

Sexual reproduction is the prerogative of eukaryotic species and involves the fusion of haploid gametes to form a diploid cell that subsequently undergoes meiosis to generate recombinant haploid forms. This process has been extensively studied in the unicellular yeast Saccharomyces cerevisiae, which exhibits separate regulatory control over mating and meiosis. Here we address the mechanism of sexual reproduction in the related hemiascomycete species Candida lusitaniae. We demonstrate that in contrast to S. cerevisiae, C. lusitaniae exhibits a highly integrated sexual program in which the programs regulating mating and meiosis have fused. Profiling of the C. lusitaniae sexual cycle revealed that gene expression patterns during mating and meiosis were overlapping, suggestive of co-regulation. This was particularly evident for genes involved in pheromone MAPK signaling, which were highly induced throughout the sexual cycle of C. lusitaniae. Furthermore, genetic analysis showed that the ortholog of IME2, a ‘diploid-specific’ factor in S. cerevisiae 3,4, and STE12, the master regulator of S. cerevisiae mating5,6, were each required for progression through both mating and meiosis in C. lusitaniae. Together, our results establish that sexual reproduction has undergone significant rewiring between S. cerevisiae and C. lusitaniae, and that a concerted sexual cycle operates in C. lusitaniae that is more reminiscent of the distantly related ascomycete, Schizosaccharomyces pombe. We discuss these results in light of the evolution of sexual reproduction in yeast, and propose that regulatory coupling of mating and meiosis has evolved multiple times as an adaptation to promote the haploid lifestyle.
Sexual reproduction is divided into two programs: gamete fusion, in which haploid cells merge to form a diploid cell, and gametogenesis, in which a specialized cell division, meiosis, generates recombinant haploid gametes. Sexual reproduction in S. cerevisiae involves mating between cells of opposite mating type, generating diploid products7 that subsequently undergo meiosis upon nutritional limitation1,8. It is unclear, however, how representative this sexual cycle is of that in other yeast species. Here, we examine regulation of sexual reproduction in the related hemiascomycete Candida lusitaniae, a member of the Candida clade of human pathogens9. C. lusitaniae was recently shown to have a complete sexual cycle despite lacking orthologs of key meiosis genes such as IME1, encoding the master transcriptional regulator of meiosis in S. cerevisiae9-11. In addition, whereas S. cerevisiae is predominantly diploid, C. lusitaniae cells preferentially exist in the haploid form and therefore exhibit a transient diploid state9,10.
Transcriptional profiling of C. lusitaniae cells progressing through mating and meiotic programs was performed and compared to those of S. cerevisiae. In total, 406 genes were induced >4-fold during C. lusitaniae mating, including highly conserved MAPK genes that regulate pheromone signaling in diverse fungal species2,12,13 (Fig. 1a and Extended Data Fig. 1). Interestingly, we also observed elevated expression of several genes that are orthologs of ‘meiosis-specific’ genes in S. cerevisiae, including SPO11, REC8, and IME23,14,15,16 (Fig. 1a). We similarly performed expression profiling on C. lusitaniae diploid a/α cells induced to enter meiosis. Cells became enlarged by 8 h, exhibited a morphological change by 12 h, and by 18-36 h dyad (2-cell) spores were formed, as is typical of this species10 (Extended Data Fig. 2a-c). Profiling revealed that a total of 618 genes were induced during C. lusitaniae meiosis, compared to 480 genes during S. cerevisiae meiosis17. In S. cerevisiae, meiotic gene expression includes early, middle, and late stages of expression17. We were similarly able to distinguish three temporal classes of meiotic gene expression in C. lusitaniae (Fig. 1b and d, and Extended Data Fig. 2d). In total, we observed increased expression (>3-fold) of 255 early genes, 307 middle genes, and 56 late genes during C. lusitaniae meiosis (Fig. 1e). Thus, despite lacking an ortholog of IME1, many downstream components of meiosis are regulated in a similar stage-specific manner in C. lusitaniae as in S. cerevisiae.
Figure 1.

Transcriptional profiling of mating and meiosis in C. lusitaniae. a, Profiling of C. lusitaniae mating. Top panel, induction of C. lusitaniae pheromone-signaling genes. Bottom panel, induction of genes characteristic of the meiotic program in S. cerevisiae. b, Stage-specific meiotic gene expression in S. cerevisiae and C. lusitaniae. Early, Middle, and Late meiosis genes in C. lusitaniae are defined as those >3 fold induced after 12, 18, and 24 h, respectively. c, Expression of MAPK pathway genes (blue text) and pheromone-processing genes (red text) during C. lusitaniae mating and meiosis. Expression changes for S. cerevisiae genes30 were enhanced 3-fold to be comparable to expression changes in C. lusitaniae genes. d, Average fold induction of early, middle, and late meiosis genes in C. lusitaniae. e, Comparison of meiosis gene expression changes in C. lusitaniae and S. cerevisiae. f, Fold induction of MAPK pathway genes during meiosis in C. lusitaniae and S. cerevisiae.
Extended Data Figure 1. Schematic showing induction of (a) pheromone-processing genes and (b) pheromone MAPK genes during both mating and meiosis in C. lusitaniae.

Expression changes highlight genes induced during co-incubation of a and α cells on PDA medium (mating), as well as during growth of diploid a/α cells on PDA medium (induces meiosis and sporulation). In contrast, these genes are mating-specific in the related yeast S. cerevisiae.
Extended Data Figure 2. Temporal analysis of meiosis in C. lusitaniae.

a, C. lusitaniae a/α cells divide as stable diploid cells on YPD medium. n=3. b, On PDA medium, a/α cells are induced to enter meiosis and dyad spores (asterisks) begin to appear at 18 h. n=3. c, Time course of meiosis in wildtype, ste12Δ/ste12Δ, and STE12 complemented C. lusitaniae strains. A morphology change (polarized growth) is evident in wildtype diploid strains grown on PDA medium starting at 12 h, while spore formation is apparent at 18-36 h. In the ste12Δ/ste12Δ mutant, both sporulation and morphology change are absent. d, Global transcriptional profile of meiosis in C. lusitaniae showing induced genes (yellow) and repressed genes (blue). C. lusitaniae diploid a/α cells (RSY432) were grown on PDA (sporulating) medium and expression changes compared to those on YPD (non-sporulating) medium. Genes changing more than 4-fold in expression are shown.
The most striking aspect of C. lusitaniae meiosis was the expression of many genes whose orthologs are expressed specifically during mating in S. cerevisiae. In particular, multiple components of the pheromone MAPK signaling cascade were highly induced, including the terminal transcription factor STE12 (Fig. 1c and f). In addition to MAPK genes, pheromone, pheromone receptor, and pheromone processing genes were also induced in C. lusitaniae meiosis (Extended Data Fig. 1). We therefore surmised that MAPK signaling might play a role in regulating C. lusitaniae meiosis, in addition to its conserved function in directing cell-cell communication and conjugation.
Given that program-specific expression of many S. cerevisiae mating and meiosis genes does not occur in C. lusitaniae orthologs, genetic experiments were performed to determine whether this transcriptional rewiring has functional consequences for the sexual cycle. First, the role of C. lusitaniae Ime2 in mating and meiosis was analyzed. S. cerevisiae Ime2 (Inducer of meiosis 2) is a conserved serine/threonine kinase that acts in tandem with the cyclin-dependent kinase Cdk1 to promote meiosis3,4,18,19. As predicted based on IME2 function in S. cerevisiae, loss of IME2 (CLUG_00015) in C. lusitaniae blocked meiosis; wildtype diploid cells formed haploid progeny with ~40% efficiency after 3 days, whereas <1% of haploids were formed by ime2Δ/ime2Δ cells (Fig. 2a). In addition, whereas wildtype diploids generated dyad spores, ime2Δ/ime2Δ mutants failed to sporulate (Fig. 2b). These results establish a conserved role for IME2 in regulating meiosis in hemiascomycete yeast, even in species for which IME1 is absent. Profiling of C. lusitaniae ime2Δ/ime2Δ mutants revealed that most early meiosis genes were still induced in this background, while induction of many middle and late meiosis genes was lost (Extended Data Fig. 3). We also note that induction of NDT80 (CLUG_05634) and genes encoding cell cycle regulators was compromised in the ime2Δ/ime2Δ mutant (Extended Data Fig. 3b). NDT80 is responsible for the induction of middle meiosis genes in S. cerevisiae, where its expression is dependent on Ime220. The diminished expression of NDT80, together with the loss of expression of cell cycle genes, is likely responsible for the inability of C. lusitaniae ime2Δ/ime2Δ cells to proceed through meiosis.
Figure 2.

IME2 is required for both mating and meiosis in C. lusitaniae. a, Deletion of IME2 blocks meiosis in C. lusitaniae (5-day time course, D1-D5). # represents p < 0.01, one way ANOVA, n=3. b , Wildtype diploid cells generate dyad spores (asterisks) after 3 days on PDA medium, whereas ime2Δ/ime2Δ diploids do not sporulate, n=3. c, RT-PCR data demonstrates induction of IME2 during C. lusitaniae mating, n=3. d, Loss of IME2 in both a and α cells leads to decreased mating (4 day time course, D1-D4). “#” represents P < 0.05, Kruskal Wallis, n=4. e, ime2Δ mutants generate mating zygotes, indicating the block to mating occurs post-cell fusion, n=4. Scale bars, 5 μm. Data represented as mean +/- s.e.m.
Extended Data Figure 3. Transcriptional profiling of C. lusitaniae wildtype and ime2Δ mutants during mating and meiosis.

a, Full profile of wildtype and ime2Δ/ime2Δ strains during meiosis indicates that many transcriptional changes occur in ime2Δ/ime2Δ mutants as in wildtype strains. b, Analysis of cell cycle regulating genes during C. lusitaniae meiosis. Several cell cycle genes are induced in wildtype cells undergoing meiosis but not in ime2Δ/ime2Δ mutants (e.g., APC4, CDC3, CDC10, and CDC14). c, Comparative expression of early (IME2, IME4, SPO11), middle (REC102, CDC3, SPS4, SPS1, NDT80, SWM1) and late (DIT1, DIT2) meiosis genes. d, In contrast to the meiotic profiles, the mating profiles of wildtype and ime2Δ strains were very similar. e, Comparison of meiosis genes induced in wildtype and ime2Δ mutants.
We next addressed the role of IME2 in C. lusitaniae mating. In contrast to S. cerevisiae where IME2 has no role in mating, C. lusitaniae IME2 was induced during mating (Fig. 2c) and was required for the efficient formation of mating products. Thus, co-incubation of C. lusitaniae a and α cells resulted in approximately 0.5% of cells forming a/α diploids, while bilateral crosses between ime2Δ a and ime2Δ α cells resulted in a mating frequency of less than 0.03% (a 16-fold decrease, Fig. 2d). Unilateral crosses between wildtype and ime2Δ mutants showed mating frequencies similar to those between wildtype partners (Fig. 2d). Complementation by reintegration of the IME2 gene into the mutant background restored mating competency in the bilateral cross (Extended Data Fig. 4). These results establish a novel role for IME2 in C. lusitaniae mating, and indicate that the ime2Δ defect is limited to bilateral crosses. The latter observation is consistent with a late role for Ime2 during mating following the fusion of a and α partner cells. Microscopic analysis of ime2Δ crosses showed the formation of zygote structures further supporting a late role for Ime2 during mating (Fig. 2e), and additional studies will be required address the precise role of Ime2 in this process.
Extended Data Figure 4. Reintegration of IME2 restores mating efficiency.

IME2 was reintegrated at the endogenous locus in an ime2Δ mutant (RSY437). Mating frequency was quantified by monitoring the formation of prototrophic products from auxotrophic parents. WT cross, RSY411 x RSY284, ime2Δ mutant cross, RSY406 x RSY437, IME2 complemented cross, RSY406 x CAY5022. Differences between the WT cross and the ime2Δ mutant cross, and between the IME2 complemented cross and the ime2Δ mutant cross were both significant. (n=7, p < 0.001, Kruskal Wallis test). Data are representative of mean +/- s.e.m.
Next, we investigated the role of pheromone MAPK signaling and the transcription factor Ste12 in the C. lusitaniae sexual cycle. First, we tested whether C. lusitaniae cells undergoing meiosis actively secrete pheromone. A pheromone reporter assay was developed in which the C. lusitaniae α pheromone receptor was heterologously expressed in the related species Candida albicans (Fig. 3a). Such heterologous expression of receptors has been shown to result in successful signaling in response to species-specific pheromones21. Co-incubation of the GFP-labeled reporter strain with C. lusitaniae cells undergoing meiosis showed induction of a morphological response in the reporter cells (Fig. 3b), demonstrating that C. lusitaniae cells are actively secreting α pheromone during meiosis.
Figure 3.

STE12 is essential for both mating and meiosis in C. lusitaniae. a, Schematic of assay to detect pheromone secretion from C. lusitaniae cells. C. lusitaniae a/α cells were co-incubated with GFP-labeled C. albicans a cells expressing the C. lusitaniae α pheromone receptor. b, C. albicans cells generate polarized mating projections (arrows) in response to C. lusitaniae pheromone, demonstrating that C. lusitaniae cells actively secrete pheromone during meiosis, n=2. c, Deletion of STE12 abolishes mating in C. lusitaniae (4 day time course, D1-D4). “#” represents P < 0.05, Kruskal Wallis, n=3. d, Mating occurs between wildtype C. lusitaniae cells (arrow) but not between ste12Δ mutant cells, n=3. e, Loss of STE12 inhibited the formation of meiotic haploid progeny (4 day time course, D1-D4). “#” represents P < 0.05, unequal variance t test, n=3. f, Absence of meiotic spores in C. lusitaniae cells lacking STE12, n=5. g, Gene expression changes when C. lusitaniae haploid, diploid, or ste12Δ/ste12Δ strains are incubated on PDA medium. Deletion of STE12 abolished most meiosis-specific gene expression changes. Where appropriate, scale bars, 5 μm; data represented as mean +/- s.em.
STE12 is the master regulator of mating and pheromone signaling in S. cerevisiae and many other hemiascomycetes, including Candida species2,12. To test the role of STE12 in the sexual cycle of C. lusitaniae we constructed haploid and diploid ste12Δ mutants. Loss of STE12 (CLUG_02576) in haploid cells led to a complete block in zygote formation and mating between a and α cells (Fig. 3c, 3d), as expected based on previous studies22. Surprisingly, however, loss of STE12 in C. lusitaniae a/α diploids also led to a block lock in meiosis and sporulation. Thus, formation of meiotic progeny was reduced ~100-fold in ste12Δ/ste12Δ mutants (Fig. 3e) and spore formation was abolished (Fig. 3f and Extended Data Fig. 2c). Reintegration of the STE12 gene into the ste12Δ/ste12Δ background restored the ability to undergo meiosis (Extended Data Fig. 5). Expression profiling revealed that the meiotic transcriptional profile was essentially absent from ste12Δ/ste12Δ mutants (Fig. 3g). Our results therefore establish that STE12 plays a critical role in regulating both mating and meiosis in C. lusitaniae, in contrast to S. cerevisiae where STE12 specifically regulates mating only.
Extended Data Figure 5. Schematic of genetically marked C. lusitaniae strains and control of meiosis by STE12.

a, C. lusitaniae mating experiments were performed between RSY284 (a strain) and RSY411 (α strain) and mating products selected based on auxotrophic makers. b, The a/α diploid strain RSY432 was induced to undergo meiosis on PDA medium and meiotic progeny identified by their red color on YPD medium (ade colonies) or by growth on medium containing cycloheximide (CHX resistant colonies). c, Diploid a/α strains of C. lusitaniae were incubated on PDA medium for 3 days and analyzed for the frequency of formation of meiotic progeny. Two independent ste12Δ mutants were constructed in the a/α background and tested for meiotic progeny using both the CHXR and ADE2 markers. Mutants lacking STE12 were unable to undergo meiosis to produce haploid progeny, while reintegration of STE12 into the mutant background restored meiosis. Differences between both wildtype strains and ste12Δ mutants, and between ste12Δ mutants and STE12-complemented strains were significant (n=3, p < 0.05, student's t test, two tailed). Data representative of mean +/- s.e.m.
Taken together, our studies demonstrate that a fundamental rewiring of the sexual cycle has occurred between the two hemiascomycetes, S. cerevisiae and C. lusitaniae. Mating and meiosis are distinct programs in S. cerevisiae, yet these programs are integrated in C. lusitaniae (Fig. 4a and Extended Data Fig. 7). This is evident both from transcriptional profiles that reveal overlap of gene expression patterns in mating and meiosis, as well as genetic analysis of key regulators of the sexual cycle. In particular, whereas STE12 and IME2 are necessary for S. cerevisiae mating and meiosis, respectively, the C. lusitaniae orthologs are required for efficient progress through both stages of the sexual cycle.
Figure 4.

Sexual regulation in yeast. a, Model comparing the sexual lifecycles of S. cerevisiae, C. lusitaniae, and S. pombe. The diploid species S. cerevisiae exhibits distinct regulation of mating and meiosis; MAPK signaling via Ste12 regulates mating, while Ime1 regulates meiosis. In contrast, S. pombe exhibits integrated control of its sexual cycle; MAPK signaling and Ste11 regulate entry into both mating and meiosis. C. lusitaniae exhibits similar control over its sexual cycle to that in S. pombe. In particular, C. lusitaniae mating and meiosis are highly coupled, and both are regulated by the transcription factor Ste12. b, C. lusitaniae is the only hemiascomycete tested that requires STE12 to undergo meiosis. Loss of the MTLα2 transcription factor specifically in the C. lusitaniae lineage could have been a key step during evolution of this regulatory control, as it would permit expression of ‘haploid-specific genes’ (including MAPK genes) in diploid cells.
Extended Data Figure 7. Rewiring of the genetic programs that control sexual reproduction in hemiascomycetes.
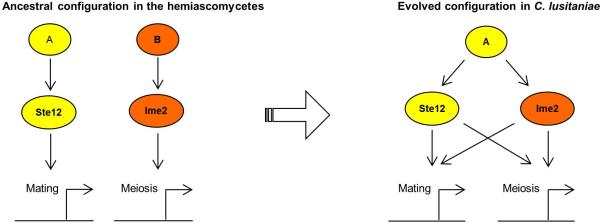
In many hemiascomycete species, including the model yeast S. cerevisiae, mating and meiosis are controlled by two distinct transcriptional programs with STE12 as the master regulator of mating and IME2 as a key regulator of meiosis. However, in the opportunistic pathogen C. lusitaniae, STE12 has retained its role in regulating mating, but has also acquired control over meiosis. Similarly, C. lusitaniae IME2 has a conserved role in regulating mating, but also has a role in promoting mating. The transcriptional programs controlling these two processes have therefore fused in C. lusitaniae, perhaps to facilitate a transient diploid state and the more efficient return to the predominant haploid state.
The regulation of sexual reproduction in C. lusitaniae exhibits striking parallels to that in the distantly related ascomycete S. pombe, even though these lineages diverged from one another more than 330 million years ago23. In S. pombe, mating and meiosis are also tightly coupled and both are dependent on pheromone MAPK signaling and the downstream transcriptional regulator, Ste1124,25. In C. lusitaniae, we show that the pheromone MAPK-associated transcription factor STE12 is similarly essential for both mating and meiosis. Thus, in both S. pombe and C. lusitaniae the programs regulating mating and meiosis are fused (Fig. 4a).
To address if Ste12 also regulates meiosis in other hemiascomycete species, we deleted the STE12 ortholog from related sexual species including S. cerevisiae, Kluyveromyces lactis, Pichia pastoris, and Yarrowia lipolytica (Extended Data Fig. 6). In each species loss of STE12 failed to block meiosis, suggesting that the meiotic function of STE12 evolved relatively recently in the C. lusitaniae lineage (Fig. 4b). In support of this hypothesis we note that C. lusitaniae, but not the other hemiascomycete species, lost the transcriptional regulator α2 during evolution9,10. The α2 gene acts to prevent expression of haploid-specific genes (including MAPK genes) in diploid a/α cells in diverse yeast species26,27. Thus, as previously hypothesized9,10, the loss of α2 could have preceded the rewiring of meiotic control in C. lusitaniae. In particular, we propose that it facilitated MAPK signaling and enabled Ste12 to assume control of meiosis in this species.
Extended Data Figure 6. Analysis of STE12 function in mating and meiosis in diverse hemiascomycete species.

The STE12 gene was deleted from haploid and diploid strains of K. lactis, P. pastoris, and S. cerevisiae. The resultant strains were tested both for mating competency and the formation of meiotic progeny. Whereas deletion of STE12 blocked mating in haploid strains of all three species, loss of STE12 from diploid a/α strains did not have a significant effect on the formation of meiotic progeny. Thus, C. lusitaniae is unique among the hemiascomycete species tested in that STE12 is essential for meiosis only in this species. K. lactis mating and meiosis, n=2, data combined for 3 independent mutants. P. pastoris mating, n=3. P. pastoris meiosis n=4. S. cerevisiae meiosis n=3. All data presented as mean +/- s.e.m.
Why have such distinct modes of sexual regulation evolved in diverse unicellular yeast? The most parsimonious explanation is that these differences reflect a species’ preference for one ploidy state over another. Both C. lusitaniae and S. pombe, despite being highly divergent, exhibit haploid lifestyles that contrast sharply with the predominantly diploid lifestyle of S. cerevisiae. Co-regulation of the transcriptional programs controlling mating and meiosis therefore ensures that cells, once mated, immediately enter meiosis and return to the haploid state. This could represent an adaptive advantage for haploid cells in both C. lusitaniae and S. pombe. Alternatively, the selective pressure to keep these two sexual programs separate may have been lost during evolution, leading to the amalgamation of these programs in the two species. While the relative advantages of haploidy and diploidy continue to be debated28,29, it is clear that distinct transcriptional circuits can evolve to promote either haploid or diploid lifestyles. That this mode of regulation evolved independently in S. pombe and C. lusitaniae further suggests that examples of fused sexual cycles will be found throughout the fungal tree of life.
Methods
Media and Reagents
Standard laboratory media were prepared as previously described31. Potato dextrose agar (PDA) was prepared as a 10% dilution of standard PDA medium (Becton Dickinson) that was solidified using 1.45% Bacto Agar (Becton Dickinson)32. Nourseothricin-resistant transformants were selected on YPD (yeast peptone dextrose) medium containing 200 μg/ml nourseothricin, as previously described33. Kanamycin-resistant transformants were selected on YPD medium supplemented with 50 ug/ml kanamycin. Liquid yeast peptone sorbitol (YPS) was prepared similar to YPD, however 2% sorbitol was used in the place of dextrose. Haploid progeny from sporulation assays were selected on YPD medium supplemented with 10 μg/ml cycloheximide10. SC-Maltose consisted of synthetic complete medium supplemented with 2% maltose. Malt medium (2% malt extract, 3% agar) was used to induce mating and meiosis in K. lactis. Pichia pastoris mating/sporulation medium consisted of sodium acetate medium (0.5% sodium acetate, 1% KCl, 1% glucose, 2% agar34). Yarrowia lipolytica sporulation was achieved using V8 sporulation medium (40% V8 juice, 0.8% yeast extract, 1.6% agar, pH 6.5).
C. lusitaniae transformations
For C. lusitaniae strains, three transformation protocols yielded mutant strains in this study. In the first protocol, cells from overnight cultures were diluted into 50-100 ml liquid YPD cultures. Cultures were grown overnight and harvested at a final OD600 of 1.7-1.8. Cells were pelleted and resuspended in a transformation buffer composed of: 1 X Tris EDTA, 0.1 M lithium acetate, 0.01 mM dithiothreitol to a final volume of 20 ml. Cells were incubated in a roller for one hour at 24°C, washed twice with ice-cold water, once with ice-cold 1 M sorbitol and resuspended in 100 μl of 1 M sorbitol. 40 μl of cells and 5 μg of construct DNA were added to a 0.2 mm electroporation cuvette (BioRad). Cells were electroporated at 1.8 kv (BioRad MicroPulser) and immediately resuspended in 1 ml ice-cold sorbitol. Cells were pelleted and allowed to recover for 4 h in a 50/50 mix of YPD and YPS. Cells were plated onto YPD supplemented with nourseothricin for transformant selection10,22,35,36.
In an alternate protocol, transformations were performed when target genes were highly expressed during sporulation or nutritional starvation. To accomplish this, 6×107 diploid C. lusitaniae cells were plated onto PDA and subsequently harvested when targeted genes were highly induced (after 10-14 h of sporulation). Cells were rinsed from PDA plates with water, pelleted, and incubated in transformation buffer. The remainder of the protocol was completed as described above. Finally, RSY575 and CAY4240 were generated using a previously described protocol37.
Strain Construction
All strains constructed for this study are listed in Supplemental Table 1, and oligonucleotides used are listed in Supplemental Table 2. Strain RSY148 was genetically marked by deleting both ARG4 and ADE2 genes. To accomplish this, the 5’ flank of ARG4 was PCR amplified with oligonucleotides RS84/RS85. The 3’ flank of ARG4 was amplified with oligonucleotides RS86/RS87. The 5’ flank was digested with ApaI and XhoI and ligated between ApaI and XhoI sites in pSFS2a33. Next, the ARG4 3’ flank was digested with PstI and NotI and ligated between the same sites within the plasmid containing the 5’ flank of ARG4. This construct (pRB243) was linearized with ApaI and NotI and transformed into RSY148 to create RSY289. Integration was checked by PCR across the 5’ and 3’ gene boundaries using oligonucleotides RS90/BL70 and BL71/RS91, respectively. Loss of the ARG4 open reading frame was confirmed by PCR using oligonucleotides RS88/RS89. RSY289 was cultured on SC-Maltose to induce expression of the MAL2 promoter resulting in expression of caFLP and excision of the SAT1 cassette at the FRT sites33 creating RSY297. Loss of the SAT1 marker from the deletion construct was confirmed by PCR analysis using RS90 and BL70.
ADE2 was deleted from RSY297 by PCR amplifying the 5’ flank of ADE2 using oligonucleotides RS230/RS231 and the product ligated between ApaI and XhoI sites in pSFS2a. The 3’ flank of ADE2 was PCR amplified using oligonucleotides RS228/RS229 and ligated into the plasmid using NotI and SacII, generating pRB243. The ADE2 deletion cassette was generated with ApaI and SacII and transformed into RSY297 to create RSY379. Correct integration of the construct was verified by PCR analysis across the 5’ and 3’ junctions using oligonucleotides RS226/BL70 and BL71/RS227. Loss of the ADE2 ORF was confirmed by PCR using oligonucleotides RS276/RS277. RSY379 was cultured on SC-Maltose to induce excision of the SAT1 cassette to generate RSY411.
IME2 deletions were constructed in a and α strains RSY411 and RSY284, respectively. The 5’ flank of IME2 was PCR amplified using oligonucleotides RS224/RS265 and cloned into pSFS2a with KpnI and ApaI. Next, the 3’ flank of IME2 was PCR amplified using oligonucleotides RS263/RS225. The pSFS2a plasmid containing the 5’ flank of IME2 and the 3’ IME2 PCR product were digested with NotI and SacII and ligated to generate pRB245. pRB245 was linearized with KpnI and SacII and transformed into RSY284 to generate ime2Δ strain RSY410. The same construct was transformed into RSY411 creating ime2Δ strains RSY406, RSY407, and RSY408. Accurate integration of the construct was confirmed by PCR across 5’ and 3’ junction boundaries using oligonucleotides RS167/BL70 and BL71/RS148. Loss of IME2 was verified by PCR analysis using oligonucleotides RS165/RS166. RSY410 was cultured on SC-Maltose to induce excision of the SAT1 cassette creating RSY437. To reintegrate IME2 at the endogenous locus the IME2 ORF, together with ~ 1 kb of promoter, was PCR amplified using primers RS281 and RS282. The PCR product was digested with KpnI and XhoI and cloned into pSFS2a to generate plasmid pRB278. pRB278 was linearized using PshAI and transformed into RSY437. Integration at the endogenous IME2 locus was checked by PCR using primers RS224/BL70. Diploid Δime2/Δime2 strains RSY427, RSY428 and RSY429 were generated by mating haploid RSY406 and RSY410 strains. Strains were verified by flow cytometry, PCR verification of MTL a/α configuration, and prototrophy.
Diploid STE12 mutant strains were constructed in RSY432. The 5’ flank of STE12 was PCR amplified using oligonucleotides RS323/RS324. The 5’ flank and pSFS2a were digested with KpnI/ApaI and ligated. The 3’ flank of STE12 was PCR amplified using oligonucleotides RS325/RS326 and integrated into this vector using NotI/SacII. The STE12 deletion construct was excised with KpnI/SacII and transformed into RSY432 to generate independent STE12/ste12Δ heterozygous strains RSY541 and RSY575. RSY541 and RSY575 were incubated on SC-Maltose to induce excision of the SAT1 cassette to generate RSY542 and CAY4160, and transformed with the STE12 deletion construct to generate RSY543 and RSY610, RSY611, RSY612, respectively. These strains were incubated on SC-Maltose to excise SAT1 to generate RSY564, CAY4240, CAY4241, and CAY4242.
Haploid ste12Δ strains RSY539, RSY535, RSY544, and RSY545 were generated by incubating RSY541 (STE12/ste12Δ heterozygote) on PDA medium for 3 days to generate haploid progeny. The MTL of these progeny was checked by PCR using oligonucleotides RS66/67 and RS68/69. PCR using primers RS399/RS400 was used to verify the absence of STE12.
The C. lusitaniae STE12 gene was reintegrated into a ste12Δ/ste12Δ strain (RSY564). The STE12 ORF, together with ~1 kb of the promoter, was PCR amplified using oligonucleotides RS401/RS402. pSFS2a and the STE12 PCR product were digested with KpnI and ApaI and ligated together. The resulting construct was linearized using BmgBI and transformed into RSY564 to generate complemented strains RSY601, RSY602, and RSY603. Accurate integration of the construct was determined by PCR using oligonucleotides RS403/RS345.
Construction of K. lactis strains
To obtain diploid K. lactis strains, CAY571 and CAY572 were mated on malt medium at 30°C and selected on medium lacking tryptophan and uracil to generate RSY467, RSY468, RSY469, RSY470, and RSY478. K. lactis mutants were generated using an electroporation protocol previously described37. To make gene deletion cassettes, STE12 5’ and 3’ homologous flanks were generated by PCR of genomic DNA using oligonucleotides BL1581/BL1568 and BL1569/BL1582 respectively. Nourseothricin and Kanamycin resistant genes were amplified by PCR using plasmids pKANmx4 and pNATmx442 with primers BL1313 and BL1414. Resistance genes were fused to target gene-flanking regions by fusion PCR43 using oligonucleotides BL1581/BL1582. This created two linear DNA constructs for transformation, STE12-NAT and STE12-KAN. STE12 double mutants were generated by two sequential rounds of transformation. RSY478 was first transformed with the STE12-KAN cassette to obtain RSY551 and RSY555. Accurate integration was verified by PCR across 5’ and 3’ junctions using oligonucleotides BL1500/BL1329 and BL1501/BL1330. RSY551 and RSY555 strains were transformed with the STE12-NAT construct to obtain ste12Δ/ste12Δ strains RSY571/RSY572 and RSY574, respectively. Loss of the STE12 ORF was verified by PCR (BL1498/BL1499). Haploid K. lactis STE12 mutants were generated by transforming CAY571 with the STE12-NAT1 cassette to generate RSY567 and RSY568. CAY572 was transformed with STE12-KAN to generate RSY550 and with STE12-NAT to generate RSY570.
Construction of P. pastoris and Y. lipolytica strains
To delete the STE12 gene in Pichia pastoris, 5’ and 3’ flanks of the gene were amplified by PCR using oligonucleotides 2357/2358 and 2359/2360. HYG or NAT1 selection markers were PCR amplified using oligonucleotides BL1309/BL1310 from plasmids pNatMX4 and pHYGMX4. The KAN marker was PCR amplified from plasmid pKANB44 using oligonucleotides 2365/2366. STE12 flanks and selection markers were joined by fusion PCR using oligonucleotides 2357/2360. PCR constructs were then transformed into P. pastoris strains JC304 and JC306 using an established protocol34.
To delete the STE12 gene in Yarrowia lipolytica, the STE12 ORF and flanking regions were first amplified by PCR using oligonucleotides BL2618/BL2619 and cloned into pCR-Blunt II-TOPO (Invitrogen) to generate pRB339. The LEU2 gene was also PCR amplified from Y. lipolytica using oligonucleotides BL2620/BL2621 and cloned into pCR-BluntII-TOPO. LEU2 was then excised from this vector using BamHI/KpnI and used to replace the STE12 ORF in pRB340. A cassette was released from this vector using HindIII/EcoRV and used to transform Y. lipolytica to delete the STE12 gene. The URA3 gene was also amplified from vector pINA156 (gift from Carmen Lisset-Flores Mauriz, Instituto de Investigaciones Biomédicas Alberto Sols CSIC-UAM, Madrid) using primers BL2622/BL2483 and cloned into pSC-B-amp/kan (Agilent). URA3 was excised from this vector and used to replace STE12 in vector pRB341. Subsequently, a cassette was released from this vector using HindIII/EcoRV and transformed into Y. lipolytica to delete STE12. Transformations in Y. lipolytica were achieved using the lithium acetate method of Barth and Gaillardin45.
Construction of S. cerevisiae strains
Diploid S. cerevisiae strains were generated by mating CAY4584 and CAY4585 on YPD medium for 24 h, then selecting for diploid products on SCD medium lacking tryptophan and adenine. The resulting mating product, CAY4595, was verified as being diploid by its ability to sporulate and PCR of the MAT locus using primers 2461/2562 (MATα1) and 2565/2566 (MATa1). Subsequent transformations were performed as described46. To delete the STE12 gene, oligonucleotides BL2468/BL2469 were used to PCR amplify selection markers from pNATMX4 and pKANMX4 and contained sequences that were complementary to the 5’ and 3’ regions of the STE12 ORF. Transformation using a NATMX4-STE12 PCR construct generated strain CAY4770, which was PCR checked using primers BL1329/BL2470 and BL1330/BL2471. CAY4770 was transformed with a KANMX4-STE12 PCR construct to generate CAY4808, a ste12Δ/ste12Δ deletion mutant. Loss of the ORF was confirmed by PCR with primers BL2472/BL2473.
Microarray Design
C. lusitaniae genomic sequences were downloaded from the Broad Institute (http://www.broadinstitute.org) to design a custom 8 × 15 K microarray using Agilent eArray. Each array had a minimum of two independent 60-mer oligonucleotides designed against the 5,941 open reading frames encoded by the C. lusitaniae genome, resulting in a total of 11,882 unique oligonucleotides on each microarray.
Cell preparation for microarray analysis
To analyze meiosis, diploid C. lusitaniae strains were grown on PDA (meiosis-inducing) or YPD (non-inducing) medium. Cells from wild type strain RSY432 were removed after 0.5, 2, 4, 8, 12, 18, 24 and 30 h on solid PDA medium and flash frozen in liquid nitrogen. For ime2Δ/ime2Δ and ste12Δ/ste12Δ profiling, cells from strain RSY428 and CAY4240 were incubated on solid PDA medium for 0, 12, 18, and 24 h and flash frozen. RSY432 cell aliquots were collected after 2 h incubation on YPD and used as a reference for all meiosis-profiling experiments. For mating profiles, 6.6 × 107 RSY284 (a) and RSY411 (α) cells were co-incubated on PDA medium for 0, 4 and 12 h and frozen. For comparison, 6.6 × 107 RSY410 (a) and RSY406 (α) cells were co-incubated on PDA medium for 4 h to profile ime2Δ mating expression patterns. RSY284 a cells incubated on YPD for 4 h were used as a reference sample for mating profiles.
Microarray analysis
RNA was isolated from cells using the Ribopure yeast RNA extraction kit (Ambion). RNA was treated with turbo DNAase (Ambion) and evaluated for quality and purity using an Agilent bioanalyzer. Approximately 10 μg of total RNA was used for each cDNA synthesis reaction. Random oligonucleotides (pdN9) as well as dT19 oligos were annealed to total RNA. cDNA was synthesized using Superscript III reverse transcriptase (Invitrogen) in reaction mixes containing 0.25 mM DTT and 0.5 mM total deoxynucleotide phosphates added in a ratio of 3:2 aminoallyl-dUTP/dNTPs. cDNA reactions were stopped after 3 h incubation and subject to 0.3 M NaOH/0.3 M EDTA hydrolysis at pH 7. Single-stranded cDNA was purified and concentrated using a Zymo Cleaner and Concentrator kit (25μg capacity columns). cDNA concentration was determined and each sample separated into 2 μg aliquots, speed vacuumed, and resuspended in 9 μL of water.
cDNA was labeled by incubation with Cy3 or Cy5 dyes in DMSO and 1 M sodium bicarbonate, and concentrated using a Zymo Cleaner and Concentrator spin column kit. Final cDNA concentration and dye-incorporation were determined by nanodrop using the “microarray” setting (Nanodrop 2000c, Thermo Scientific). Approximately 300 ng of labeled experimental cDNA was pooled with an equal amount of reference cDNA to a final volume of 25 μl. Pooled cDNA was denatured at 95°C, mixed with 2x GE Hybridization buffer (HI-RPM buffer, Agilent) and applied to the array. Arrays were incubated for 18 h at 65°C and sequentially washed in Agilent Wash Buffer I, Agilent Wash Buffer II, acetonitrile (Sigma-Aldrich) and Agilent drying and stabilization buffer.
Slides were scanned on an Axon 4000B slide scanner (GenePix) and fluorescence quantified using GENE Pix Pro software. Data was normalized using Goulphar (http://transcriptome.ens.fr/goulphar)38. Normalized gene expression data was averaged for all array replicates and used to generate heatmaps. Pair wise average linkage clustering was performed by Cluster (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm) and heatmaps visualized using treeview (http://jtreeview.sourceforge.net/)39,40. All microarray data has been deposited into the NCBI Gene Expression Omnibus (GEO) portal under accession number GSE51794.
Genetic assays for C. lusitaniae mating, meiosis, and recombination
Throughout this study we utilize genetic crosses between C. lusitaniae strains RSY284 (MTLa) and RSY411 (MTLα). These strains carry different auxotrophic markers that allows for selection of mating products. The diploid product of mating, RSY432, is heterozygous for the cycloheximide recessive genetic marker (chxR). This allows for selection of haploid (meiotic) progeny by growth on cycloheximide, whereas diploid parents (chxR/chxS) do not grow on this medium (see Extended Data Fig. 5). The formation of haploid meiotic progeny was also defined by determining the fraction of ade- colonies due to loss of the ADE2 gene on chromosome 5.
Mating Assays
C. lusitaniae strains were incubated in liquid YPD medium overnight and washed in water. Aliquots of 2×107 cells of each mating type were mixed and cultured on PDA medium at 24°C for 1-5 days. Aliquots of cells were removed and plated for selection of parents as well as diploid products. Percentage of diploid formation = 100 x (# of colonies on double selection medium/# minimal parents on single selection medium). Double selection medium was SCD –uracil –arginine, while single selection medium was SCD – arginine and SCD – uracil. Subsets of cells were screened for ploidy by flow cytometry and MTLa/α alleles were verified by PCR using oligonucleotides RS68/RS69 and R66/RS67. Experiments were repeated in three biological replicates for ste12Δ assays and four biological replicates for ime2Δ assays. Wildtype crosses: RSY284 x RSY411, ime2Δ cross: RSY406 x RSY410. IME2 complemented cross: RSY406 x CAY5022. ste12Δ cross, RSY539 x RSY545.
K. lactis strains were incubated in liquid YPD medium overnight. Aliquots of 2×107 cells of each mating type were mixed and cultured on malt medium at 30°C for 3 days. Aliquots of cells were removed and plated for selection of parents as well as diploid products. Diploid formation was quantified as for C. lusitaniae. Diploid selection media was SC –tryptophan –adenine, while single selection media was SC- tryptophan and SC – adenine. Experiments were repeated in two biological replicates with four independent ste12Δ strains. WT crosses: CAY571 x CAY572. ste12Δ crosses: RSY550 x RSY567, RSY570 x RSY568.
P. pastoris strains were incubated in YPD medium overnight. Aliquots of 2×107 cells of each mating type were washed, mixed, and cultured on sodium acetate medium at 24°C for 5 days. Cells were removed and plated for selection of parents as well as diploid products. Diploid formation was quantified as for C. lusitaniae. Diploid selection medium was SC-histidine-arginine, while single selection media was SC-histidine and SC –arginine. Experiments were repeated in three biological replicates. WT cross: JC304 x JC306. WT x ste12Δ crosses: RBY4440 x JC306, RBY4441 x JC306.
Meiosis Assays
For C. lusitaniae assays, strains were incubated in YPD overnight, washed in water, and 6×107 cells spread onto PDA plates. Cells were incubated at 24°C and then plated onto YPD and haploid selection media (YPD+ cycloheximide). Sporulation percentage was calculated as 100 x (cells on haploid selection media x 2)/( cells on YPD). Experiments were repeated in three biological replicates. Wildtype sporulation: RSY432. ime2Δ/ime2Δ sporulation: #1,RSY427, #2, RSY248, #3, RSY429. ste12Δ/ste12Δ sporulation: RSY543 (Fig. 3) and CAY4240 (Extended Data Fig. 5c).
For K. lactis assays, diploid strains were incubated on malt medium at 30°C for 3 days to induce meiosis. Cells were removed and approximately 200 cells plated onto YPD. Sporulation percentage was calculated as 100 x (ade- cells x 2/ total cells). Meiosis assays were performed in two biological replicates with three independent ste12Δ/ste12Δ mutants. For S. cerevisiae meiosis assays, strains were incubated in YPA medium (2% peptone, 1% yeast extract, 1% KOAc) at 30°C to an OD600 of 1.5-2.5. Cells were washed and resuspended in sporulation medium (1% KOAc, 0.02% raffinose) to an OD600 of 1.5. Cells were incubated in sporulation medium at 30°C for 24 hours and then plated onto YPD. Sporulation was calculated as 100 x (ade- cells x 2/ total cells). The experiment was performed in three biological replicates. For P. pastoris meiosis assays, diploid cells were incubated on sodium acetate medium for 6 days. Cells were recovered and approximately 200 cells plated onto YPD medium. The sporulation percentage was defined as 100 x (ade- cells x 2/ total cells). The experiment was performed in four biological replicates. To induce meiosis in Y. lipolytica, cells were grown on V8 medium at 24°C for 7 days. Meiotic spores were subsequently identified by staining cells with a 5% solution of malachite green41.
Statistical Analysis of Mating and Meiosis Data
Analysis of C. lusitaniae IME2 meiosis data was performed on log-transformed data. Data were normally distributed (p>0.05, Anderson Darling test) and homoscedastic (p>0.05, Levene’s test for homogeneity of variance), and one way ANOVA was performed. In ime2Δ mating and ste12Δ mating experiments, the data was not normally distributed or displayed heteroscedasticity, and a Kruskal-Wallis test was performed to compare across wildtype, unilateral, and bilateral crosses. In ste12Δ/ste12Δ meiosis experiments, pairwise comparisons were made between wildtype and ste12Δ/ste12Δ mutants using an unequal variance t-test.
Pheromone Detection Assay
A wildtype C. lusitaniae diploid strain and C. albicans CAY3705 strain were cultured in SCD medium at 24°C overnight. CAY3705 contains a chimeric STE2 pheromone receptor; the majority of the receptor sequence is derived from C. lusitaniae Ste2, while the intracellular C-terminal tail is from C. albicans Ste2. Previous studies have shown that C. albicans cells expressing the chimeric receptor can respond to synthetic C. lusitaniae α pheromone21. CAY3705 is also expressing a fluorescent nuclear reporter (a histone HTB-YFP fusion protein). Cells were washed in water and 4×107 C. lusitaniae cells mixed with 0.4×107 CAY3705 cells and plated onto PDA plates at 24°C. Cells were removed from PDA plates after 14-18 h incubation and imaged using a Zeiss Observer Z1 microscope.
Microscopy
Differential interference contrast (DIC) and fluorescent images of cells were captured using a Zeiss Inverted Microscope (Axio Observer. Z1) fitted with an AxioCam HR. Images were processed with AxioVision Rel. 4.8 (Zeiss, Germany). For analysis of C. lusitaniae cell morphology during meiosis, diploid a/α cells were incubated on PDA medium at 24°C and samples taken at the indicated time points in Extended Data Fig. 2c and examined microscopically. At least 300 cells per strain were examined for a change in morphology (defined as polarized growth of the cell) or for spore formation.
Defining C. lusitaniae Gene Expression Changes During Meiosis
Fold induction of C. lusitaniae meiotic genes demonstrating expression patterns characteristic of early, middle and late meiosis were determined by evaluating microarray data. Genes were characterized as being induced during meiosis if they exhibited >3-fold induction after 12, 18, or 24 h on PDA and also demonstrated < 3-fold induction in haploid a and α cells incubated on PDA medium for 30 min, 2 h, or 24 h. This analysis generated a set of 618 genes. The resulting list of genes was then compared to genes induced during ime2Δ/ime2Δ meiosis data to determine the number of meiosis-specific genes dependent on Ime2 (Extended Data Fig. 3e). This same set of 618 genes was used to compare to gene induction during meiosis of ste12Δ/ste12Δ mutant cells.
Analyzing MAPK gene expression during C. lusitaniae meiosis
Average fold gene induction was calculated by averaging expression changes of S. cerevisiae MAPK genes (STE20, STE11, STE7, FUS3, STE12, STE2, STE3, GPA1, STE4, STE18, MFALPHA and KSS1) or their C. lusitaniae orthologs during meiosis. S. cerevisiae gene expression data was from Roberts et al.30 and was enhanced 3 fold to be comparable to expression changes in C. lusitaniae genes.
RT-PCR
WT strains RSY411 and RSY284 were mated on PDA at 24°C. Aliquots of mating mixes were snap frozen after 0 min, 30 min, 2 h, 4 h and 24 h of co-incubation. pdN9 was annealed to 2 μg of RNA extracted from the mating mixes. First strand cDNA was synthesis was performed with GoScript reverse transcriptase (Promega). 75 ng of the resulting cDNA was PCR amplified for IME2 and ACT1 expression using oligonucleotides RSY183/RSY184 and RS333/RS334.
Supplementary Material
Acknowledgements
We thank Joseph Heitman, Carmen Lisset-Flores Mauriz, Thierry Noel, Neil Hunter, and Jennifer Reedy for gifts of strains and plasmids, and Shail Kabrawala and Nayimisha Balmuri for help with strain construction. We also thank Trevor Sorrells, Liam Holt, and members of the Johnson and Bennett labs for comments on the paper, and Stephen Jones for help with statistical analysis. This work was supported by National Science Foundation Grant MCB1021120 (to R.J.B), by National Institutes of Health R01 Grant AI081704 (to R.J.B.), by T32GM007601 (to C.M.S.), by F31AI075607 (To R.K.S.), and by an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund (to R.J.B.).
Footnotes
Author Contributions
R.K.S. and C.S. constructed strains, analyzed phenotypes, and performed transcriptional profiling experiments. S.T. and R.J.B. constructed strains and analyzed phenotypes. R.K.S., C.S., and R.J.B were involved in study design and writing of the manuscript.
References
- 1.van Werven FJ, Amon A. Regulation of entry into gametogenesis. Philos Trans R Soc Lond B BiolSci. 2011;366:3521–3531. doi: 10.1098/rstb.2011.0081. doi:366/1584/3521[pii]10.1098/rstb.2011.0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong Sak Hoi J, Dumas B. Ste12 and Ste12-like proteins, fungal transcription factors regulating development and pathogenicity. Eukaryot Cell. 2010;9:480–485. doi: 10.1128/EC.00333-09. doi:EC.00333-09 [pii]10.1128/EC.00333-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irniger S. The Ime2 protein kinase family in fungi: more duties than just meiosis. MolMicrobiol. 2011;80:1–13. doi: 10.1111/j.1365-2958.2011.07575.x. doi:10.1111/j.1365-2958.2011.07575.x. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida M, et al. Initiation of meiosis and sporulation in Saccharomyces cerevisiae requires a novel protein kinase homologue. Mol Gen Genet. 1990;221:176–186. doi: 10.1007/BF00261718. [DOI] [PubMed] [Google Scholar]
- 5.Dolan JW, Kirkman C, Fields S. The yeast STE12 protein binds to the DNA sequence mediating pheromone induction. Proc Natl Acad Sci U S A. 1989;86:57035707. doi: 10.1073/pnas.86.15.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Errede B, Ammerer G. STE12, a protein involved in cell-type-specific transcription and signal transduction in yeast, is part of protein-DNA complexes. Genes Dev. 1989;3:1349–1361. doi: 10.1101/gad.3.9.1349. [DOI] [PubMed] [Google Scholar]
- 7.Souza CA, Silva CC, Ferreira AV. Sex in fungi: lessons of gene regulation. Genetics and Molecular Research : GMR. 2003;2:136–147. [PubMed] [Google Scholar]
- 8.Govin J, Berger SL. Genome reprogramming during sporulation. IntJ Dev Biol. 2009;53:425–432. doi: 10.1387/ijdb.082687jg. doi:082687jg [pii]10.1387/ijdb.082687jg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butler G, et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–662. doi: 10.1038/nature08064. doi:nature08064[pii]10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reedy JL, Floyd AM, Heitman J. Mechanistic plasticity of sexual reproduction and meiosis in the Candida pathogenic species complex. Curr Biol. 2009;19:891–899. doi: 10.1016/j.cub.2009.04.058. doi:S0960-9822(09)01057-4 [pii]10.1016/j.cub.2009.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gargeya IB, Pruitt WR, Simmons RB, Meyer SA, Ahearn DG. Occurrence of Clavispora lusitaniae, the teleomorph of Candida lusitaniae, among clinical isolates. J Clin Microbiol. 1990;28:2224–2227. doi: 10.1128/jcm.28.10.2224-2227.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lengeler KB, et al. Signal transduction cascades regulating fungal development and virulence. Microbiol Mol Biol Rev. 2000;64:746–785. doi: 10.1128/mmbr.64.4.746-785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler G. Fungal sex and pathogenesis. Clin Microbiol Rev. 2010;23:140–159. doi: 10.1128/CMR.00053-09. doi:23/1/140 [pii]10.1128/CMR.00053-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keeney S. In: Recombination and Meiosis Vol. 2 Genome Dynamics and Stability. Lankenau D, editor. Springer; Berlin/Heidelberg: 2007. [Google Scholar]
- 15.Klein F, et al. A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell. 1999;98:91–103. doi: 10.1016/S0092-8674(00)80609-1. doi:S0092-8674(00)80609-1 [pii]10.1016/S0092-8674(00)80609-1. [DOI] [PubMed] [Google Scholar]
- 16.Smith HE, Mitchell AP. A transcriptional cascade governs entry into meiosis in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:2142–2152. doi: 10.1128/mcb.9.5.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu S, et al. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 18.Guttmann-Raviv N, Martin S, Kassir Y. Ime2, a meiosis-specific kinase in yeast, is required for destabilization of its transcriptional activator, Ime1. Mol Cell Biol. 2002;22:2047–2056. doi: 10.1128/MCB.22.7.2047-2056.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holt LJ, Hutti JE, Cantley LC, Morgan DO. Evolution of Ime2 phosphorylation sites on Cdk1 substrates provides a mechanism to limit the effects of the phosphatase Cdc14 in meiosis. Mol Cell. 2007;25:689–702. doi: 10.1016/j.molcel.2007.02.012. doi:S1097-2765(07)00112-8 [pii]10.1016/j.molcel.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sopko R, Raithatha S, Stuart D. Phosphorylation and maximal activity of Saccharomyces cerevisiae meiosis-specific transcription factor Ndt80 is dependent on Ime2. Mol Cell Biol. 2002;22:7024–7040. doi: 10.1128/MCB.22.20.7024-7040.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin CH, Choi A, Bennett RJ. Defining pheromone-receptor signaling in Candida albicans and related asexual Candida species. Mol Biol Cell. 2011;22:49184930. doi: 10.1091/mbc.E11-09-0749. doi:mbc.E11-09-0749 [pii]10.1091/mbc.E11-09-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young LY, Lorenz MC, Heitman J. A STE12 homolog is required for mating but dispensable for filamentation in Candida lusitaniae. Genetics. 2000;155:17–29. doi: 10.1093/genetics/155.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sipiczki M. Where does fission yeast sit on the tree of life? Genome Biol. 2000;1:1011.1011–1011.1014. doi: 10.1186/gb-2000-1-2-reviews1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugimoto A, Iino Y, Maeda T, Watanabe Y, Yamamoto M. Schizosaccharomyces pombe ste11+ encodes a transcription factor with an HMG motif that is a critical regulator of sexual development. Genes Dev. 1991;5:1990–1999. doi: 10.1101/gad.5.11.1990. [DOI] [PubMed] [Google Scholar]
- 25.Kjaerulff S, Lautrup-Larsen I, Truelsen S, Pedersen M, Nielsen O. Constitutive activation of the fission yeast pheromone-responsive pathway induces ectopic meiosis and reveals Ste11 as a mitogen-activated protein kinase target. Mol Cell Biol. 2005;25:2045–2059. doi: 10.1128/MCB.25.5.2045-2059.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herskowitz I. A regulatory hierarchy for cell specialization in yeast. Nature. 1989;342:749–757. doi: 10.1038/342749a0. doi:10.1038/342749a0. [DOI] [PubMed] [Google Scholar]
- 27.Booth LN, Tuch BB, Johnson AD. Intercalation of a new tier of transcription regulation into an ancient circuit. Nature. 2010;468:959–963. doi: 10.1038/nature09560. doi:nature09560 [pii]10.1038/nature09560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otto SP. The evolutionary consequences of polyploidy. Cell. 2007;131:452–462. doi: 10.1016/j.cell.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 29.Otto SP, Gerstein AC. The evolution of haploidy and diploidy. Curr Biol. 2008;18:R1121–1124. doi: 10.1016/j.cub.2008.09.039. doi:S0960-9822(08)01268-2 [pii]10.1016/j.cub.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 30.Roberts CJ, et al. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 2000;287:873–880. doi: 10.1126/science.287.5454.873. [DOI] [PubMed] [Google Scholar]
- 31.Guthrie C, Fink GR. Guide to Yeast Genetics and Molecular Biology. Academic Press; San Diego: 1991. [Google Scholar]
- 32.Gargeya IB, Pruitt WR, Simmons RB, Meyer SA, Ahearn DG. Occurrence of Clavispora lusitaniae, the teleomorph of Candida lusitaniae, among clinical isolates. J. Clin. Microbiol. 1990;28:2224–2227. doi: 10.1128/jcm.28.10.2224-2227.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reuß O, Vik A, Kolter R, Morschhauser J. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene. 2004;341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 34.Cregg JM, et al. In: Expression in the Yeast Pichia pastoris, in Methods Enzymology. Burgess RR, Deutscher MP, editors. Vol. 463. Elsevier; 2009. pp. 169–189. [DOI] [PubMed] [Google Scholar]
- 35.El-Kirat-Chatel S, Dementhon K, Noël T. A two-step cloning-free PCR-based method for the deletion of genes in the opportunistic pathogenic yeast Candida lusitaniae. Yeast. 2011;28:321–330. doi: 10.1002/yea.1836. [DOI] [PubMed] [Google Scholar]
- 36.François F, Chapeland-Leclerc F, Villard J, Noël T. Development of an integrative transformation system for the opportunistic pathogenic yeast Candida lusitaniae using URA3 as a selection marker. Yeast. 2004;21:95–106. doi: 10.1002/yea.1059. [DOI] [PubMed] [Google Scholar]
- 37.Kooistra R, Hooykaas PJ, Steensma HY. Efficient gene targeting in Kluyveromyces lactis. Yeast. 2004;21:781–792. doi: 10.1002/yea.1131. [DOI] [PubMed] [Google Scholar]
- 38.Lemoine S, Combes F, Servant N, Le Crom S. Goulphar: rapid access and expertise for standard two-color microarray normalization methods. BMC bioinformatics. 2006;7:467. doi: 10.1186/1471-2105-7-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 40.de Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 41.Flores CL, Gancedo C, Petit T. Disruption of Yarrowia lipolytica TPS1 gene encoding trehalose-6-P synthase does not affect growth in glucose but impairs growth at high temperature. PLoS One. 2011;6:e23695. doi: 10.1371/journal.pone.0023695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wach A, Brachat A, Pohlmann R, Philippsen P. Dominant marker vectors for selecting yeast mating products. Yeast. 2008;52:595–599. doi: 10.1002/yea.1604. [DOI] [PubMed] [Google Scholar]
- 43.Noble SM, Johnson AD. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell. 2005;4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin-Cereghino J, et al. Direct selection of Pichia pastoris expression strains using new G418 resistance vectors. Yeast. 2008;25:293–299. doi: 10.1002/yea.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barth G, Gaillardin C. Physiology and genetics of the dimorphic fungus Yarrowia lipolytica. FEMS microbiology reviews. 1997;19:219–237. doi: 10.1111/j.1574-6976.1997.tb00299.x. [DOI] [PubMed] [Google Scholar]
- 46.Schiestl RH, et al. Methods: A Companion to Meth. Enzy. 1993;5:79–85. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.