the effectiveness of nuclease-based methods in editing genomes, safe in vivo gene correction in human patients remains difficult. Most notably, the introduction of a double-strand break or even a nick at the wrong off-target site can lead to unexpected mutations or rearrangements that may culminate in oncogenesis. Site-specific recombinase (and potentially transposase) enzymes present fewer problems by tightly controlling generation of double-strand breaks to coordinate donor-target coupling. By fusing the catalytic domain of a small serine recombinase44 to Cas9, analogous to previous zinc finger and TAL fusions45, it may be possible to create an RNA-guided recombinase enzyme. Because the activity of such retargeted fusion recombinases is generally low, extensive directed evolution may be necessary to produce a useful RNA-guided recombinase.
Controlling sgRNA expression and localization
Although Cas9 expression can be readily modulated, current formats of sgRNA expression rely on the use of polymerase III promoters46. These are by nature constitutive promoters, and transcribed RNAs have limited total lengths and short half-lives. Successful expression of sgRNAs using polymerase II promoters could enable coordinated and inducible control over multiple aspects of cellular behavior as well as production of multiple sgRNAs from a single transcript. Unfortunately, most polymerase II transcripts are rapidly exported to the cytoplasm. Potential methods of ensuring proper nuclear localization include removing the 5′ cap and 3′ tail with ribozymes47 or endogenous RNases, embedding the sequences in a stabilized intron48 or expressing them in a circularized format49,50. Tightly controlling the dose and duration of Cas9-sgRNA expression will also be critical for tuning targeting specificity.
Enhanced genome editing
The Streptococcus pyogenes Cas9 generates a blunt-ended double-stranded break 3 base pairs upstream of the 3′ end of the protospacer in a process mediated by two catalytic domains: an HNH nuclease domain that cleaves the strand complementary to the sgRNA and a RuvC-like nuclease domain that cleaves the non-complementary strand. If one of the two nuclease domains is inactivated, Cas9 will function as a nickase33. Both modalities have been demonstrated to induce nonhomologous end joining (NHEJ)-mediated disruption of the genome and homologous recombination (HR)-mediated modification of the genome in eukaryotic systems5,15,21. However, the intricacies of this process remain poorly understood, as exemplified by the wide range of efficiencies and specificities observed in many systems.
Determining targeting biases
Spacer sequences vary dramatically in their targeting efficiencies in both eukaryotic and prokaryotic systems, and PAM sequences also have a role in targeting21. For example, a spacer targeting the unc-119 locus yielded gene disruptions in only 1.7% of progeny in Caenorhabditis elegans, whereas a spacer targeting klp-12 reproducibly exhibited efficiencies over 77% (ref. 51). In eukaryotes, we observed the frequency of insertions versus deletions during NHEJ to vary substantially between target sites15. Similar confounding aspects were also found when working with nickase versions of Cas9. In particular, use of the nickase did not result in any detectable NHEJ at a large majority of target sites5,21,52 but induced a very high rate at certain target sequences for unknown reasons15. More generally, the extent to which underlying chromatin structure, DNA modifications or cell type–specific contributions affect Cas9-sgRNA targeting remains unknown. Rigorous quantification of all these influences is urgently needed to construct predictive models of Cas9 targeting, especially as we begin to design large sgRNA libraries for genetic screens.
Improving specificity
An increasingly recognized constraint limiting Cas9-mediated genome engineering applications concerns their specificity of targeting. The sgRNA-Cas9 complexes are in general tolerant of 1–3 mismatches in their target and occasionally more, with the actual specificity being a function of the Cas9 ortholog, the sgRNA architecture, the targeted sequence, the PAM, and also the relative dose and duration of these reagents21,52–54. Although imperfect Cas9 specificity is a major reason for concern, there are several methods of potentially improving this. Broadly, these include requiring multiple sgRNA-Cas9 complexes for activity21, reducing affinity while increasing cooperativity, establishing competition between inactive and active forms, discovering improved natural orthologs55, engineering improved variants and judiciously choosing targeting sgRNAs5,15,21,52.
Requiring cooperativity
Obligate Cas9 cooperativity, i.e., the requirement for two or more Cas9-sgRNA complexes to achieve effector function, can be achieved in numerous ways. The most straightforward option for genome-editing purposes is to employ nickase enzymes rather than nucleases21,56–58. Two offset nicking events can be used to create a double-strand break with a defined overhang rather than using a single nucleolytic event to produce a blunt cut21. Because a majority of nicks do not result in NHEJ events, only the coordinate nicks at the targeted site will initiate a genome editing event. By tailoring the overhangs generated, this approach can potentially be used to steer the genome repair machinery toward HR or NHEJ as desired. Another route to improving specificity would couple dimerization-dependent nuclease domains, such as FokI, to a nuclease-null Cas9, thereby requiring coordinate binding by two adjacent Cas9-sgRNA complexes to dimerize (otherwise weakly interacting) FokI monomers and hence effect double-strand breaks59. Finally, coupling a monomeric nuclease such as I-Tev that additionally requires a separate motif (5′ -CNNNG-3′ for I-Tev) to effect cleavage could also increase the overall specificity of the Cas9-sgRNA- nuclease complex60.
Discovering or evolving improved Cas9 proteins
It is possible that certain Cas9 orthologs might prove more specific than the Cas9 from S. pyogenes. The specificity of natural Cas9 proteins is likely determined by the evolutionary fitness cost of genome cutting by new and existing spacers. Consequently, it is unlikely that Cas9 proteins with longer PAM requirements will exhibit greater overall specificity, as the net selective pressure for accurate recognition of the combined spacer and PAM remains constant. However, Cas9 proteins from species with larger genomes may be somewhat more specific, and those that have undergone frequent horizontal gene transfer along with their CRISPR loci and consequently been selected for avoidance of multiple host genomes are likely the most specific of all.
The best Cas9 proteins identified in nature might be improved by rational design, directed evolution or ideally a combination of the two. Our ability to rationally modify the enzyme is presently constrained by the lack of a crystal structure, a deficit that must be remedied. One attractive strategy for improving specificity is to reduce the basal Cas9 affinity for DNA, which could be mitigated at target sites by employing two cooperatively binding sgRNAs with complementary 3′ overhangs that target adjacent protospacers. Alternatively, the PAM might be changed to expand the range of targetable sites or enlarged to increase specificity, although such alteration may not be accessible by rational design alone.
PAM alteration and more complex modifications might be accessible using directed evolution, including increasing the overall specificity of each Cas9 monomer. Such an experiment must be designed to select for activity at a perfectly matched protospacer and against activity at mismatched sites, preferably those identified as problematic by specificity measurement assays. Ideally, the process would result in selection against many mismatched protospacers at any one time, and the process would be repeated over many rounds of selection, rendering automated methods of directed evolution particularly well-suited to this challenge61.
Target-site selection
Judiciously choosing the targeting sgRNAs themselves will also be critical to achieving highly specific modifications. Several studies demonstrated that the ‘seed’ region consisting of the 10–12 bases closer to the 3′ end of the spacer provides much of the specificity in targeting, so choosing sites predicted to have the fewest off-target sequences vis-a-vis the seed region is important30,33,62, although bases outside the seed region also contribute to specificity21,52. Moreover, we and others observed dramatic differences in the extent to which different spacers tolerate mismatches, suggesting that multiple candidate spacers should be empirically tested for applications that require great specificity. Ideally, these differences would be predicted computationally, but additional experiments are needed to elucidate the rules governing spacer-dependent specificity21,52–54.
Multiplexing Cas9-mediated activities
Unlike the case with previous methods for sequence-specific DNA targeting, the simplicity of sgRNA design readily permits multiplexed localization. One can target multiple genes simultaneously and also harness synergies between multiple activators or repressors. In principle, array-based oligonucleotide synthesis15,63 could be used to produce nearly 106 designed sgRNAs at once, a library capable of targeting every gene in the human genome 20 times.
Careful multistep methods for creating libraries could allow each synthesized oligonucleotide to encode multiple sgRNAs for synergistic regulation. For example, a 2-sgRNA library would involve cloning each oligonucleotide into a vector containing flanking promoters and adding 3′ sgRNA tails in the oligonucleotide sequence in a second insertion step. Combined with appropriate screening methodologies, these library-based approaches could identify individual genes or even combinations that regulate a variety of phenotypes in eukaryotic organisms and cells (Fig. 3). Coupled with tools that enable multiplexed monitoring of the resulting changes64–66, this multiplexing ability of the sgRNA-Cas9 system15 will have potentially far-ranging implications for our ability to understand and control the factors governing cellular differentiation and behavior.
Figure 3.

Platform for multiplex biological screens. To modulate multiple genomic sites, sgRNA libraries can be generated and delivered into target cells that also express orthogonal Cas9 effectors (nucleases, activators and/or repressors). This format enables multiplex ex vivo and in vivo genetic screens via targeted genome editing and/ or regulation.
Given the abundance of potential Cas9-mediated functions, it will be necessary to develop methods for independent targeting such that each function exclusively responds to its own set of guide RNAs. By carefully choosing and characterizing Cas9 orthologs with widely disparate repeat sequences, it is possible to identify fully orthogonal sets of sgRNA-Cas9 pairings and hence to simultaneously execute multiple functionalities by engineering each ortholog with a custom effector domain67. For example, three orthogonal Cas9 proteins would allow activation, repression and editing to be performed simultaneously at independent target sites in the same cell.
Toward Cas9 therapeutics
Given the tremendous utility of Cas9 for the regulation and modification of complex biological systems, might the Cas9 system prove equally useful as a basis for new therapeutics? We envision two primary routes to Cas9-mediated therapeutic interventions (Fig. 4). The first entails targeted genome editing to correct genetic disorders68–70 and possibly to disrupt invading viral genomes. The second will use Cas9nuclease-null fusions for targeted genome regulation in a manner akin to the use of small-molecule drugs, except that both repression and activation modalities would be available. One can imagine using such an approach to correct epi-genetic misregulation of gene expression, to control inflammation and autoimmunity, and also to repress transcription of viral genes or even viral co-receptors in vulnerable cell types.
Figure 4.
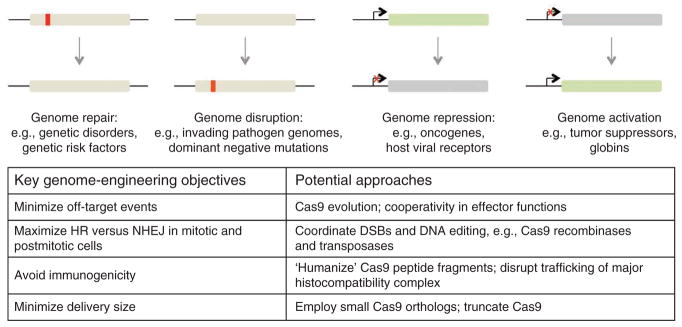
Cas9 therapeutics. Potential Cas9-mediated therapeutic approaches include targeted genome editing to correct genetic disorders and targeted genome regulation to modify endogenous protein levels (top). Technical hurdles and potential routes to achieve these objectives are listed (bottom).
However, multiple technical hurdles must be addressed before Cas9-based therapeutics become a reality. First, Cas9-encoding cassettes must be efficiently delivered to target cells in vivo. Unfortunately, Cas9 proteins are quite large; the commonly used Cas9 protein from S. pyogenes is 1,368 amino acids in length. Employing smaller Cas9 orthologs, several of which are <1,100 amino acids or engineering a minimal Cas9 by removing domains extraneous to the intended purpose could enable efficient packaging into limited-capacity viral vectors (adeno-associated viruses, adenoviral vectors and lentiviruses)71–74 for direct in vivo delivery. The development of tightly regulated expression vectors or modalities that enable both transient and controlled release of targeting reagents will be critical to restricting the resulting Cas9-mediated functions to specific tissues.
Apart from the problem of gene delivery, the foremost obstacle to therapeutic applications is the comparatively poor specificity of Cas9 binding. Because off-target activity results in a risk of oncogenesis and even highly specific methodologies will inevitably lead to disastrous outcomes when applied to sufficient numbers of cells, addressing this problem is of the utmost importance. We have outlined several prospective approaches, most notably requiring cooperativity through offset nicking and biasing repair outcomes toward HR versus NHEJ. Co-localizing target and donor DNA via direct guide RNA tethering or Cas9 recombinases and transposases could also be highly useful for this endeavor, especially in targeting post-mitotic cells where function of endogenous HR machinery is expected to be highly diminished.
Avoiding an adverse immune response is also critical. Classical immune suppression for the duration of treatment is one option for mitigating the immune response, but this strategy will be less useful for long-term regulatory modifications. A more promising approach would involve analyzing the immunogenicity of Cas9 and ‘humanizing’ the relevant peptide fragments as has been accomplished for antibody therapeutics75. Finally, the regulatory control afforded by Cas9 may permit the mimicry of strategies used by viruses such as disrupting the major histocompatibility complex trafficking machinery76.
Fully realizing the therapeutic potential of Cas9 proteins will require the development of additional synergistic technologies, most notably efficient, targeted and safe in vivo gene-delivery vehicles. If ex vivo genome targeting proves effective (for instance, in hematopoietic stem cells), then tools that enable extremely rapid retrieval and screening of modified cells in a population will be critical.
Looking forward, the versatility and ease of use afforded by Cas9 coupled with its singular ability to bring together RNA, DNA and protein in a fully programmable fashion will form the basis of a powerful toolset for the perturbation, regulation and monitoring of complex biological systems.
Acknowledgments
We thank J. Aach and L. Yang for insightful discussions, and W.L. Chew for insightful discussions and critical reading of the manuscript. This work was supported by US National Institutes of Health NHGRI grant P50 HG005550, Department of Energy grant DE-FG02-02ER63445 and the Wyss Institute for Biologically Inspired Engineering (K.M.E.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 2.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 3.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 4.Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 5.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. This work is one of the first demonstrations of engineering the type II CRISPR-Cas system to function in eukaryotes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiCarlo JE, et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013;41:4336–4343. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Q, et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedland AE, et al. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10:741–743. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gratz SJ, et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–1035. doi: 10.1534/genetics.113.152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang WY, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jinek M, et al. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D, et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013;31:681–683. doi: 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- 13.Li JF, et al. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol. 2013;31:688–691. doi: 10.1038/nbt.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, Teng F, Li T, Zhou Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol. 2013;31:684–686. doi: 10.1038/nbt.2652. [DOI] [PubMed] [Google Scholar]
- 15.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. This work is one of the first demonstrations of engineering the type II CRISPR-Cas system to function in eukaryotes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nekrasov V, Staskawicz B, Weigel D, Jones JD, Kamoun S. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:691–693. doi: 10.1038/nbt.2655. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maeder ML, et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013 Jul 25; doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013 Aug 1; doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez-Pinera P, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013 Jul 25; doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bikard D, et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013 Jun 12; doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14:321–327. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. This paper describes deciphering the function of the CRISPR locus. [DOI] [PubMed] [Google Scholar]
- 26.Brouns SJ, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. This work is a demonstration of role of guide RNAs in CRISPR targeting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hale CR, et al. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell. 2012;45:292–302. doi: 10.1016/j.molcel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–571. doi: 10.1038/nature08703. This work is a demonstration of how CRISPR-Cas systems distinguish self versus non-self. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semenova E, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci USA. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. This is a report of the discovery and demonstration of tracrRNA function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. This paper reported the characterization of the Cas9-crRNA-tracrRNA complex in vitro; it also demonstrated that the complex was sufficient for DNA targeting, and showed that Cas9 could be guided using single chimeric guide RNAs (sgRNAs) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horvath P, et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 2008;190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, et al. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell. 2013;50:488–503. doi: 10.1016/j.molcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beerli RR, Dreier B, Barbas CF., III Positive and negative regulation of endogenous genes by designed transcription factors. Proc Natl Acad Sci USA. 2000;97:1495–1500. doi: 10.1073/pnas.040552697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang F, et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol. 2011;29:149–153. doi: 10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maeder ML, et al. Robust, synergistic regulation of human gene expression using TALE activators. Nat Methods. 2013;10:243–245. doi: 10.1038/nmeth.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez-Pinera P, et al. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods. 2013;10:239–242. doi: 10.1038/nmeth.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fusco D, et al. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr Biol. 2003;13:161–167. doi: 10.1016/s0960-9822(02)01436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanctot C, Cheutin T, Cremer M, Cavalli G, Cremer T. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet. 2007;8:104–115. doi: 10.1038/nrg2041. [DOI] [PubMed] [Google Scholar]
- 42.Deng W, et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–1244. doi: 10.1016/j.cell.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothemund PW. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 44.Groth AC, Calos MP. Phage integrases: biology and applications. J Mol Biol. 2004;335:667–678. doi: 10.1016/j.jmb.2003.09.082. [DOI] [PubMed] [Google Scholar]
- 45.Gaj T, Mercer AC, Sirk SJ, Smith HL, Barbas CF. 3rd A comprehensive approach to zinc-finger recombinase customization enables genomic targeting in human cells. Nucleic Acids Res. 2013;41:3937–3946. doi: 10.1093/nar/gkt071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 47.Kruger K, et al. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 48.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 49.Memczak S, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 50.Hansen TB, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 51.Friedland AE, et al. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10:741–743. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013 Jul 21; doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu Y, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013 Jun 23; doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pattanayak V, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013 Aug 11; doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Makarova KS, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim E, et al. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res. 2012;22:1327–1333. doi: 10.1101/gr.138792.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramirez CL, et al. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res. 2012;40:5560–5568. doi: 10.1093/nar/gks179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, et al. Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res. 2012;22:1316–1326. doi: 10.1101/gr.122879.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kleinstiver BP, et al. Monomeric site-specific nucleases for genome editing. Proc Natl Acad Sci USA. 2012;109:8061–8066. doi: 10.1073/pnas.1117984109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wiedenheft B, et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc Natl Acad Sci USA. 2011;108:10092–10097. doi: 10.1073/pnas.1102716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kosuri S, et al. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nat Biotechnol. 2010;28:1295–1299. doi: 10.1038/nbt.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Livet J, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- 66.Mali P, et al. Barcoding cells using cell-surface programmable DNA-binding domains. Nat Methods. 2013;10:403–406. doi: 10.1038/nmeth.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Esvelt KM, et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. doi: 10.1038/nmeth.2681. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 69.Lombardo A, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 70.Li H, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verma IM, Somia N. Gene therapy–promises, problems and prospects. Nature. 1997;389:239–242. doi: 10.1038/38410. [DOI] [PubMed] [Google Scholar]
- 72.Grieger JC, Samulski RJ. Adeno-associated virus as a gene therapy vector: vector development, production and clinical applications. Adv Biochem Eng Biotechnol. 2005;99:119–145. [PubMed] [Google Scholar]
- 73.Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci USA. 1997;94:10319–10323. doi: 10.1073/pnas.94.19.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yeh P, Perricaudet M. Advances in adenoviral vectors: from genetic engineering to their biology. FASEB J. 1997;11:615–623. doi: 10.1096/fasebj.11.8.9240963. [DOI] [PubMed] [Google Scholar]
- 75.Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature. 1988;332:323–327. doi: 10.1038/332323a0. [DOI] [PubMed] [Google Scholar]
- 76.Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science. 2013;340:701–706. doi: 10.1126/science.1233028. [DOI] [PMC free article] [PubMed] [Google Scholar]