

Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by mutant huntingtin (Htt) with an expanded polyglutamine tract. It has been reported that Htt regulates autophagy. However, it remains unclear whether mutant Htt affects chaperone-mediated autophagy (CMA). Our study aimed to investigate the effect of mutant Htt on CMA activity in cultured HEK293T cells. A HEK293T cell model of HD was produced by transient transfection of wild-type (20Q) or mutant (120Q) Htt plasmids. The effect of mutant Htt on two CMA components, lysosomal-associated membrane protein 2a (Lamp2a) and heat-shock cognate protein 70 (Hsc70), was determined by western blotting and immunofluorescent staining. We observed that mutant Htt did not significantly alter the expression of Lamp2a and Hsc70 when compared to normal Htt. These findings suggest that mutant Htt does not reduce CMA activity and that enhancing CMA activity to clear mutant Htt may be a novel strategy for the management of HD.
Keywords: Huntington’s disease, chaperone-mediated autophagy, lysosomal-associated membrane protein 2a, heat-shock cognate protein 70
Introduction
Huntington’s disease (HD) is a progressive, autosomal dominant, neurodegenerative disorder caused by the expansion of a CAG trinucleotide repeat (>35 repeats) in the huntingtin (Htt) gene, which is translated into an expanded polyglutamine tract in the N-terminus of the Htt protein (1). The most obvious characteristic of HD is uncontrolled movement, dementia, emotional disturbance and early death, which often occur in middle age (2,3). These symptoms are associated with neuronal loss that preferentially occurs in medium-sized spiny neurons of the striatum and also extends to other brain regions during the late stages of the disease.
Wild-type Htt is considered to possess a number of cellular functions, such as protein transport, vesicle trafficking and cytoskeletal anchoring. When Htt is mutated with an expanded polyglutamine tract (>36 glutamines), Htt becomes misfolded to induce gain of toxic function and may also lose its normal function. Mutant Htt appears to affect a variety of cellular functions, including intracellular signaling pathways (4), the protein degradation pathway (5) and the nuclear transcription and axon transport (6,7), resulting in degeneration of neuronal cells in the striatum and other brain regions.
Clearing mutant Htt may be of value in preventing the pathogenesis and development of HD. In eukaryotic cells, there exist two systems of protein degradation, the ubiquitin-proteasome system and autophagy. Autophagy is a non-specific bulk degradation pathway for long-lived cytoplasmic proteins, protein complexes, or damaged organelles. There are three types of autophagy, namely macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). Macroautophagy is considered to promote the degradation of Htt and clearance of aggregates. This is supported by several lines of evidence that rapamycin (an activator of autophagy) (8) and its several small molecular enhancers (9–11) may enhance the degradation of mutant Htt and reduce its toxicity. It is also important to investigate whether mutant Htt can damage autophagic function. It was reported that mutant Htt did not alter the expression of microtubule-associated protein 1 light chain 3, suggesting the activity of macroautophagy is not changed by Htt (12).
CMA is another type of autophagy, which is distinct from macroautophagy and microautophagy, as a subset of long-lived cytosolic soluble proteins are directly delivered to the lysosomal lumen via specific receptors (13). The basic machinery consists of at least three types of proteins, including i) chaperone proteins, which are responsible for recognizing substrates based on their specific motifs and delivering them to the lysosomes; ii) receptor proteins, which bind and transport/pull substrates into the lysosomal lumen; and iii) substrate proteins, which are a subset of soluble cytosolic proteins containing specific motifs associated with KFERQ. When CMA is activated, the expression levels of two important proteins, lysosomal-associated membrane protein 2a (Lamp2a) and heat-shock cognate protein 70 (Hsc70) are significantly increased (14–16). It is known that heat-shock protein β-8 may participate in protein quality control through a non-chaperone-like mechanism, which requires the phosphorylation of eIF2α (17). It was recently demonstrated that CMA contributes to the degradation of mutant Htt in cultured cells (18). However, it remains unclear whether mutant Htt alters CMA activity. Elucidating this issue may help determine whether modulating CMA activity is an effective therapeutic approach for the management of HD.
In this study, we aimed to investigate the effect of mutant Htt on the basal activity of CMA in cultured cells and determine whether enhancing CMA activity is an effective way to reduce the toxicity of mutant Htt.
Materials and methods
Plasmids, antibodies and reagents
Enhanced green fluorescent protein (EGFP)-Htt-exon1-20Q and EGFP-Htt-exon1–120Q plasmids were gifts from Professor Xiaojiang Li from the Department of Human Genetics at Emory University School of Medicine (Atlanta, GA, USA). XL1-Blue Competent cells were obtained from Beijing TransGen Biotech Co., Ltd., Beijing, China; the Plasmid Mini kit was purchased from Omega Bio-Tek, Inc., Norcross, GA, USA; the Endotoxin-Free Plasmid Maxi kit was purchased from Tiangen Biotech Co., Ltd., Beijing, China; and the BamHI and EcoRI restriction nucleotide enzymes used in this study were purchased from Fermentas, Inc., Glen Burnie, MD, USA.
HEK293T cells were purchased from ATTC (Teddington, UK) and cultured with high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, Logan, USA) supplemented with penicillin-streptomycin, fetal bovine serum (FBS) and 0.25% trypsin-EDTA (Gibco-BRL, Carlsbad, CA, USA). SuperFectin™ In Vitro DNA Transfection reagent (Shanghai Pufei Biotech Co., Ltd, Shanghai, China) was used for DNA transfection.
The following antibodies were used in the study: mouse monoclonal anti-green fluorescent protein (GFP) antibody (1:20,000, Proteintech Group, Inc., Chicago, IL, USA); rabbit polyclonal anti-Lamp2a antibody (1:600; Abcam, Cambridge, MA, USA); rabbit monoclonal anti-Hsc70 antibody (1:500; Abcam); mouse anti-β-actin antibody (1:2,000; Kangwei Century Biology Ltd., Co., Beijing, China). Enhanced chemiluminescence (ECL) developer (Tiangen Biotech Co.) was used for western blotting.
Cell culture and Htt transfection
Two plasmids (EGFP-Htt-exon1-20Q and EGFP-Htt-exon1–120Q) were used for transfection of HEK293T cells. The HEK293T cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin sulfate at 37°C in an atmosphere of 5% CO2 and 95% humidity. The cells were seeded onto 6-well or 12-well plates and, after reaching a confluency of 80%, they were transiently transfected with 0.75 or 1.0 μg wild-type (20Q) or mutant (120Q) Htt plasmids using SuperFectin™ In Vitro DNA Transfection reagent. After 24, 36 and 48 h of culture, the cells were used for western blotting and fluorescence microscopy analysis.
Western blot analysis and immunofluorescent microcopy
After 36 or 48 h of transfection, the cultured cells were rinsed with phosphate-buffered saline (PBS) three times at room temperature and 1× SDS loading buffer (250 μl) was added immediately to the cell pellet. The cell lysates were sonicated for 5–8 sec and heated at 100°C for 5 min in 1× gel-loading buffer containing 2% SDS for western blot analysis. The samples (20 μg of protein) were separated on a 10% (for Htt and other proteins) Tris-glycine SDS-polyacrylamide gel. The proteins were transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The nitrocellulose membranes were blocked with 5% non-fat dry milk/PBS with 0.1% Tris-buffered saline with Tween-20 (TBST) for 30 min and the blots were rinsed with 1× TBST and incubated with the primary antibodies overnight at 4°C. Secondary horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG were incubated with the blot in 5% non-fat dry milk/PBS for 1 h at room temperature. The ECL developer was then used to reveal immunoreactive bands on the blots. To verify the protein loading amounts, the blots were also re-probed with mouse anti-β-actin antibody.
After 24 h of transfection, immunofluorescent staining of cultured cells with antibodies against Htt and Lamp2a or Hsc70 was performed. Non-transfected and Htt-transfected HEK293T cells were fixed for 7 min with 4% paraformaldehyde/PBS and then examined under a immunofluorescent microscope. The cells were also stained with Hoechst dye (blue) for the nuclei and the ratios of Lamp2a (red) or Hsc70 (red) to blue color were quantified. These ratios in Htt-transfected (green) or non-transfected cells (not green) were compared to determine whether mutant Htt was able to reduce Lamp2a or Hsc70. For the control, HEK293T cells were also transfected with normal Htt (20Q).
Statistical analysis
All the data are expressed as means ± standard error of the mean. The statistical results were analyzed by GraphPad Prism software, version 5 (GraphPad Software, Inc., San Diego, CA, USA) and the statistical significance (P<0.05) was assessed using the Student’s t-test or one-way analysis of variance, followed when appropriate by a post hoc analysis using the Dunnett’s test.
Results
High-efficiency expression of Htt in HEK293T cells
The expression of wild-type or mutant Htt was achieved by transient transfection of wild-type Htt (EGFP-Htt-exon1-20Q) and mutant Htt (EGFP-Htt-exon1–120Q) in HEK293T cells. We performed western blotting using GFP antibody to detect wild-type and mutant Htt expression. As expected, wild-type Htt migrated at 50 kDa, while mutant Htt migrated at 70 kDa, as it carries a larger polyglutamine repeat (Fig. 1).
Figure 1.
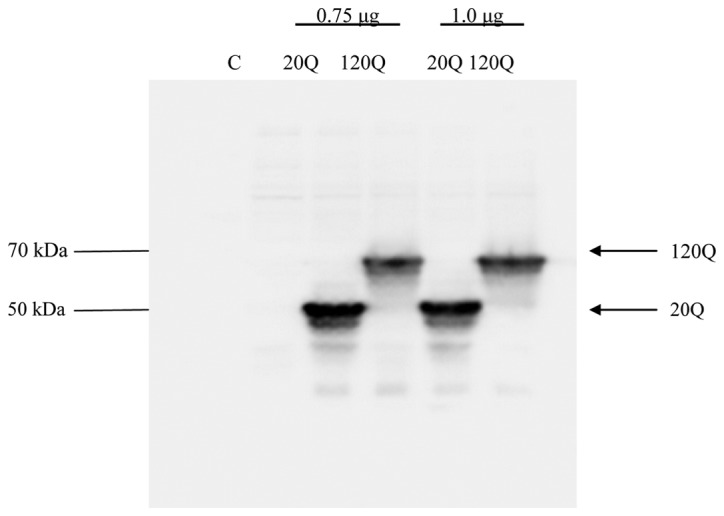
Expression of huntingtin (Htt) in HEK293T cells. The HEK293T cells were transiently transfected with 0.75 or 1.0 μg enhanced green fluorescent protein (EGFP)-Htt-exon1-20Q or EGFP-Htt-exon1–120Q and harvested 36 h later for western blot analysis. Both wild-type and mutant Htt were expressed at the expected molecular mass.
Mutant Htt did not reduce the activity of CMA
The expression levels of two important proteins, Lamp2a and Hsc70, represents CMA activation. When CMA is activated, the expression levels of Lamp2a and Hsc70 are significantly increased (14–16). We used the ratios of these proteins to β-actin on the western blots to assess their levels and observed no significant difference in the Lamp2a/β-actin ratio between the cells transfected with Htt-20Q and those transfected with Htt-120Q at a dose of 0.75 and 1.0 μg (Fig. 2A). In addition, there was no significant difference in the Hsc70/β-actin ratio between the cells transfected with Htt-20Q and those transfected with Htt-120Q (Fig. 2B). We also performed immunofluorescent double staining of the transfected cells and did not identify significant differences in the staining signals for Hsc70 and Lamp2a between Htt-20Q- and Htt-120Q-transfected cells (data not shown). These results suggest that the expression of mutant Htt did not reduce the CMA activity in HEK293T cells.
Figure 2.
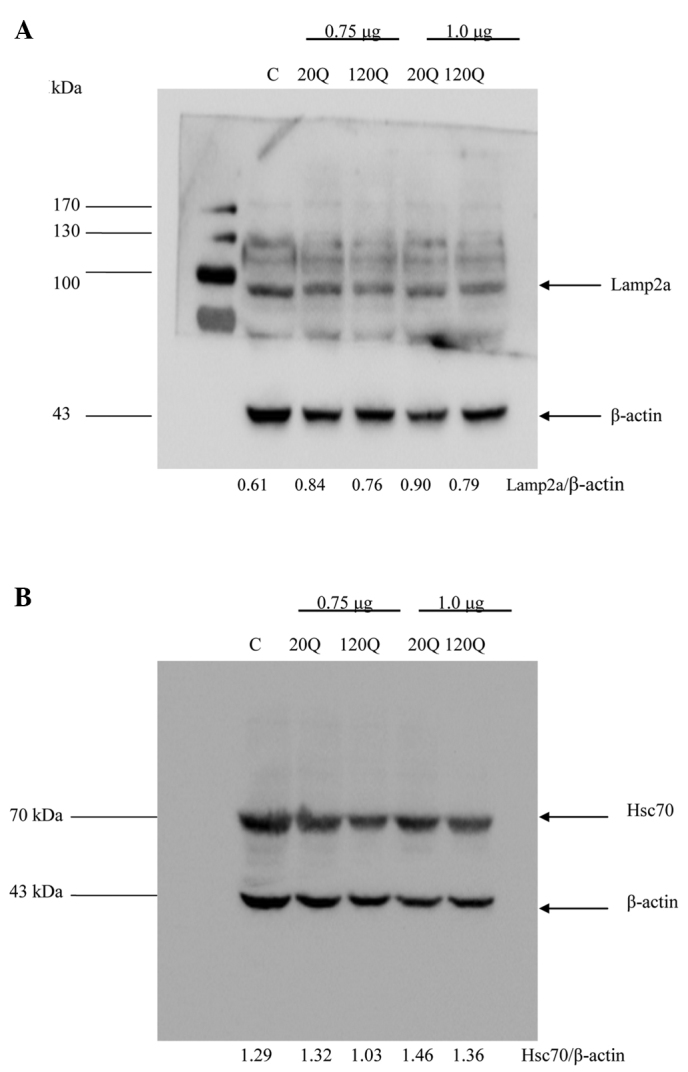
Lysosomal-associated membrane protein 2a (Lamp2a) and heat-shock cognate protein 70 (Hsc70) expression in cultured HEK293T cells transfected with huntingtin (Htt). (A and B) HEK293T cells were transfected with PRK-green fluorescent protein-exon1 Htt containing 20Q or 120Q at 0.75 or 1.0 μg. The total cell lysates were collected after 36 h of transfection for western blotting with antibodies to (A) Lamp2a and (B) Hsc70. Representative western blots are shown and the ratios of Lamp2a or Hsc70 to β-actin are noted below the blots. There was no significant difference in the Lamp2a/β-actin or the Hsc70/β-actin ratio between the cells transfected with Htt-20Q and those transfected with Htt-120Q.
Discussion
HD is one of nine polyQ expansion diseases that shares the age-dependent and selective neurodegeneration characteristic with Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (19). All these diseases are characterized by late-onset accumulation of toxic proteins that are misfolded and form aggregates. Thus, clearance of misfolded proteins is key to effectively treating these neurodegenerative diseases.
Autophagy is an important intracellular metabolic pathway that includes macroautophagy, microautophagy and CMA. All the substrates are degraded by hydrolytic enzymes. It was reported that the activity of CMA displayed constitutive compensatory upregulation in different cellular and transgenic mouse models of HD (20). Further investigations indicated that the Lamp2a and Hsc70 proteins played an important role in clearing Htt through the CMA pathway (21,22). Although the involvement of CMA in Htt metabolism has been suggested by other investigators, the essential effect of mutant Htt on CMA remains to be elucidated. Thus, our study aimed to provide evidence regarding the effect of Htt on CMA.
Using HEK293T cells transfected with wild-type and mutant Htt, we were able to define whether mutant Htt specifically affects CMA activity. Our results suggested that mutant Htt did not significantly alter the activity of CMA. Consistent with a recent finding that mutant Htt does not affect the basal levels of autophagy in the hypothalamus of HD transgenic mice (23), it appears that the expression of mutant Htt may not affect the basal activity of CMA in cells. Thus, enhancing CMA activity may effectively remove the toxic and misfolded Htt and alleviate its toxicity.
Acknowledgements
This study was supported by a grant from the Natural Science Foundation of Jiangxi Province (no. 20122BAB215034). We would like to thank Professor Xiaojiang Li for providing the Htt plasmids and Xiaoyu Wang, De Kang and Qiang Qiu for their technical assistance.
References
- 1.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. No authors listed. [DOI] [PubMed] [Google Scholar]
- 2.Gusella JF, MacDonald ME, Ambrose CM, Duyao MP. Molecular genetics of Huntington’s disease. Arch Neurol. 1993;50:1157–1163. doi: 10.1001/archneur.1993.00540110037003. [DOI] [PubMed] [Google Scholar]
- 3.Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Kim YJ, Yi Y, Sapp E, et al. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci USA. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho LW, Carmichael J, Swartz J, Wyttenbach A, Rankin J, Rubinsztein DC. The molecular biology of Huntington’s disease. Psychol Med. 2001;31:3–14. doi: 10.1017/s0033291799002871. [DOI] [PubMed] [Google Scholar]
- 6.Gauthier LR, Charrin BC, Borrell-Pages M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 7.Morton AJ, Edwardson JM. Progressive depletion of complexin II in a transgenic mouse model of Huntington’s disease. J Neurochem. 2001;76:166–172. doi: 10.1046/j.1471-4159.2001.00059.x. [DOI] [PubMed] [Google Scholar]
- 8.Berger Z, Ravikumar B, Menzies FM, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 9.Floto RA, Sarkar S, Perlstein EO, Kampmann B, Schreiber SL, Rubinsztein DC. Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington’s disease models and enhance killing of mycobacteria by macrophages. Autophagy. 2007;3:620–622. doi: 10.4161/auto.4898. [DOI] [PubMed] [Google Scholar]
- 10.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 11.Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Wang CE, Huang S, Xu X, Li XJ, Li H, Li S. Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum Mol Genet. 2010;19:2445–2455. doi: 10.1093/hmg/ddq127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci. 2011;68:749–763. doi: 10.1007/s00018-010-0565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. 1995;269:C1200–C1208. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- 15.Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997;137:825–834. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000;1:570–583. doi: 10.1034/j.1600-0854.2000.010707.x. [DOI] [PubMed] [Google Scholar]
- 17.Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J Biol Chem. 2009;284:5523–5532. doi: 10.1074/jbc.M807440200. [DOI] [PubMed] [Google Scholar]
- 18.Bauer PO, Goswami A, Wong HK, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010;28:256–263. doi: 10.1038/nbt.1608. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Li H, Li XJ. Intracellular degradation of misfolded proteins in polyglutamine neurodegenerative diseases. Brain Res Rev. 2008;59:245–252. doi: 10.1016/j.brainresrev.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J Neurosci. 2011;31:18492–18505. doi: 10.1523/JNEUROSCI.3219-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One. 2012;7:e46834. doi: 10.1371/journal.pone.0046834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi L, Zhang XD. Role of chaperone-mediated autophagy in degrading Huntington’s disease-associated huntingtin protein. Acta Biochim Biophys Sin (Shanghai) 2014;46:83–91. doi: 10.1093/abbs/gmt133. [DOI] [PubMed] [Google Scholar]
- 23.Baldo B, Soylu R, Petersen A. Maintenance of basal levels of autophagy in Huntington’s disease mouse models displaying metabolic dysfunction. PLoS One. 2013;8:e83050. doi: 10.1371/journal.pone.0083050. [DOI] [PMC free article] [PubMed] [Google Scholar]