

Automation is the response to overcoming the crystallization bottleneck in biological crystallography. This review provides a summary of the current methods and technologies applied in automated platforms for the setup of initial and follow-up crystallization experiments.
Keywords: crystallization, automation
Abstract
Crystallization remains the bottleneck in the crystallographic process leading from a gene to a three-dimensional model of the encoded protein or RNA. Automation of the individual steps of a crystallization experiment, from the preparation of crystallization cocktails for initial or optimization screens to the imaging of the experiments, has been the response to address this issue. Today, large high-throughput crystallization facilities, many of them open to the general user community, are capable of setting up thousands of crystallization trials per day. It is thus possible to test multiple constructs of each target for their ability to form crystals on a production-line basis. This has improved success rates and made crystallization much more convenient. High-throughput crystallization, however, cannot relieve users of the task of producing samples of high quality. Moreover, the time gained from eliminating manual preparations must now be invested in the careful evaluation of the increased number of experiments. The latter requires a sophisticated data and laboratory information-management system. A review of the current state of automation at the individual steps of crystallization with specific attention to the automation of optimization is given.
1. Introduction
The crystallization of biological macromolecules dates back to a time when little to nothing was known about the intricate ways in which proteins and nucleic acids perform their many tasks in living organisms (Hünefeld, 1840 ▶). The intended purpose of crystallization in early-day chemistry was one of purification. Probably more importantly, the very fact that at least some biological macromolecules had the ability to crystallize demonstrated that they had a common and defined shape. It then took more than another 100 years before the full value of biological crystals came to light (Kendrew et al., 1958 ▶): the ability to determine the three-dimensional structures and therefore to understand the functions and modi operandi of nature’s tools and building blocks at atomic resolution. This success ushered in the field of biological X-ray crystallography. Since then, close to 100 000 structures of biological macromolecules, proteins, nucleic acids or complexes between them, ranging in size from a few hundred daltons to over 1.5 MDa, have been determined and deposited in structural repositories such as the Protein Data Bank (PDB, http://www.pdb.org; Berman et al., 2000 ▶). Despite the arrival of new and important methods for deriving structural information from biological macromolecules, crystallography remains the method of choice, and is responsible for close to 90% of the data deposited in the PDB. As the name of the method indicates, all matter examined by crystallography is crystalline: no crystals, no crystallography.
An examination of the individual steps that lead from a gene to the three-dimensional crystallographic structure of its encoded protein shows a remarkable pattern (Table 1 ▶). The average rate of survival as a protein moves from one step to the next is two out of three (i.e. one protein in every three is dropped at each stage). There is one exception: attempts to crystallize purified targets are successful for only one in every seven candidates (14.2%; http://sbkb.org/metrics/milestonestables.html).
Table 1. Success rates for steps in crystallographic structure determination.
Percentages are given in relation to the previous step.
| Total | Cloned | Expressed | Purified | Crystallized | Structures |
|---|---|---|---|---|---|
| 54744 | 35893 | 24306 | 16833 | 2390 | 1711 |
| 100% | 65.6% | 67.7% | 69.3% | 14.2% | 71.6% |
The present-day understanding of the fundamental laws that govern macromolecular crystallization and the associated difficulties are detailed in an earlier article in this series (McPherson & Gavira, 2014 ▶).
The most important approaches to overcome the biological crystallization bottleneck have been the discovery of suitable precipitants, new methods of preparing samples of macromolecules, new methods of executing crystallization experiments (including the use of pre-prepared random screens) and the reduction of cost and increase in efficiency of biological crystallization through automation. The latter is founded on the realisation that the inherent inability to predict conditions which are conducive to the formation of biological crystals is best overcome by screening a variety (tens) of constructs of a target of interest against a large number (hundreds) of precipitant combinations. This process entails the execution of the same kind of experiment from a limited amount (∼200 µl) of very pure sample. Robots are predestined for such tasks.
These efforts have resulted in the establishment of several academic (Heinemann et al., 2000 ▶; Watanabe et al., 2002 ▶; Luft et al., 2003 ▶; Albeck et al., 2005 ▶; Mueller-Dieckmann, 2006 ▶; Mueller et al., 2012 ▶) and industrial (Peat et al., 2002 ▶; Hosfield et al., 2003 ▶) high-throughput facilities.
2. The process of biomolecular crystallization
All biological crystallization experiments occur in solution. The laws of thermodynamics dictate that the formation of crystals from a solute sample can only occur from a state of supersaturation. To this end, hundreds of precipitants, chemical compounds of inorganic and organic nature, which manipulate the solubility of the sample, have found their way into biological crystallization. The path towards supersaturation can be achieved by different means (McPherson et al., 2003 ▶). They all share the preparation of mixtures of sample and precipitant(s), optionally supported by additional ways to further increase sample and precipitant concentration by the removal of water, e.g. vapour diffusion. The sheer number of possible combinations of precipitants (together with variations of the pH value or the ambient temperature of the solution) is overwhelming. To make matters worse, difficult targets often require subdivision of proteins into individual domains, truncations at the natural termini, internal deletions or complex formation with small ligands or macromolecular binding partners (protein or RNA/DNA) to improve either stability or solubility. Online services that analyse the amino-acid sequences of target proteins attempt to estimate the likelihood that the proteins in question will crystallize (Prilusky, Felder et al., 2005 ▶; Slabinski et al., 2007 ▶; Kurgan et al., 2009 ▶). However, it is not possible to rationally generate sample constructs or complexes that are guaranteed to crystallize, nor to predict conducive crystallization cocktails. Crystallographers are therefore forced to design and conduct large numbers of experiments (∼500 drops per unique sample) before identifying favourable conditions for the formation of crystals. Nota bene, there is no guarantee that any number of experiments will ever result in crystals!
The setup of crystallization experiments by hand is not only tedious but it is, in the face of the enormous number of experiments, also an inefficient use of the time of qualified staff or students. At the same time, robotics are well placed to handle liquids efficiently, to recombine them accurately into new formulations and to generate large numbers of experiments, even in small volumes (<100 nl). The latter is important because the high demand for sample homogeneity in crystallization usually limits the amount of starting material to a few hundred microlitres. This again limits the amount of sample per experiment (drop) to a few hundred nanolitres or even less.
After the preparation of the sample, the individual steps of a biological crystallization experiment include
(i) the preparation of stock solutions of pure precipitants and buffers;
(ii) the production of crystallization cocktails from stock solutions;
(iii) dispensing these cocktails onto appropriate crystallization devices;
(iv) combining small volumes of purified sample with cocktail solution in appropriate reaction chambers;
(v) storage and retrieval of the experiments in a controlled environment;
(vi) regular imaging of the individual experiments to monitor their progress;
(vii) the administration and user-friendly provision of all critical data pertaining to the experiments.
With the exception of the preparation of stock solutions, each of the steps has been automated for different crystallization methods with the required throughput (Fig. 1 ▶).
Figure 1.

Data and sample flow in automated crytallization. (1) Users submit their samples and enter instructions for their initial screening experiments, which are set up by a crystallization robot. (2) Experiments are set up according to the instructions (and the capabilities of the facility). (3) Crystallization plates are commited to an imager, which records the development of the individual droplets over time. (4) Users access the facility’s database and evaluate the outcome of the individual experiments. (5) Based on the results of previous rounds of experiments, follow-up/optimization experiments are designed.
The ability of biological crystals to diffract X-rays can vary greatly, not only between different experiments but also within the same droplet or reaction chamber. In addition to random fluctuations during the formation of crystal lattices, inappropriate cryoprotection prior to cooling crystals (a necessary means of extending the lifetime of a crystal in the high-energy synchrotron beam) is considered to be responsible for this phenomenon. Crystal harvesting and mounting is another source of interference. This is one of the reasons that automatic crystal handling prior to data collection has been attempted, since it can remove the inconsistent results of manual manipulation (Deller & Rupp, 2014 ▶).
Whether or not a biological crystal diffracts X-rays well enough to answer the scientific question at hand can usually only be determined by the diffraction experiment itself. It is therefore advisable and customary to first screen targets of interest broadly, in order to identify as many different crystallization conditions per construct as possible. This process has been dubbed initial screening. Subsequent refinement of initial crystallization conditions is almost invariably necessary and may or may not improve crystal quality. Therefore, the decision of whether to forward an initial crystallization hit for further refinement should be based on rational grounds, such as the disparity of the underlying parameters (e.g. the chemical composition of the precipitant cocktails or the sample variation) and not on appearance (such as the crystal size or morphology). Obviously, accepting more initial lead conditions into the optimization process increases the chances of eventual success. Automation supports this strategy by enabling scientists to be generous in their initial selection and cast a large net over early lead conditions. Nowadays, many optimization strategies and protocols are available to systematically explore the parameter space around initial lead conditions and to guide the experimenter.
2.1. Crystallization methods
The need for structural information stimulated the development of a number of techniques to achieve supersaturation, which is one of the prerequisites for crystallization (another prerequisite is effective crystal nucleation, as discussed below in §2.4.4). This evolution was based on the realisation that success in biological crystallization depended not only on the right combination of precipitants, but also on the path chosen to get there. The four most important and principally different crystallization methods are dialysis, batch, interface diffusion and vapour diffusion.
Dialysis allows modifications to the sample environment by exposing a dialysis bag containing the sample to different precipitant solutions. Solutes smaller than the selected cutoff value of the dialysis membrane can then diffuse in or out of the dialysis bag according to the prevailing concentration gradients. The disadvantages of this method are twofold. Firstly, it requires comparatively large amounts of sample. Secondly, dialysis is not easy to automate.
As a consequence, only batch, interface-diffusion and vapour-diffusion crystallization have been automated (see §2.3 and following sections).
2.2. Automation of crystallization
2.2.1. Production of crystallization cocktails
The selection of initial screening conditions undoubtedly influences the likelihood of identifying promising lead conditions. A systematic approach through the available parameter space – precipitants, pH and temperature, to name the most significant – is ruled out by a lack of time and sample. Instead, an intelligent ‘shotgun’ strategy is employed, which attempts to distribute initial conditions in the multivariate parameter space such that they are either randomly distributed or concentrated in regions that have been exceptionally productive in the past (sparse matrix). Today, crystallographers can choose from thousands of pre-formulated and commercially available crystallization solutions. They are sold in a variety of formats, ranging from several millilitres in test tubes to smaller volumes (∼1 ml) in SBS-format deep-well blocks (DWBs). Pre-filled crystallization plates are also available. Convenience comes at a price, which increases from simple tubes to pre-filled crystallization plates. Acquisition of cocktails in tubes or DWBs still requires transfer of the solutions to their final destination, the crystallization plate. This process is referred to as reformatting. Although technically simple and achievable by many robots, this step, like many that follow, harbours the grave risk of cross-contamination. The presence of even the smallest amount of a chemical in biological crystallization can make the difference between crystals or no crystals. The importance of thorough and rigorous washing of pipetting tips cannot be overemphasized. Rinsing is in fact the most time-consuming step in most automated routines. Contamination can be overcome through the use of disposable tips, which is a safe but expensive solution.
Alternatively, liquid-handling robots can produce crystallization cocktails from stock solutions in situ. The advantage of home-made crystallization cocktails rests mainly on the issue of reproducibility. The issue of cost is partially offset by investment in the necessary equipment and the need to produce the stock solutions. By preparing cocktails from home-made stock solutions, the final parameters of any individual cocktail become unambiguous. A good example of this point is the pH value of commercial crystallization solutions. Its definition varies from the pH of the 1 M buffer solution before the addition of precipitants and before dilution to the final concentration (in most cases 0.1 M) to the pH of the actual final cocktail. Frequently, the pH values of precipitants such as acetate or malonate, which are salts of weak acids (and thus are buffers in themselves), are not defined. Unless the composition tables specifically state the pH of the final solution, or each ingredient, users have to determine the pH values themselves if they want to reproduce the experiment in their home laboratories.
In the case of crystallization cocktails prepared from home-made stock solutions, the subsequent process of optimization becomes much more reproducible not only because the starting point is exactly defined, but also because the same stock solutions can be put to use in the production of optimization screens. Naturally, this approach hinges on the conscientious preparation and quality tracking of the stock solutions. Accurate and reliable preparation of stock solutions must include the definition of standard protocols, preferably also recording quality criteria such as pH, refraction index or conductivity. The final stock solutions have to be stored appropriately. Polyethylene glycol (PEG) solutions, for example, like other oxidation-prone compounds, need to be protected, for example through storage in a freezer to prevent their facile oxidation. Moreover, solutions that cannot be sterile-filtered must be protected against contamination by appropriate means, for example the addition of azide.
A liquid-handling robot suitable for the production of initial screens has to provide storage and access to a multitude (50–100) of stock solutions. During a pipetting operation where solutions are aspirated and dispensed, difficulties arise from the wide range of wetting capacities, surface tensions, viscosities and osmolarities of the stock solutions, which are usually highly concentrated. Only liquid-handling units with sophisticated hardware and software where a multitude of liquid-handling parameters can be adjusted, including aspiration and dispensing speeds, air gaps, liquid-level detection and tracking or tip touch off to remove small residual droplets, are compatible with the high demands on composition accuracies for crystallization cocktails. The initial alignment of such a unit is accordingly challenging and its operation and maintenance requires appropriate know-how. A proper determination of the coefficients of variation (CVs) at several points along the desired range of pipetting volumes, e.g. between 50 and 1000 µl for the preparation into DWB (or 1 and 50 µl for the preparation into crystallization plates), and across different liquid classes, such as water, ethanol, high salt or high PEG, are mandatory. These values should be of the order of less than 3% in the middle and upper volume range and should not exceed 10% at its very low end.
The high demands on the pipetting accuracies during the composition of a single deep-well block with 96 different crystallization cocktails entails preparation times of 1–3 h. This includes a thorough mixing of the final cocktails to prevent concentration gradients within individual cavities. Some liquid-handling units allow the preparation of several DWBs in parallel, which reduces the time per block accordingly. It is also worth mentioning that the ∼1.5 ml of cocktail per well of a typical SBS DWB is sufficient for about 25 individual 96-well crystallization plates in a standard vapour-diffusion experiment. Other liquid-handling robots offer the preparation of precipitant solutions straight into crystallization plates, bypassing intermediate steps (such as DWBs). In this scenario, the final volumes decrease by about an order of magnitude and the preparation times shorten to less than 10 min per single crystallization plate with 96 wells. The decision for large-scale or small-scale preparation of crystallization cocktails depends on the intended throughput capacities.
The time for the preparation of an optimization screen with, say, two precipitants and one buffer is faster in both cases because there is less washing and logistics required in between individual pipetting steps. DWBs containing optimization screens can usually only be used once or twice because new hits are spread randomly throughout the initial screens and the same condition is unlikely to crop up several times during the lifetime of the solutions. An elegant solution to this problem is different additive screens, where small amounts of the additive are combined with the mother liquor of an initial lead condition (see §2.4.6).
2.3. Setup of initial crystallization experiment
As already mentioned, the number of precipitant/buffer/temperature combinations and therefore the number of possible crystallization experiments is virtually unlimited. The number of construct variations of a given sample is comparatively small but multiplies the number of initial screening experiments with each construct to be screened. The efficiency with which different crystallization methods have been automated and sample the phase diagram differ, however.
2.3.1. Batch crystallization
People may feel that microbatch is hard work, but this need not be true. In batch crystallization, the protein and the precipitant solution are combined and left undisturbed. In the microbatch setup the experiments are performed under paraffin oil, which seals and immediately protects the droplets from evaporation. The same concentration of protein can be used in microbatch and vapour diffusion. Protein and precipitant can be dispensed simultaneously (without oil) before being covered automatically by oil (Shah et al., 2005 ▶). Other systems automatically dispense the aqueous solutions directly into the paraffin oil, with small amounts of sample being dispensed first, followed by the cocktail solution. In this case, centrifuging the plates may be needed to coalesce the drops and form the experimental droplets (Luft et al., 2003 ▶; Albeck et al., 2005 ▶). An advantage of this system is the possibility of accurately dispensing very small volumes (<100 nl) into a liquid (the paraffin oil). Therefore, this method lent itself to automation easily early on, because dispensing of small droplets on dry surfaces, as is necessary in vapour-diffusion experiments, could be bypassed.
It is widely believed that microbatch experiments sample less of the phase diagram of a target protein than vapour-diffusion experiments since vapour diffusion provides slow equilibration of the drop with the reservoir. This assumption is misleading for two reasons. Firstly, the most popular precipitant is PEG, and PEG drives equilibration very slowly (Luft & DeTitta, 1995 ▶). Luft and DeTitta showed that equilibration in high-PEG conditions is normally driven by small concentrations of salt that are also present, but this equilibration is relatively slow, so that crystallization often takes place in vapour diffusion before equilibration is complete. Secondly, microbatch can be modified by mixing the paraffin oil with silicone oil (D’Arcy et al., 1996 ▶). This speeds up evaporation from the drops, giving a scanning effect that is similar to vapour diffusion.
However, unlike in vapour diffusion there is no end-point, and the drops continue to evaporate until they reach equilibrium with the atmosphere and may dry out completely. Thus, the phase diagram can be fully scanned in a few weeks. This approach increases the number of hits, but the cost is that more salt crystals are found, so that a method of distinguishing salt crystals from protein crystals (such as UV or second-order nonlinear optical imaging) becomes essential.
2.3.2. Free-interface crystallization
The concept of crystallization by free-interface diffusion (FID) dates back to the 1970s (Salemme, 1972 ▶). Here, a capillary was filled with sample and one end was put into direct contact with precipitant solution. The system was then allowed to equilibrate by diffusion across the common free interface. The generation of concentration gradients along a liquid column exposes the sample to a wide range of precipitant concentrations and thereby very effectively samples the phase diagram. The equilibration kinetics can be very different, depending on the size and design of the interface area and the length of the sample column.
A very convenient version of this method is commercially available and is sold under the name Granada Crystallization Box (García-Ruiz et al., 2002 ▶). Because of its unique setup, this technique is usually referred to as counter-diffusion (CD). Since it cannot be automated, it will not be discussed further here.
2.3.3. Vapour-diffusion crystallization
Crystallization by vapour diffusion is by far the most widespread method. This prevalence is owing to a favourable combination of circumstances. Vapour diffusion combines the ability to use small sample volumes (>50 nl) with a broad (albeit smaller than FID- or CD-based) search of the phase diagram. The method has been completely automated for sitting-drop experiments. There are many established procedures to optimize initial lead conditions. Last but not least, crystals can be harvested from vapour-diffusion experiments for X-ray diffraction experiments comparatively easily. Crystal harvesting is a critical intervention in the process of collecting the best possible data from a crystal. Ironically, it is not automated at all, owing to its intrinsic delicacy. A previous article in this series addresses this topic (Deller & Rupp, 2014 ▶).
Based on empirical and theoretical considerations, it can be shown that 300–600 initial screening experiments per construct justify terminating further efforts on a given construct (Rupp & Wang, 2004 ▶). Rather than continuing to perform more experiments with the same sample, the same number of experiments performed on a different version of a sample (either a different construct or a ligand-bound or otherwise complexed form of the sample) is more likely to result in crystals. This rule, by the by, can be used as a guideline by the users to define the drop volume of their initial vapour-diffusion experiment setup. 200 µl of sample at a suitable concentration is sufficient for five SBS crystallization plates (480 conditions), for example using two 200 nl droplets of sample per condition or one 400 nl droplet of sample per experiment.
The question of the ideal drop size per experiment is still a matter of discussion. The old credo smaller is faster is better, however, is certainly not true (Newman et al., 2007 ▶). Since the equilibration rate is an important parameter in nucleation and therefore crystal growth (Vekilov & Vorontsova, 2014 ▶), users must strike a balance between experiment volume and the appropriate number of experiments (see above). Good crystallization robots should therefore allow this parameter to be varied within reasonable limits, say between 50 and 1000 nl per drop.
Particularly at smaller drop volumes, the setup of the crystallization droplets on a plate has to be either fast (<2 min) or appropriate measures have to be taken to prevent evaporation. Obviously this is not an issue when setting up batch crystallization experiments under oil (which was one of the main reasons for the implementation of microbatch in the early days of automation).
There are two kinds of crystallization robots available (for the setup of vapour-diffusion experiments). Both can set up drops by combining small volumes of sample and precipitant solution, but some can additionally transfer mother liquor from DWBs into crystallization plates, i.e. they include the reformatting step. In both cases the ratio of protein and sample can can be varied, e.g. 1 (sample):2 (reservoir). Robots that include the reformatting step are less flexible (because the volume of protein and reservoir solution in the droplet cannot be varied across the plate) but more convenient (because the entire crystallization plate can be produced on demand in one go). Robots that do not carry out the reformatting step can set up gradients, where each row or column can be treated differently. With this approach, however, the reformatting step has to be performed in an additional step either by hand or by another robot. Prefilled plates can then be stored, properly sealed, until they are needed. Naturally, this approach requires a higher level of organization and planning.
2.3.4. Lipidic cubic phase crystallization
Membrane proteins can be crystallized from a solution containing lipids and protein in the lipidic cubic phase (LCP). This approach can give crystallization of membrane proteins that could not otherwise be crystallized, and membrane proteins that do crystallize in normal aqueous experiments may give increased resolution when crystallized in LCP (Cherezov, 2011 ▶). LCP is a semisolid material similar to grease or toothpaste (it is not simply a liquid with high viscosity) and it cannot be dispensed by normal liquid-handling techniques. An important advantage of LCP crystallization is that the protein sample is immobilized within the LCP, so that very small quantities of protein can be dispensed into larger volumes of aqueous solution without the need for great accuracy and with very low protein wastage. LCP can be dispensed between specially made glass (or plastic) sheets or into standard sitting-drop crystallization plates. It is difficult to harvest crystals from all-glass crystallization plates, but the imaging of crystals is very good, and it is not necessary to use UV imaging. LCP has a very high refractive index, which makes it difficult to image crystals in other systems, since the rough surface of the semi-solid material refracts light strongly. Therefore, crystals in sitting-drop setups must be imaged using UV illumination (by detecting either fluorescence or absorption of UV by protein crystals in transmission mode). In all commercially available systems for this purpose, LCP is dispensed straight from a small-diameter syringe using a short hollow steel needle which is moved over the crystallization plate. (The small diameter allows high pressures to be generated that can move the semisolid material through the needle.) Some automatic dispensers such as the Gryphon LCP (Art Robbins) rapidly dispense LCP boluses to all 96 wells of a plate from a single syringe, then cover them with aqueous solutions using 96 separate needles. Others such as the NT8 (Formulatrix) and the Mosquito LCP (TTP Labtech) dispense LCP to one column of a plate at a time, then dispense aqueous solutions to these eight wells together. The first drop to be dispensed is exposed for about 10 s before being covered by aqueous solution. The Oryx LCP (Douglas Instruments) delivers one LCP bolus at a time, then covers it with aqueous solution within 1 s. This system can also dispense LCP to regular cover slides for hanging-drop experiments (giving improved viewing compared with sitting drops). The NT8 and the ProCrys Meso Plus (ZinsserAnalytic) systems have built-in humidifiers. Videos of all of these systems are available at http://www.youtube.com.
2.4. Automation of optimization experiments
Occasionally, well diffracting crystals can be harvested straight from an initial screen. In the majority of cases, however, adjustments to the concentrations of the macromolecule, precipitant or additives are needed to give diffracting crystals. Note, however, that random microseed matrix screening (rMMS) may considerably reduce and even avoid the need for optimization (D’Arcy et al., 2007 ▶). The method helps in three ways. (i) It increases the number of hits by generating crystals in wells that could support crystallization but where there is a nucleation problem. (ii) It increases the likelihood of growing crystals in the metastable zone of the crystallization phase diagram. The best-diffracting crystals often grow in this region. (iii) It allows the crystallizer to control the number of crystals per drop by diluting the seed stock (Shaw Stewart et al., 2011 ▶).
A very simple method of improving the quality of crystals without optimization is to repeat the original hit condition 10–20 times. Small pipetting errors and variations in crystal nucleation often give improved crystals in some of the drops (Newman et al., 2007 ▶)
2.4.1. Liquid-handling hardware
A large number of liquid-handling approaches have been used for automatic crystal optimization. Generally, a sophisticated liquid-handling system is used to combine and mix the reservoir solutions and a second system sets up the sample droplets (i.e. two separate ‘robots’ are used). An exception to this is the Oryx8 robot from Douglas Instruments, which routinely sets up droplets but can also mix reservoir solutions for optimization experiments (Shah et al., 2005 ▶). Other systems use special proprietary hardware for dispensing solutions, such as the Formulator by Formulatrix, which uses patented ‘chips’ with 96 microfluidic valve clusters that can accurately dispense viscous and nonviscous liquids, and the Alchemist (Rigaku Automation Inc., USA), which uses ‘Birdfeeder’ technology that eliminates cross-contamination.
Liquid-handling systems designed for protein crystallization such as the Oryx, the Formulator, the Alchemist, the Dragonfly from TTP Labtech and the Scorpion Screen Builder from Art Robbins Instruments come with dedicated software applications to generate the instructions for the setup of crystallization plates with intricate composition patterns.
General-purpose liquid-handling stations such as the MICROLAB STAR line from Hamilton and the Freedom EVO from Tecan work by aspirating and dispensing solutions from and to the experimental deck. Sophisticated hardware is available, but it may be difficult or expensive to obtain versatile software for biological crystallization.
2.4.2. The optimum sequence of experiments
Traditionally, macromolecular crystallization was carried out using random screening followed by simple optimization experiments, typically two-dimensional grids in which one parameter was varied against another (see below). Today, many high-throughput laboratories still use random screens followed immediately by two-dimensional grids. Only if these techniques do not yield diffracting crystals will they try other techniques such as random microseed matrix screening (rMMS) or ‘targeted screens’ (see below). It would be more logical to use rMMS before two-dimensional grids because it often generates new hits. Moreover, rMMS is very easy to set up because the original screening solutions can be reused (this gives a control experiment since crystals should reappear in the conditions where they grew in the first round). Similarly, targeted screens should be set up before two-dimensional grids since they can find the optimum combination of ingredients, which can subsequently be further optimized if necessary. Using rMMS and targeted screens early in a project can help researchers to keep an open mind and to switch to new conditions if progress with the first condition chosen is slow.
In small laboratories where most optimization experiments must be set up by hand, we suggest the following sequence: (i) random screening, (ii) rMMS, (iii) microseed dilution experiments and (iv) two-dimensional grids. High-throughput laboratories with extensive robotics that are tackling difficult projects (such as the determination of the structures of mammalian proteins) might consider a more powerful sequence: (i) random screening, (ii) rMMS, (iii) targeted screens, (iv) microseed dilution experiments and (v) multivariate optimization. All steps after the first can include microseeding to increase the likelihood of crystallization in the metastable zone. Unfortunately, very few high-throughput studies of rMMS have been published so the statistical effectiveness of the method for all classes of protein is not known. Direct comparisons of rMMS and other optimization techniques for statistically significant numbers of proteins would be very helpful to the field.
2.4.3. Grids with two-dimensional gradients
The conventional approach to optimization is to make a small grid of wells (often a 6 × 4 block) in which one parameter, such as precipitant concentration, is varied against another, such as pH (Weber, 1990 ▶). Grids where precipitant concentration is varied against the concentration of the macromolecule or an additive are also popular. Many liquid-handling systems have software and hardware to construct such grids. A simple and popular approach is to write a script defining a sequence of commands for the robot and to import a text file into the script that contains an array of numbers. The numbers correspond to the volumes of reagents to be dispensed, and users can define new experiments by generating new text files, for example with a spreadsheet. Other liquid-handling stations have special software to generate the grids directly, which reduces the need to train the user. A third approach uses fixed scripts that use solution labels such as ‘A, B, C, D’ etc. At the time of setting up the experiment the user makes up solutions that give the desired concentrations for a hit that needs to be optimized. For example, solutions A, B, C and D could be placed at the four corners of a grid, and the intermediate wells would be filled by interpolation. Here, the script and the liquid-handling parameters stay the same, but the solutions vary to achieve the desired crystallization conditions.
Grids have the advantage that they are easy to understand and set up, but they are relatively inefficient, wasting samples and materials. This is because the points are relatively close to each other and may be confined to one surface within the multidimensional crystallization space that needs to be explored.
2.4.4. Random microseed matrix screening
The random microseed matrix screening (rMMS) method (D’Arcy et al., 2007 ▶) has the potential to roughly double productivity (see below) but it is still not used routinely in the majority of laboratories. It involves adding crushed seed crystals to random crystallization screens. This allows crystal nucleation in conditions where crystal growth would not otherwise occur, and its effectiveness suggests that many conditions in a typical screen are capable of supporting crystallization but crystallization does not occur because there is a nucleation problem. Note also that when the typical volumes are used (300 nl protein sample with 200 nl reservoir solution and 100 nl seed stock) roughly one-third of the precipitant comes from the seed stock (St John et al., 2008 ▶; Fig. 3).
The method should ideally be used before traditional optimization methods in order to make available as many crystallization leads as possible. Very few high-throughput laboratories outside industry use the method as soon as the first batch of crystals stop growing, which is the approach recommended by D’Arcy and coworkers. Obmolova and coworkers used the method routinely in a small industrial laboratory; of 70 structures produced by the group in roughly three years, 38 benefited from the rMMS method, including 80% of the structures of complexes that were produced (Obmolova et al., 2010 ▶). Not all robots can perform the method. It works well with the Mosquito, the Oryx, the NT8 and the Crystal Gryphon LCP, all of which use a ‘contact’ dispensing method where the tip touches the plate when dispensing seed stock.
The rMMS method helps to generate diffracting crystals in three ways. (i) It increases the number of hits by generating crystals in wells that could support crystallization but where nucleation is a problem. (ii) It increases the likelihood of growing crystals in the metastable zone of the crystallization phase diagram. The best-diffracting crystals often grow in this region. (iii) It allows the crystallizer to control the number of crystals per drop by diluting the seed stock (Shaw Stewart et al., 2011 ▶). The method is reviewed in a forthcoming article in this series on seeding by D’Arcy and coworkers.
2.4.5. Combining several hits: ‘targeted’ screens and ‘combinatorial’ experiments
An effective optimization strategy that is often overlooked is recombining the ingredients from several hits. If an initial screen picks up several hits, a new random screen can be made that uses only the set of ingredients that were present in the hits. For example, imagine a set of hits that contain a variety of precipitants, buffers, salts and other additives. The best diffraction may come from mixing, say, the precipitant from hit 1 with the buffer from hit 2. Some liquid-handling systems have software and hardware for making such ‘targeted screens’. For example, Obmolova and coworkers found four hit conditions for their target Fab fragment called H2L6 (using microseeding; Obmolova et al., 2010 ▶). Using an automatic liquid-handling system, they made a random screen comprising of the most promising salt/PEG 3350 combinations (24 conditions). Two conditions, both containing the ammonium salts of organic acids, gave X-ray-quality crystals.
Similar results can be obtained by a ‘combinatorial’ approach (Till et al., 2013 ▶) where precipitants are arranged in the columns of a plate while buffers and additives are added to the rows (in the drops only). Every combination of precipitant and additive used appears somewhere on the plate.
2.4.6. Liquid multivariate experimental designs
Crystallization experiments can be mapped into a multi-dimensional space. This space has as many dimensions as the number of ingredients in all of the hits to be optimized. In addition, all (macromolecular) crystallization experiments have a macromolecule concentration, a temperature, a pH, a volume and a plate geometry. All of these parameters need to be explored. Textbooks of experimental design recommend that such multidimensional spaces be searched with multivariate designs rather than with simple two-dimensional grids (Atkinson et al., 2007 ▶). The key point is that all of the important experimental parameters should be varied in each experimental run (whereas only two parameters, such as precipitant and pH, are varied in simple two-dimensional grids). Imagine the following example: a hit is found in a condition that could be optimized by, say, decreasing the precipitant concentration, increasing the salt concentration, eliminating an additive and decreasing the pH. You can imagine that it may be hard to find that optimum. Moreover, crystallization variables typically ‘interact’ with each other, that is to say adjustment of one variable may affect the optimum levels of the others. The resulting confusion in interpreting results can be avoided by appropriate experimental design.
The pitfalls of poor experimental design in protein crystallization and their resolution have been reviewed by Shaw Stewart & Baldock (1999 ▶). Several multivariate approaches can be used, ranging from the most rigorous to the informal. Carter used a minimum integrated variance design matrix with four factors and 20 wells to crystallize bacterial tryptophanyl-tRNA synthetase (Carter & Yin, 1994 ▶). Well known formal designs found in textbooks include the central composite and the Box–Behnken designs. The central composite, shown in Fig. 2 ▶, is regarded as the most efficient general-purpose design (Box & Hunter, 1957 ▶). Numerous designs with similar properties exist that make use of other geometrical points (Shaw Stewart & Baldock, 1999 ▶). Douglas Instruments’ XSTEP software can set up multivariate designs with up to seven dimensions, which can be used for vapour-diffusion, microbatch-under-oil and lipidic cubic phase crystallization (freely available from the company on request).
Figure 2.
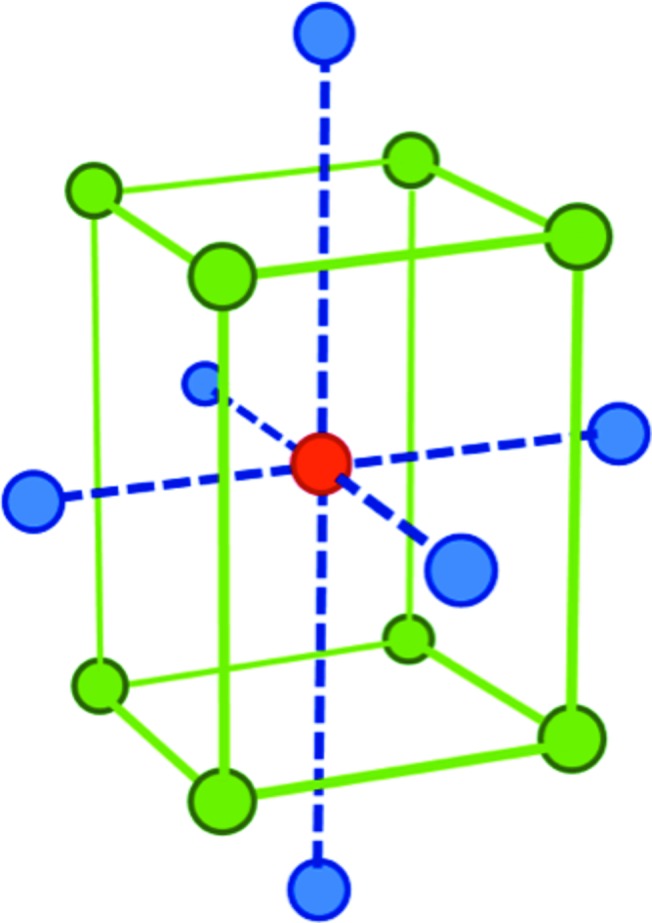
The central composite experimental design (Box & Hunter, 1957 ▶) shown in three dimensions. This is one of several well known multivariate designs that are recommended for optimizing processes that have several important experimental parameters. For example, protein concentration, temperature, pH, precipitant concentration, additive concentration etc. need to be optimized in protein crystallization experiments. Ideally, all of these parameters should be varied in each experimental run, and the central composite efficiently achieves this goal. This can find the best direction to move in, since several parameters may need to be adjusted simultaneously. The design comprises one or more centre points (red), which are the crystallizer’s ‘best guess’ for the best crystallization conditions (for example, a hit from a screening experiment). These points are surrounded by a set of ‘factorial’ points (green) and ‘axial’ points (blue). The details of the experiment are not important: the important principle is that the points surround the central point reasonably evenly in the multidimensional space. A three-dimensional version is shown in Fig. 2 ▶, but higher numbers of dimensions can be used. For example, six-dimensional central composites have been used in crystallization (Shaw Stewart & Baldock, 1999 ▶). Less formal designs that occupy several dimensions in the crystallization hyperspace can achieve similar results (see text for examples), although they may be more wasteful of time and materials.
Many groups use less formal bespoke designs that also occupy several dimensions of the crystallization space. For example, the Structural Genomics Consortium (Oxford, England) uses a standard approach for conditions that have three components (e.g. PEG, buffer and salt). A 96-well plate is divided into quadrants, with zero, low, medium and high concentrations of salt. For example, if a hit contained 0.2 M NaCl, the four quadrants would contain 0, 0.1, 0.2 and 0.3 M NaCl. Each quadrant contains a two-dimensional grid, with precipitant varying by ±5% and pH varying by ±0.3 pH units. The drop size is usually doubled when using a optimization screen and three drop ratios are investigated, 150 + 150 nl (1:1), 100 + 200 nl (1:2) and 200 + 100 nl (2:1), in a three-subwell sitting-drop plate.
Where the standard approach is unsuccessful, more unusual approaches focusing on screening around the hit condition might be recommended. These may include seeding or screening for additives or small molecules that might enhance crystallization or produce new crystal forms. The Collaborative Crystallization Center (C3) in Melbourne, Australia uses several different multidimensional techniques for optimization. These include both random screens and incomplete factorial designs around one or more hits, additive screens (see below) and microseeding combined with fine screening or additives and additive screens.
Like more formal designs, bespoke designs that occupy three or more dimensions can quickly find the best direction to move in, although they are likely to be less efficient in terms of sample and materials than more formal multivariate designs. Another successful approach is the use of additive experiments, where a hit condition that needs to be optimized is mixed with a random screen (or possibly a special ‘additive’ screen), giving for example 96 points that are close to the hit condition but distributed in many dimensions, as shown schematically in Fig. 3 ▶.
Figure 3.

A schematic representation of screening and additive experiments (including rMMS). (a) An initial screen can be depicted as a cloud of points in the multidimensional crystallization space (represented here as points in three-dimensional space). (b) If a hit is obtained this can be used as the centre point of an optimization experiment (red circle) by adding small quantities of solutions from a random screen to the initial hit condition. This gives a smaller cloud of points close to the hit, increasing the chance of obtaining diffracting crystals (Shaw Stewart et al., 2011 ▶).
2.5. Storage and retrieval
An efficient, computer-based system to store, handle and administrate crystallization experiments (i.e. the crystallization plates or chips) fulfils two functions. Firstly, the system keeps track of individual experiments, which can be retrieved on demand. Secondly, large-scale HT-X units are equipped with automated imaging and the storage and retrieval system commits the experiments to the imager for optical recording on a regular schedule. The former establishes reliable record keeping and supports data confidentiality, which is important when there are large numbers of experiments from many different users or user groups. The second allows the evolution in time of crystallization experiments to be studied (see below). The association of image, sample construct and crystallization composition can be preserved with a very high degree of certainty, and because all data are stored electronically, users may access their data at any convenient time and place via the internet. More advanced systems also transmit information concerning, for example, temperature control or power or computational failures to the system administrators. Additionally, such systems simplify the interpretation of experiments by the user by providing a consistent and stable environment with respect to temperature and exposure to vibration and acceleration.
2.6. Automated imaging
High-throughput facilities are capable of generating tens of thousands of individual crystallization experiments per day. The process of screening and evaluating these experiments manually under a microscope is laborious and time-consuming. Additionally, it is mandatory to inspect the crystallization experiments repeatedly and at regular time points in order to record overall trends (such as the fraction of experiments with precipitated sample or the onset of crystallogenesis) and to register transitory crystal formation. Because crystal formation is an intricate and initially stochastic process, the total time period of observation is long: up to six months or more. To reduce the need for manual inspection, automated crystallization facilities regularly image each and every experiment and archive the results. This guarantees a record of the evolution of the experiment over time, which is electronically stored and remotely accessible. Users may now access this information from any computer at their convenience. Perusal of the snapshots taken as the experiments developed through time (the drop history) can be very informative. The appearance of crystals within 24 h of setup often indicates the presence of crystals of nonbiological material, which form more readily than protein crystals. Air bubbles (which often shrink during the experiment) and accidentally included small dust particles in the initial setup may appear to be crystalline in nature.
Since initial screening serves the purpose of identifying as many conditions as possible that are capable of supporting crystallogenesis (however far away that may be from those conditions producing single, large, untwinned and well diffracting crystals), image quality is of the utmost importance. The highest optical resolution of an imager is obtained when the numerical aperture of the objective is increased. However, this reduces the depth of field that is in focus. ‘Slicing’ refers to the technique of taking several pictures per drop along the perpendicular of the image plane at intervals that correspond to the depth of field. The best image is then obtained as a composite image by selecting the pixels that are most highly focused from the different slices of each series.
In addition to high resolution and a finely tuned focusing mechanism, the imaging has to be fast. A typical SBS-format plate contains 96 wells and every plate should be imaged several times in the first two weeks (say four to six times), when most changes occur owing to equilibration or nucleation. Afterwards the imaging frequency can be reduced to about once per week for 4–6 weeks. The total residence time of a plate in the imager depends on the capacity of the facility. A period of less than 6–8 weeks, however, simply returns the duty of experiment surveillance to the user. It is therefore not feasible at facilities that offer their services to remote users.
Nonetheless, there are hundreds to thousands of images per individual project to inspect. As a consequence, much effort has been put into attempts to develop and implement automatic scoring and classification algorithms (Zuk & Ward, 1991 ▶; Spraggon et al., 2002 ▶; Cumbaa et al., 2003 ▶; Bern et al., 2004 ▶; Watts et al., 2008 ▶). These approaches use many different mathematical models. They also use different classification schemes, which consist of subjective descriptions such as clear drop, denatured precipitate, amorphous material, micro-crystals, phase separation, single crystal, crystal cluster etc. One fundamental problem for automatic image classification in biological crystallization is the low agreement rate about the assessments, even among trained and experienced crystallographers (Watts et al., 2008 ▶). Since the development of these algorithms depends on properly defined training sets of example images, it is not surprising that no reliable solution that would fit any imager or platform has emerged.
The distinction between biological and nonbiological crystals (i.e. crystals of the various precipitants, especially salts) is difficult. UV imaging is one means of doing so and it is now a well developed and established method. The source of fluorescence is no longer confined to the protein (which relied on the comparatively rare and occasionally absent amino acid tryptophan), but may instead be provided by dyes (Groves et al., 2007 ▶; Dierks et al., 2010 ▶; Sigdel et al., 2013 ▶). Particularly helpful are images taken with UV light and with visible light close in time. By comparing both images, it is usually straightforward to distinguish crystals of precipitant juxtaposed with crystals of biological material. Another advantage of UV imaging is the vastly increased contrast between crystal and background (Watts et al., 2008 ▶). Because the refractive index of biological crystals is very close to the refractive index of the surrounding liquid, it is easy to overlook them, particularly if they are small or hidden by precipitate. The power of this method has also been demonstrated in automated image recognition. While this requires a difficult and lengthy training process with images taken in visible light, UV images are much easier to classify (Watts et al., 2010 ▶).
2.7. Service by and access to high-throughput crystallization facilities
A fully automated high-throughput crystallization facility is still expensive to set up and maintain. They are therefore typically shared by a consortium and/or publicly funded and hence open to the national or international user community. Productive access for remote user groups (even across a campus) requires efficient and fast data transfer between the facility and the users, along with a straightforward and user-friendly GUI. The GUI should allow users to design their initial or follow-up experiments taking full advantage of the platform’s capabilities, as well as to inspect the results of their crystallization experiments (Fig. 1 ▶).
Typical parameters of initial screening experiments to be set up by the users are as follows.
(i) Crystallization method.
(ii) Number and choice of initial screens.
(iii) Individual experiment volume and incubation temperature.
(iv) Ratio of sample to crystallization cocktail.
Not every facility offers different crystallization methods, but users can usually find one that either offers microbatch crystallization, as at the Hauptman–Woodward Medical Institute in Buffalo, New York, or vapour diffusion, as at the Oxford Protein Production Facility (OPPF) in Oxford or at the EMBL in Hamburg or Heidelberg. Often, these facilities are even publicly supported and can offer their services inexpensively or even free of charge to the user community. The scope of services offered by such facilities may vary depending on their size and mission. It is usually easy to obtain this information either on a website or from the scientist in charge and to choose a facility that best fits the individual demands of the scientist or project.
2.8. LIMS
Without the parallel improvement of all steps leading from a gene to a structure, the amazing increase in structural information over the past 15 years would not have taken place. By the time crystallization experiments commence, a plethora of experiments have already been performed, commonly by several scientists or collaborators from different laboratories. As presented in the previous sections, crystallization experiments generate large amounts of data themselves. The number of experiments per project may exceed one thousand. Furthermore, because of the wide spread of quality among crystals from different setups or even from the same experiment, the number of crystals exposed to X-ray radiation before a structure has been solved averages about 100 (Elsliger et al., 2010 ▶; http://www.jcsg.org). High-throughput crystallization facilities therefore have to deal with a large stream of incoming data, link it to individual experiments and pass this information initially on to (remote) users and eventually to the point of data collection, in most cases a synchrotron facility.
Obviously, data recording and tracking with the traditional laboratory notebook is inadequate and inefficient. Instead, electronic and internet-based data-management systems, dubbed laboratory information-management systems (LIMS), have found their way into modern-day structural biology. Attempts to define a common LIMS, or even a common database format, have failed and today a diverse range of commercially available and academically funded LIMS are in use: LISA (Haebel et al., 2001 ▶), XTRACK (Harris & Jones, 2002 ▶), SESAME (Zolnai et al., 2003 ▶), CLIMS (Fulton et al., 2004 ▶), HALX (Prilusky, Oueillet et al., 2005 ▶), MOLE (Morris et al., 2005 ▶) and PiMS (Morris et al., 2011 ▶; Savitsky et al., 2011 ▶). Small and medium-sized laboratories often use only the LIMS that is provided with their imagers, but large high-throughput laboratories need to develop their own laboratory-specific LIMS, as reflected by the long list of LIMS above.
Integration of the crystallization process into a LIMS presents the following challenges.
(i) Crystallization experiments contain data from different pieces of equipment, such as pipetting robots, crystallization robots and imagers. If all of the instruments are from the same manufacturer, there is usually a common database schema from which data can be extracted. If they are not, data retrieval from and data exchange between the individual pieces of equipment accordingly becomes more difficult. In some instances manufacturers are reluctant to permit access to the underlying DB management system out of proprietary or data-integrity concerns.
(ii) The advantages of an automated imaging system have been stated in §2.6. The high-resolution images of crystallization drops require fast and high-bandwidth connections from the crystallization platform over the intranet and internet to the end user. One solution is that users are routinely provided with a medium-resolution image and request high-resolution data only when they consider it to be appropriate.
(iii) Advanced high-throughput crystallization facilities also offer the preparation of follow-up experiments, ideally to the point where single, reasonably sized crystals (>10 µm in each direction) grow reproducibly. This ability allows users to test all, or at least a significant portion of, initial lead conditions for their potential to produce single crystals. Ideally, several optimization protocols are provided in order to systematically explore the surrounding parameter space. The corresponding protocols are then directed towards different parts of the platform. Fine grids, for example, would be prepared by a pipetting robot, while microseeding would be executed by a crystallization robot (see §2.4.1).
(iv) When data are collected from crystals at an X-ray source, usually a synchrotron, information on the crystal, such as the underlying sequence, the presence of ligands or post-translational modifications, should be available. Because most synchrotrons offer robotics to automatically load and align premounted crystals, complete data sets may be generated within minutes. A seamless connection between crystallization and synchrotron facility to transmit relevant information for data processing at the synchrotron facility will become necessary.
(v) Last not least are the results of crystallization experiments, which, as long as they have been properly annotated, are an incredibly valuable source not only to guide the optimization procedure of the project at hand but also to discover and unravel the rules that govern biological crystallization per se. The design and implementation of a system that captures this information and is subsequently capable of being ‘mined’ to extract these trends is a task that remains essentially unsolved (Gilliland et al., 2002 ▶; Peat et al., 2002 ▶).
3. Outlook
Crystallization remains the only gateway to high-precision three-dimensional structural information of biological macromolecules at atomic resolution. At the same time, crystallization continues to be the bottleneck in the process leading from a gene to a three-dimensional model. As McPherson & Gavira (2014 ▶) point out in their review, ‘there is no comprehensive theory, or even a very good base of fundamental data, to guide [a crystallographer’s] efforts’ to obtain well diffracting crystals. Crystal growth is still largely empirical in nature.
Automation of biological crystallization, the response to overcoming the bottleneck, has helped to improve the process of crystallization in two ways: (i) a dramatic reduction of the costs per experiment and (ii) the ability to test many target variations in small volumes and hundreds of experiments at high speed. The latter enables a much more comprehensive search of crystallization space than could be performed manually. It is therefore increasingly difficult for scientists without access to automated crystallization to succeed with the ambitious projects of modern-day crystallography.
One danger of automation is a thoughtless reliance on the numbers game along these lines: put enough material through the pipeline and eventually crystals will appear. No amount of automation, however, will overcome the truth in ‘garbage in, garbage out’. As ever, diligent and meticulous characterization of the sample before crystallization begins and the thorough and attentive analysis of all of the results (the images) are obligatory (Meijers & Mueller-Dieckmann, 2011 ▶). After all, the sample is the single most important ingredient in any crystallization experiment and it cannot be improved without a rigorous understanding of its properties.
Another downside of automation is the enormous flood of data (in the form of images of the individual experiments) which is being generated. Reliable image-recognition software for the automatic classification of crystallization experiments is far from being mature or generally available. Where it exists, it has required considerable development efforts with the specific local circumstances in mind. Therefore, in the majority of cases this critical step has to be executed manually and relies on the experience of the individual researcher. It is difficult to foresee to what extent this will change in the future.
Initial screening experiments attempt to map uncharted territory (the crystallization space with its many dimensions) and hence are a journey into the unknown. With this in mind, the significance of properly recording the outcome of each performed experiment becomes obvious. It is just as important to know where crystallogenesis has occurred as it is to know which conditions are unfavourable for crystal formation. In contrast to initial screening experiments, optimization of the conditions is a rational and well characterized process.
Combining the processes of experiment classification and optimization in automation has great potential. The hardware and software tools for generating new crystallization cocktails from stock solutions exist. Studies of how best to optimize crystallization conditions based on data on the benefit or harm of precipitants, pH and temperature have begun (Ménétrey et al., 2007 ▶), but more work needs to be performed along these lines. Hence, a feedback loop of experimental data (based on automatic classification or on manual input) to liquid handling and/or crystallization robots (see above) has the potential of driving an iterative optimization process autonomously until a defined end point (such as single crystals of >10 µm in each direction) has been reached. The next step of characterizing biological crystals in situ, i.e. without the need of manipulation, in an X-ray source has already been addressed (e.g. the In situ-1 crystallization plate from MitTeGen; Soliman et al., 2011 ▶). The future result of automatic crystallization may therefore be a list with averaged quality standards (e.g. resolution, mosaicity, isomorphism or twinning ratio) for different target constructs or target formulations from a variety of optimized crystallization conditions.
References
- Albeck, S., Burstein, Y., Dym, O., Jacobovitch, Y., Levi, N., Meged, R., Michael, Y., Peleg, Y., Prilusky, J., Schreiber, G., Silman, I., Unger, T. & Sussman, J. L. (2005). Three-dimensional structure determination of proteins related to human health in their functional context at The Israel Structural Proteomics Center (ISPC). Acta Cryst. D61, 1364–1372. [DOI] [PubMed]
- Atkinson, A. C., Donev, A. N. & Tobias, R. D. (2007). Optimum Experimental Designs, With SAS Oxford University Press.
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Bern, M., Goldberg, D., Stevens, R. C. & Kuhn, P. (2004). Automatic classification of protein crystallization images using a curve-tracking algorithm. J. Appl. Cryst. 37, 279–287.
- Box, G. E. P. & Hunter, J. S. (1957). Multi-factor experimental designs for exploring response surfaces. Ann. Math. Statist. 28, 195–241.
- Carter, C. W. & Yin, Y. (1994). Quantitative analysis in the characterization and optimization of protein crystal growth. Acta Cryst. D50, 572–590. [DOI] [PubMed]
- Cherezov, V. (2011). Lipidic cubic phase technologies for membrane protein structural studies. Curr. Opin. Struct. Biol. 21, 559–566. [DOI] [PMC free article] [PubMed]
- Cumbaa, C. A., Lauricella, A., Fehrman, N., Veatch, C., Collins, R., Luft, J. R., DeTitta, G. & Jurisica, I. (2003). Automatic classification of sub-microlitre protein-crystallization trials in 1536-well plates. Acta Cryst. D59, 1619–1627. [DOI] [PubMed]
- D’Arcy, A., Elmore, C., Stihle, M. & Johnston, J. E. (1996). A novel approach to crystallising proteins under oil. J. Cryst. Growth, 168, 175–180.
- D’Arcy, A., Villard, F. & Marsh, M. (2007). An automated microseed matrix-screening method for protein crystallization. Acta Cryst. D63, 550–554. [DOI] [PubMed]
- Deller, M. C. & Rupp, B. (2014). Approaches to automated protein crystal harvesting. Acta Cryst. F70, 133–155. [DOI] [PMC free article] [PubMed]
- Dierks, K., Meyer, A., Oberthür, D., Rapp, G., Einspahr, H. & Betzel, C. (2010). Efficient UV detection of protein crystals enabled by fluorescence excitation at wavelengths longer than 300 nm. Acta Cryst. F66, 478–484. [DOI] [PMC free article] [PubMed]
- Elsliger, M.-A., Deacon, A. M., Godzik, A., Lesley, S. A., Wooley, J., Wüthrich, K. & &Wilson, I. A. (2010). The JCSG high-throughput structural biology pipeline. Acta Cryst. F66, 1137–1142. [DOI] [PMC free article] [PubMed]
- Fulton, K. F., Ervine, S., Faux, N., Forster, R., Jodun, R. A., Ly, W., Robilliard, L., Sonsini, J., Whelan, D., Whisstock, J. C. & Buckle, A. M. (2004). CLIMS: Crystallography Laboratory Information Management System. Acta Cryst. D60, 1691–1693. [DOI] [PubMed]
- García-Ruiz, J. M., González-Ramírez, L. A., Gavira, J. A. & Otálora, F. (2002). Granada Crystallisation Box: a new device for protein crystallisation by counter-diffusion techniques. Acta Cryst. D58, 1638–1642. [DOI] [PubMed]
- Gilliland, G. L., Tung, M. & Ladner, J. E. (2002). The biological macromolecule crystallization database: crystallization procedures and strategies. Acta Cryst. D58, 916–920. [DOI] [PubMed]
- Groves, M. R., Müller, I. B., Kreplin, X. & Müller-Dieckmann, J. (2007). A method for the general identification of protein crystals in crystallization experiments using a noncovalent fluorescent dye. Acta Cryst. D63, 526–535. [DOI] [PubMed]
- Haebel, P. W., Arcus, V. L., Baker, E. N. & Metcalf, P. (2001). LISA: an intranet-based flexible database for protein crystallography project management. Acta Cryst. D57, 1341–1343. [DOI] [PubMed]
- Harris, M. & Jones, T. A. (2002). Xtrack – a web-based crystallographic notebook. Acta Cryst. D58, 1889–1891. [DOI] [PubMed]
- Heinemann, U., Frevert, J., Hofmann, K.-P., Illing, G., Maurer, C., Oschkinat, H. & Saenger, W. (2000). An integrated approach to structural genomics. Prog. Biophys. Mol. Biol. 73, 347–362. [DOI] [PubMed]
- Hosfield, D., Palan, J., Hilgers, M., Scheibe, D., McRee, D. E. & Stevens, R. C. (2003). A fully integrated protein crystallization platform for small-molecule drug discovery. J. Struct. Biol. 142, 207–217. [DOI] [PubMed]
- Hünefeld, F. L. (1840). Der Chemismus in der thierischen Organisation, p.160. Leipzig University, Germany.
- Kendrew, J. C., Bodo, G., Dintzis, H. M., Parrish, R. G., Wyckhoff, H. & Phillips, D. C. (1958). A three dimensional model of the myoglobin molecule obtained by X-ray analysis. Nature (London), 199, 662–666. [DOI] [PubMed]
- Kurgan, L., Razib, A. A., Aghakhani, S., Dick, S., Mizianty, M. & Jahandideh, S. (2009). CRYSTALP2: sequence-based protein crystallization propensity prediction. BMC Struct. Biol. 9, 50. [DOI] [PMC free article] [PubMed]
- Luft, J. R., Collins, R. J., Fehrman, N. A., Lauricella, A. M., Veatch, C. K. & DeTitta, G. T. (2003). A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J. Struct. Biol. 142, 170–179. [DOI] [PubMed]
- Luft, J. R. & DeTitta, G.T. (1995). Chaperone Salts, Polyethylene Glycol and Rates of Equilibration in Vapor Diffusion Crystallization. Acta Cryst. D51, 780–785. [DOI] [PubMed]
- McPherson, A., Cudney, R. & Patel, S. (2003). The Crystallization of Proteins, Nucleic Acids and Viruses for X-ray Diffraction Analysis. Biopolymers, Vol. 8, edited by S. R. Fahnenstock & A. Steinbüchel, pp. 427–468. Berlin: Wiley-VCH.
- McPherson, A. & Gavira, J. A. (2014). Introduction to protein crystallization. Acta Cryst. F70, 2–20. [DOI] [PMC free article] [PubMed]
- Meijers, R. & Mueller-Dieckmann, J. (2011). Advances in High-Throughput Crystallization. eLS. Chichester John Wiley & Sons. 10.1002/9780470015902.a0023171.
- Ménétrey, J., Perderiset, M., Cicolari, J., Houdusse, A. & Stura, E. A. (2007). Improving Diffraction from 3 to 2 Å for a Complex between a Small GTPase and Its Effector by Analysis of Crystal Contacts and Use of Reverse Screening. Cryst. Growth Des. 7, 2140–2146.
- Morris, C. et al. (2011). The Protein Information Management System (PiMS): a generic tool for any structural biology research laboratory. Acta Cryst. D67, 249–260. [DOI] [PMC free article] [PubMed]
- Morris, C., Wood, P., Griffiths, S. L., Wilson, K. S. & Ashton, A. W. (2005). MOLE: a data management application based on a protein production data model. Proteins, 58, 285–289. [DOI] [PubMed]
- Mueller, U., Darowski, N., Fuchs, M. R., Förster, R., Hellmig, M., Paithankar, K. S., Pühringer, S., Steffien, M., Zocher, G. & Weiss, M. S. (2012). Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Rad. 19, 442–449. [DOI] [PMC free article] [PubMed]
- Mueller-Dieckmann, J. (2006). The open-access high-throughput crystallization facility at EMBL Hamburg. Acta Cryst. D62, 1446–1452. [DOI] [PubMed]
- Newman, J., Xu, J. & Willis, M. C. (2007). Initial evaluations of the reproducibility of vapor-diffusion crystallization. Acta Cryst. D63, 826–832. [DOI] [PubMed]
- Obmolova, G., Malia, T. J., Teplyakov, A., Sweet, R. & Gilliland, G. L. (2010). Promoting crystallization of antibody–antigen complexes via microseed matrix screening. Acta Cryst. D66, 927–933. [DOI] [PMC free article] [PubMed]
- Peat, T., de La Fortelle, E., Culpepper, J. & Newman, J. (2002). From information management to protein annotation: preparing protein structures for drug discovery. Acta Cryst. D58, 1968–1970. [DOI] [PubMed]
- Prilusky, J., Felder, C. E., Zeev-Ben-Mordehai, T., Rydberg, E. H., Man, O., Beckmann, J. S. & Sussman, J. L. (2005). FoldIndex©: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics, 21, 3435–3438. [DOI] [PubMed]
- Prilusky, J., Oueillet, E., Ulryck, N., Pajon, A., Bernauer, J., Krimm, I., Quevillon-Cheruel, S., Leulliot, N., Graille, M., Liger, D., Trésaugues, L., Sussman, J. L., Janin, J., van Tilbeurgh, H. & Poupon, A. (2005). HalX: an open-source LIMS (Laboratory Information Management System) for small- to large-scale laboratories. Acta Cryst. D61, 671–678. [DOI] [PubMed]
- Rupp, B. & Wang, J. (2004). Predictive models for protein crystallization. Methods, 34, 390–407. [DOI] [PubMed]
- Salemme, F. R. (1972). A free interface diffusion technique for the crystallization of proteins for X-ray crystallography. Arch. Biochem. Biophys. 151, 533–539. [DOI] [PubMed]
- Savitsky, M., Diprose, J. M., Morris, C., Griffiths, S. L., Daniel, E., Lin, B., Daenke, S., Bishop, B., Siebold, C., Wilson, K. S., Blake, R., Stuart, D. I. & Esnouf, R. M. (2011). Recording information on protein complexes in an information management system. J. Struct. Biol. 175, 224–229. [DOI] [PMC free article] [PubMed]
- Shah, A. K., Liu, Z.-J., Stewart, P. D., Schubot, F. D., Rose, J. P., Newton, M. G. & Wang, B.-C. (2005). On increasing protein-crystallization throughput for X-ray diffraction studies. Acta Cryst. D61, 123–129. [DOI] [PubMed]
- Shaw Stewart, P. D. & Baldock, P. F. M. (1999). Practical experimental design techniques for automatic and manual protein crystallization. J. Cryst. Growth, 196, 665–673.
- Shaw Stewart, P. D., Kolek, S. A., Briggs, R. A., Chayen, N. E. & Baldock, P. F. M. (2011). Random microseeding: a theoretical and practical exploration of seed stability and seeding techniques for successful protein crystallization. Cryst. Growth Des. 11, 3432–3441.
- Sigdel, M., Pusey, M. L. & Aygun, R. S. (2013). Real-Time Protein Crystallization Image Acquisition and Classification System. Cryst. Growth Des. 13, 2728–2736. [DOI] [PMC free article] [PubMed]
- Slabinski, L., Jaroszewski, L., Rychlewski, L., Wilson, I. A., Lesley, S. A. & Godzik, A. (2007). XtalPred: a web server for prediction of protein crystallizability. Bioinformatics, 23, 3403–3405. [DOI] [PubMed]
- Soliman, A. S. M., Warkentin, M., Apker, B. & Thorne, R. E. (2011). Development of high-performance X-ray transparent crystallization plates for in situ protein crystal screening and analysis. Acta Cryst. D67, 646–656. [DOI] [PMC free article] [PubMed]
- Spraggon, G., Lesley, S. A., Kreusch, A. & Priestle, J. P. (2002). Computational analysis of crystallization trials. Acta Cryst. D58, 1915–1923. [DOI] [PubMed]
- St John, F. J., Feng, B. & Pozharski, E. (2008). The role of bias in crystallization conditions in automated microseeding. Acta Cryst. D64, 1222–1227. [DOI] [PubMed]
- Till, M., Robson, A., Byrne, M. J., Nair, A. V., Kolek, S. A., Shaw Stewart, P. D. & Race, P. R. (2013). Improving the success rate of protein crystallization by random microseed matrix screening. J. Vis. Exp., 10.3791/50548. [DOI] [PMC free article] [PubMed]
- Vekilov, P. G. & Vorontsova, M. A. (2014). Nucleation precursors in protein crystallization. Acta Cryst. F70, 271–282. [DOI] [PMC free article] [PubMed]
- Watanabe, N., Murai, H. & Tanaka, I. (2002). Semi-automatic protein crystallization system that allows in situ observation of X-ray diffraction from crystals in the drop. Acta Cryst. D58, 1527–1530. [DOI] [PubMed]
- Watts, D., Cowtan, K. & Wilson, J. (2008). Automated classification of crystallization experiments using wavelets and statistical texture characterization techniques. J. Appl. Cryst. 41, 8–17.
- Watts, D., Müller-Dieckmann, J., Tsakanova, G., Lamzin, V. S. & Groves, M. R. (2010). Quantitive evaluation of macromolecular crystallization experiments using 1,8-ANS fluorescence. Acta Cryst. D66, 901–908. [DOI] [PubMed]
- Weber, P. C. (1990). A protein crystallization strategy using automated grid searches on successively finer grids. Methods, 1, 31–37.
- Zolnai, Z., Lee, P. T., Li, J., Chapman, M. R., Newman, C. S., Phillips, G. N., Rayment, I., Ulrich, E. L., Volkman, B. F. & Markley, J. L. (2003). Project management system for structural and functional proteomics: Sesame. J. Struct. Funct. Genomics, 4, 11–23. [DOI] [PubMed]
- Zuk, W. M. & Ward, K. B. (1991). Methods of analysis of protein crystal images. J. Cryst. Growth, 110, 148–155.