

High-resolution crystal structures of cyclophilin D in complex with PEG 400 in primitive orthorhombic and primitive tetragonal forms are reported.
Keywords: cyclophilin D
Abstract
Cyclophilin D (CypD) is a key mitochondrial target for amyloid-β-induced mitochondrial and synaptic dysfunction and is considered a potential drug target for Alzheimer’s disease. The high-resolution crystal structures of primitive orthorhombic (CypD-o) and primitive tetragonal (CypD-t) forms have been determined to 1.45 and 0.85 Å resolution, respectively, and are nearly identical structurally. Although an isomorphous structure of CypD-t has previously been reported, the structure reported here was determined at atomic resolution, while CypD-o represents a new crystal form for this protein. In addition, each crystal form contains a PEG 400 molecule bound to the same region along with a second PEG 400 site in CypD-t which occupies the cyclosporine A inhibitor binding site of CypD. Highly precise structural information for CypD should be extremely useful for discerning the detailed interaction of small molecules, particularly drugs and/or inhibitors, bound to CypD. The 0.85 Å resolution structure of CypD-t is the highest to date for any CypD structure.
1. Introduction
Cyclophilins are a family of proteins with peptidyl-prolyl isomerase activity, which facilitates protein folding and catalyzes the isomerization of proline residues between cis and trans isoforms (Fischer et al., 1989 ▶; Galat & Metcalfe, 1995 ▶; Takahashi et al., 1989 ▶; Price et al., 1991 ▶; Valasani et al., 2014 ▶). Proteins in the cyclophilin family are found in a variety of organisms from prokaryotes to eukaryotes (Trandinh et al., 1992 ▶). In humans, 17 cyclophilin isoforms have been identified, including cyclophilin D (CypD; Davis et al., 2010 ▶). In addition to the traditional peptidyl-prolyl isomerase activity, CypD is implicated in the mitochondrial permeability transition. Generally located in the mitochondrial matrix, CypD translocation to the inner mitochondrial membrane occurs upon calcium overload or oxidative stress in cells (Tanveer et al., 1996 ▶; Du et al., 2008 ▶, 2011 ▶). This translocation initiates the opening of the mitochondrial permeability transition pore (mPTP), classifying CypD as a modulatory component (Baines et al., 2005 ▶). The mitochondrial form of CypD is encoded by the PPIF (peptidyl-prolyl isomerase F) gene, with the end product as a protein composed of 178 amino acids (Bergsma et al., 1991 ▶).
The mitochondrial permeability transition is described as an increase in inner mitochondrial membrane permeability to molecules smaller than 1500 Da. This phenomenon is initiated by opening of the mPTP in cells undergoing stress, and has been linked to neuronal cell apoptosis and necrosis (Halestrap et al., 2002 ▶). Opening of the mPTP leads to decreased membrane potential, disruption of calcium balance and release of apoptotic signaling molecules from the mitochondria to the cytosol to activate the apoptosis pathway. Mitochondrial swelling, outer membrane rupture and cell death via necrosis are common features observed following mPTP induction (Crompton, 2004 ▶; Halestrap et al., 2002 ▶; Halestrap, 2005 ▶; Valasani, Chaney et al., 2013 ▶; Valasani, Hu et al., 2013 ▶; Valaasani et al., 2014 ▶). Initially, CypD was thought to be involved in mPTP formation because of studies involving the immunosuppressive compound cyclosporine A (CsA). As CsA binds to and inhibits CypD, it desensitizes mPTP opening, resulting in reduced mitochondrial swelling, inhibition of apoptotic factor release and overall decreased cell-death induction (Halestrap & Davidson, 1990 ▶; Takahashi et al., 1989 ▶; Broekemeier et al., 1989 ▶; Du et al., 2008 ▶). The involvement of CypD in the mPTP was later confirmed with CypD-deficient mice as the animals were no longer susceptible to mitochondrial permeability transition induced by the addition of calcium (Schinzel et al., 2005 ▶; Basso et al., 2005 ▶; Du et al., 2008 ▶; Du & Yan, 2010 ▶). Of all the proposed constituents of the mPTP, CypD is the only genetically confirmed component to have a significantly protective effect on calcium-induced, oxidative-stress-induced and amyloid-β (Aβ)-induced cell death (Du et al., 2008 ▶, 2011 ▶, 2013 ▶; Guo et al., 2013 ▶; Baines et al., 2005 ▶; Nakagawa et al., 2005 ▶). Several other possible candidates have been postulated, including the translocator protein (TSPO), the voltage-dependent anion channel (VDAC) and the adenine nucleotide translocase (ANT) (Zamzami et al., 2005 ▶; Crompton et al., 2002 ▶; Halestrap, 2006 ▶). While TSPO shows promise as a structural component (Sileikyte et al., 2011 ▶), VDAC and ANT may not be essential components of the mPTP as several studies have demonstrated that cells lacking VDAC or ANT do not protect against mPTP-involved cell death (Baines et al., 2007 ▶; Kokoszka et al., 2004 ▶; McCommis & Baines, 2012 ▶). Although the roles of the unconfirmed constituents remain to be fully clarified in relation to the mPTP, the repositioning event of CypD has been established as a key regulator of the mPTP structural opening (Halestrap et al., 1997 ▶; Connern & Halestrap, 1994 ▶; Andreeva et al., 1999 ▶; Pastorino et al., 1998 ▶; Crompton et al., 1998 ▶; Du et al., 2008 ▶; Rao et al., 2013 ▶).
Recent reports suggest that a chemical alteration in the CypD configuration may fortify a cell during the event of calcium overload or oxidative stress (Baines et al., 2005 ▶; Nakagawa et al., 2005 ▶; Basso et al., 2005 ▶). Furthermore, resistance to ischemia has been observed in both the brain (Schinzel et al., 2005 ▶) and heart (Nakagawa et al., 2005 ▶; Baines et al., 2005 ▶) of CypD knockout mice. In addition to the role of mPTP formation in ischemia, it has been implicated in a variety of diseases, including neurodegeneration, such as Parkinson’s disease (PD; Gandhi et al., 2009 ▶), Huntington’s disease (HD; Brustovetsky et al., 2003 ▶), amyotrophic lateral sclerosis (ALS; Martin et al., 2009 ▶) and Alzheimer’s disease (AD; Du et al., 2008 ▶). In all of these diseases many mitochondrial alterations have been discovered which aid in mPTP induction. For instance, mutations in mitochondrial DNA and proteins have been identified in PD research, alterations in striatal mitochondrial CypD levels compared with cortical mitochondria in HD pathogenesis, and changes in mitochondrial respiratory-chain enzymes and cell-death proteins have been found in ALS research. Similarly, the involvement of the mPTP is demonstrated in AD by modifications to enzymes involved in oxidative phosphorylation, oxidative stress and mitochondrial binding of Aβ. Aβ excess within neuronal mitochondria is a common feature among AD patients, and Aβ aggregation promotes events of high oxidative stress. Notably, the absence of CypD allows neuronal cell protection in an environment rich in Aβ-induced oxidative stress (Du et al., 2008 ▶). Thus, lack of CypD prevents cell death, reduces impaired cognitive function (learning and memory) and diminishes synaptic dysfunction (Du et al., 2011 ▶). Our published findings seem to support the concept that CypD is a key cellular target for Aβ-induced synaptic dysfunction. In the light of this, it is likely that the chemical inhibition of CypD could provide an alternative treatment for AD.
High-resolution structural information for these proteins would therefore be very useful to discern the binding mode of lead compounds, particularly in the early stages of drug discovery and development. Several crystal structures of the cyclophilin proteins, including that of CypD, have previously been determined (Mikol et al., 1993 ▶, 1994a ▶,b ▶; Schlatter et al., 2005 ▶; le Maire et al., 2011 ▶; Kajitani et al., 2008 ▶) but to lower resolution compared with the high-resolution structures presented here. Since higher resolution structural detail is always desirable, we have determined the crystal structures of two different forms (primitive tetragonal, CypD-t, and primitive orthorhombic, CypD-o) of CypD bound with PEG 400 to 1.45 Å resolution and to the ultrahigh resolution of 0.85 Å. CypD-t is isomorphous with a previously reported structure (PDB entry 2bit, Schlatter et al., 2005 ▶) in which the authors conducted an elegant series of protein mutagenesis/engineering experiments to identify a construct (K133I) that could be reproducibly crystallized as the wild-type form failed to yield crystals. However, this structure was reported to lower resolution (1.7 Å), although the sample used to determine the 2bit structure clearly diffracted to higher resolution based on the 〈I/σ(I)〉 in the high-resolution shell (∼10). The lower resolution reported for the structure 2bit is likely to be a consequence of hardware limitations of the in-house instrumentation used for data collection. The 0.85 Å resolution structure of CypD-t is the highest to date for any CypD structure. The second and new crystal form (CypD-o) crystallized in a primitive orthorhombic lattice and was determined to 1.45 Å resolution.
2. Materials and methods
2.1. Protein expression and purification
Escherichia coli BL21 (DE3) cells were transformed with a pET-21a-tCypD plasmid containing the truncated human peptidyl-prolyl cis–trans-isomerase F (CypD) with a K133I mutation to facilitate crystallization. The plasmid was a generous gift from Schlatter and coworkers (Schlatter et al., 2005 ▶). Cultures were grown in Luria–Bertani (LB) medium to an A 600 of 0.5 and were then induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). The cells were then grown overnight and harvested by centrifugation at 4000 rev min−1 for 30 min the following day. The cell pellet was resuspended in 50 ml 100 mM Tris–HCl pH 7.8, 2 mM EDTA, 2 mM DTT (per 2 l of medium) and were then lysed using sonication.
Lysates were centrifuged at 17 000 rev min−1 for 45 min. The soluble portion was purified on an SP-Sepharose FF column followed by anion exchange on a Q-Sepharose HP column and was then finally subjected to gel filtration on a Superdex 200 column (GE Healthcare Life Sciences). The SP column was equilibrated with 100 mM Tris–HCl pH 7.8, 2 mM EDTA, 2 mM DTT and the CypD protein was then eluted using an increasing gradient of 0–500 mM NaCl. Fractions containing the protein were collected, concentrated using ultrafiltration and then loaded onto the Q-Sepharose HP column. The Q-Sepharose HP column was equilibrated using 100 mM Tris–HCl pH 7.8, 2 mM EDTA, 2 mM DTT. CypD-containing fractions were collected from the flowthrough, concentrated using ultrafiltration and then applied onto a Superdex 200 column for size exclusion. The Superdex 200 column was equilibrated using 50 mM potassium phosphate, 100 mM NaCl, 2 mM EDTA. Peak fractions were collected and run on an SDS–PAGE gel to analyze the purity of the CypD K133I protein. The gel is shown in Fig. 1 ▶(a).
Figure 1.

Purification and crystallization of CypD. (a) SDS–PAGE of purified CypD protein stained with Coomassie Brilliant Blue. Lane 1, 7.5 µg CypD protein; lane 2, 15 µg CypD protein; lane 3, 30 µg CypD protein. (b) CypD-t crystals obtained from ProPlex condition D1. (c) CypD-o cystals obtained from Wizard 3–4 condition H4.
2.2. Crystallization and data collection
Recombinant CypD (K133I) protein was concentrated to 30 mg ml−1 in 50 mM potassium/sodium phosphate pH 7.3, 100 mM NaCl, 2 mM EDTA for crystallization. Screening was conducted in Compact Jr (Emerald Bio) sitting-drop vapor-diffusion plates at 18°C using equal volumes of protein and crystallization solutions. Tetragonal crystals (CypD-t) displaying a prismatic morphology were obtained in 1 d from condition D1 [25%(w/v) PEG 4000, 100 mM HEPES pH 7.5, 200 mM NaCl] of the ProPlex screen (Molecular Dimensions). Orthorhombic crystals (CypD-o) grew as needle clusters after 1 d from condition H4 [20%(w/v) PEG 8000, 100 mM Tris–HCl pH 8.5, 200 mM MgCl2, 20%(v/v) PEG 400] of the Wizard 3–4 screen (Emerald Bio). Representative examples of these crystals are shown in Figs. 1 ▶(b) and 1 ▶(c). Single crystals of CypD-t were transferred into a fresh drop containing 75% ProPlex condition D1 and 25%(v/v) PEG 400 and cooled in liquid nitrogen prior to data collection. Crystals of CypD-o were transferred to a fresh drop of the crystallization solution (Wizard 4 condition H4), which served as the cryoprotectant for data collection. X-ray diffraction data were collected on the Advanced Photon Source IMCA-CAT beamline 17ID using a Dectris Pilatus 6M pixel-array detector. Coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 4o8h (CypD-t) and 4o8i (CypD-o).
3. Results and discussion
3.1. Structure solution and refinement
Intensities were integrated using XDS (Kabsch, 2010a ▶,b ▶), and the Laue-class analysis and data scaling were performed with AIMLESS (Evans, 2011 ▶; Evans & Murshudov, 2013 ▶), which suggested that the highest probability Laue classes were 4/mmm (space group P41212) for CypD-t and mmm (space group P212121) for CypD-o. Data were truncated to a resolution where appreciable signal [I/σ(I) ≃ 2.0] could be observed and the CC1/2 was above 50% as indicated by the output from AIMLESS. The lower completeness in the outer resolution shell for CypD-t is largely a result of the instrumental setup as the high-angle diffraction was observed near the corners of the detector. Therefore, a decrease in the data completeness from 94% (0.88–0.86 Å shell) to 60% (0.86–0.85 Å shell) was observed. Structure solution for CypD-t was conducted by molecular replacement with Phaser (McCoy et al., 2007 ▶) using a previously determined isomorphous structure of CypD (PDB entry 2bit) as the search model. All space groups with 422 point symmetry were tested and the top solution was obtained in P41212, which was used from this point forward. For CypD-o, the same model was used for molecular-replacement searches. All space groups with 222 point symmetry were tested and the top solution was obtained in P212121. Structure refinement with anisotropic atomic displacement parameters for all atoms was carried out for CypD-t with PHENIX (Adams et al., 2010 ▶). TLS refinement (Painter & Merritt, 2006 ▶) was incorporated in the final stages of refinement with PHENIX for CypD-o to model anisotropic atomic displacement parameters. Manual model building was performed using Coot (Emsley et al., 2010 ▶). Structure validation was conducted with MolProbity (Chen et al., 2010 ▶) and figures were prepared using the CCP4mg package (Potterton et al., 2004 ▶). Crystallographic data are provided in Table 1 ▶.
Table 1. Crystallographic data for the CypD structures.
Values in parentheses are for the highest resolution shell.
| CypD-t | CypD-o | |
|---|---|---|
| Data collection | ||
| Unit-cell parameters (Å) | a = b = 57.02, c = 87.16 | a = 40.56, b = 57.01, c = 57.34 |
| Space group | P41212 | P212121 |
| Resolution (Å) | 47.72–0.85 (0.86–0.85) | 40.56–1.45 (1.47–1.45) |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Temperature (°C) | −173 | −173 |
| Observed reflections | 1755792 | 155848 |
| Unique reflections | 123449 | 24241 |
| 〈I/σ(I)〉 | 30.7 (1.8) | 12.4 (1.8) |
| Completeness (%) | 97.7 (60.2) | 99.9 (100) |
| Multiplicity | 14.2 (1.7) | 6.4 (6.5) |
| R merge † (%) | 4.9 (34.2) | 9.7 (112.0) |
| R meas ‡ (%) | 5.0 (45.3) | 10.6 (121.8) |
| R p.i.m. ‡ (%) | 1.1 (29.4) | 4.2 (47.1) |
| CC1/2 § | 0.999 (0.766) | 0.999 (0.715) |
| Refinement | ||
| Resolution (Å) | 28.51–0.85 (0.86–0.85) | 33.12–1.45 (1.51–1.45) |
| Reflections (working/test) | 117150/6189 (1964/108) | 22858/1231 (2421/142) |
| R factor/R free ¶ (%) | 10.7/11.9 (26.8/30.2) | 16.0/19.0 (23.8/27.9) |
| No. of atoms | ||
| Protein | 1266 | 1237 |
| PEG 400 | 16 | 10 |
| Water | 303 | 133 |
| Model quality | ||
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.015 | 0.008 |
| Bond angles (°) | 1.482 | 0.909 |
| Average B factor (Å2) | ||
| All atoms | 8.1 | 19.0 |
| Protein | 5.7 | 17.8 |
| PEG 400 | 11.1 | 34.6 |
| Water | 17.5 | 29.5 |
| Coordinate error (maximum likelihood) (Å) | 0.05 | 0.19 |
| Ramachandran plot | ||
| Most favored (%) | 97.0 | 96.4 |
| Additionally allowed (%) | 3.0 | 3.6 |
R
merge = 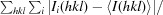
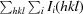 , where Ii(hkl) is the intensity measured for the ith reflection and 〈I(hkl)〉 is the average intensity of all reflections with indices hkl.
, where Ii(hkl) is the intensity measured for the ith reflection and 〈I(hkl)〉 is the average intensity of all reflections with indices hkl.
R meas is the redundancy-independent (multiplicity-weighted) R merge (Evans, 2006 ▶, 2011 ▶). R p.i.m. is the precision-indicating (multiplicity-weighted) R merge (Diederichs & Karplus, 1997 ▶; Weiss, 2001 ▶).
CC1/2 is the correlation coefficient of the mean intensities between two random half-sets of data (Karplus & Diederichs, 2012 ▶; Evans, 2012 ▶).
R factor = 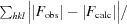
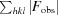 ; R
free is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
; R
free is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
3.2. Analysis
The unit-cell dimensions for CypD-t were nearly identical to those of the previously determined structure of CypD (PDB entry 2bit). However, given that the diffraction resolution for these crystals was the highest to date (0.85 Å) for any CypD structure, we analyzed the data/structure for any additional features. Superposition of 2bit with CypD-t using GESAMT (Krissinel, 2012 ▶) yielded an r.m.s.d. of 0.07 Å between Cα atoms (164 residues), indicating that the structures were virtually identical (Fig. 2 ▶ a).
Figure 2.
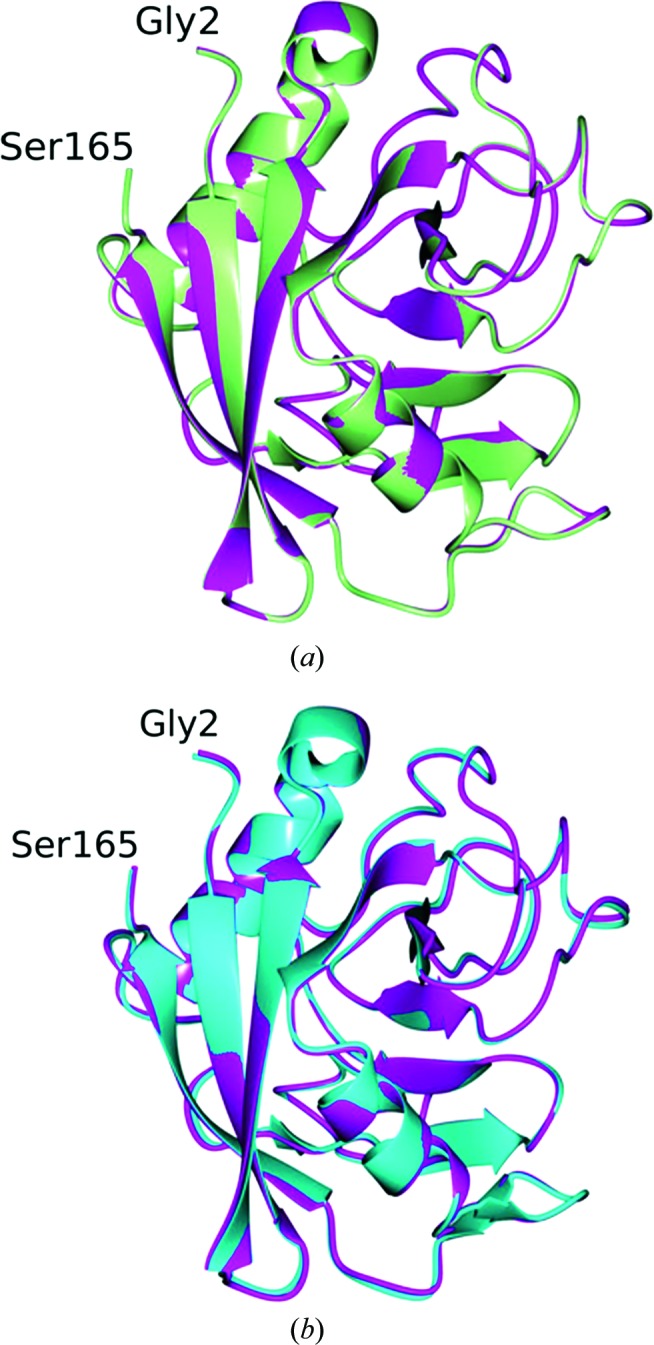
(a) Superposition of CypD-t (magenta) with 2bit (green) drawn as ribbons. The r.m.s.d. of 0.07 Å (164 residues, Cα atoms) indicated that the structures were basically identical. (b) Superposition of CypD-t (magenta) with CypD-o (cyan) drawn as ribbons.
Examination of the electron-density maps revealed a large region of positive difference density that was ultimately assigned as a PEG 400 molecule (Fig. 3 ▶ a) from the cryoprotectant that was bound to CypD. This PEG 400 is positioned near a hydrophobic region spanning Val93–Val97 and fits within a hydrophobic pocket on the CypD surface (Fig. 3 ▶ b). In addition, a second PEG 400 fragment (partially disordered) was modeled as shown in Fig. 4 ▶(a) that forms hydrogen bonds to Arg55 and Gln63 (Fig. 4 ▶ b), and as was observed for the ordered PEG 400 molecule, was positioned within another relatively hydrophobic cavity. Interestingly, this partially disordered PEG 400 fragment binds to the same region as was observed for CsA-bound CypD (Fig. 5 ▶) and occupies a position similar to Mva4 of the CsA inhibitor (Kajitani et al., 2008 ▶). This is not too surprising given the similar hydrophobic properties of both CsA and PEG 400.
Figure 3.
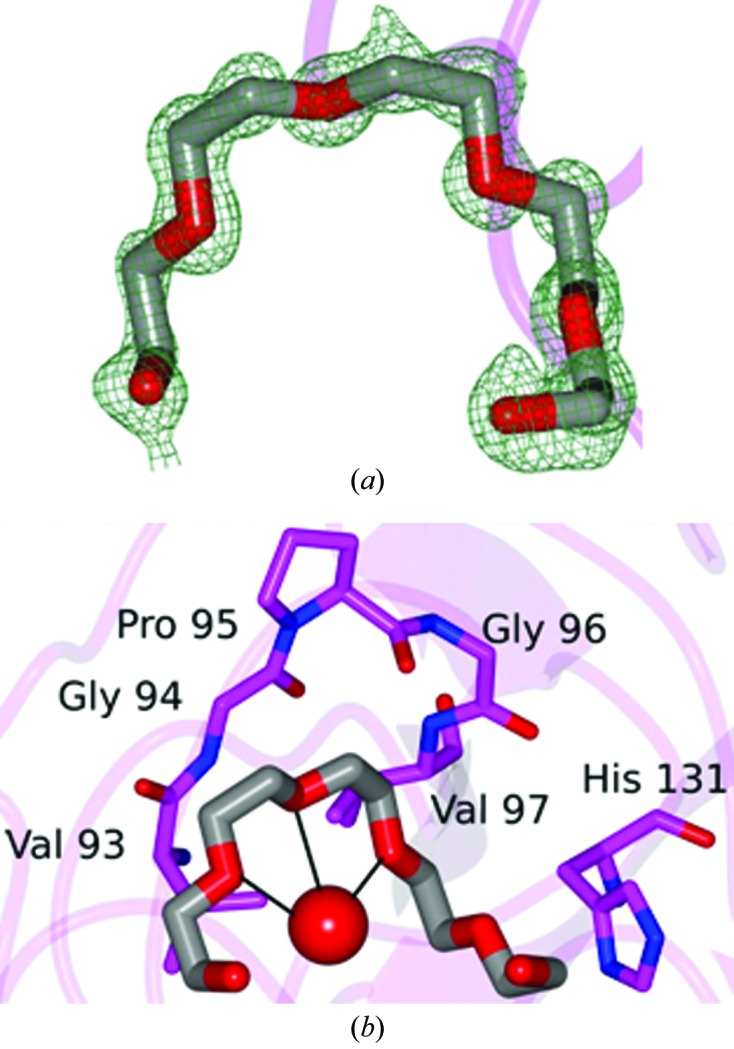
PEG 400 molecule bound to CypD-t. (a) F o − F c electron-density map contoured at 3σ. (b) Hydrophobic residues that are in proximity (<4 Å) to the PEG 400 molecule. A water molecule (red sphere) is within hydrogen-bonding distance as indicated by the black lines.
Figure 4.
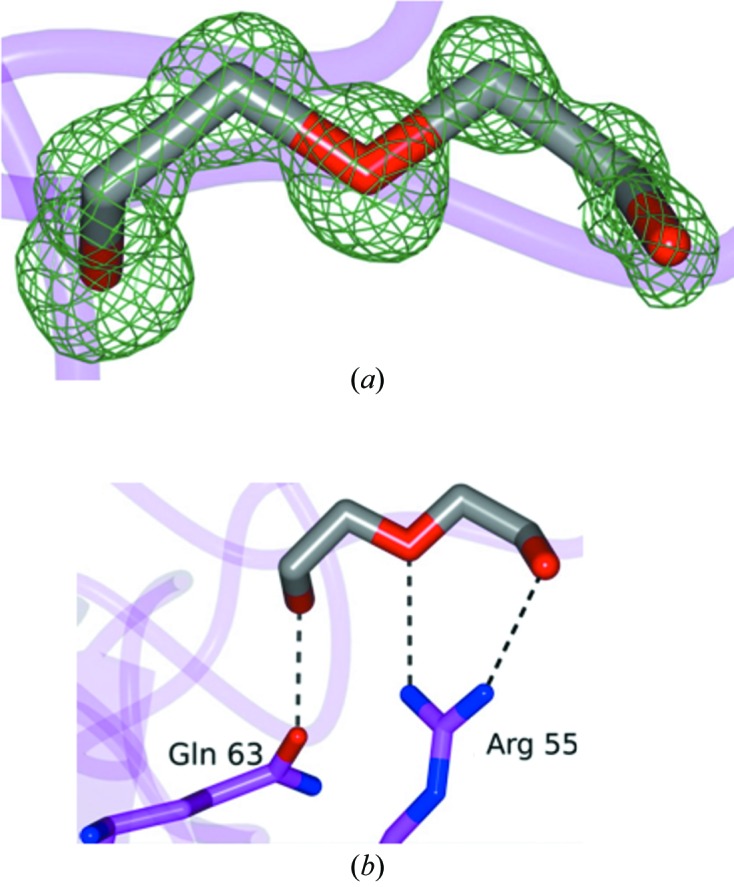
The second PEG 400 molecule fragment bound to CypD-t. (a) F o − F c electron-density map contoured at 3σ. (b) Hydrogen bond (dashed line) between the PEG 400 fragment and CypD.
Figure 5.
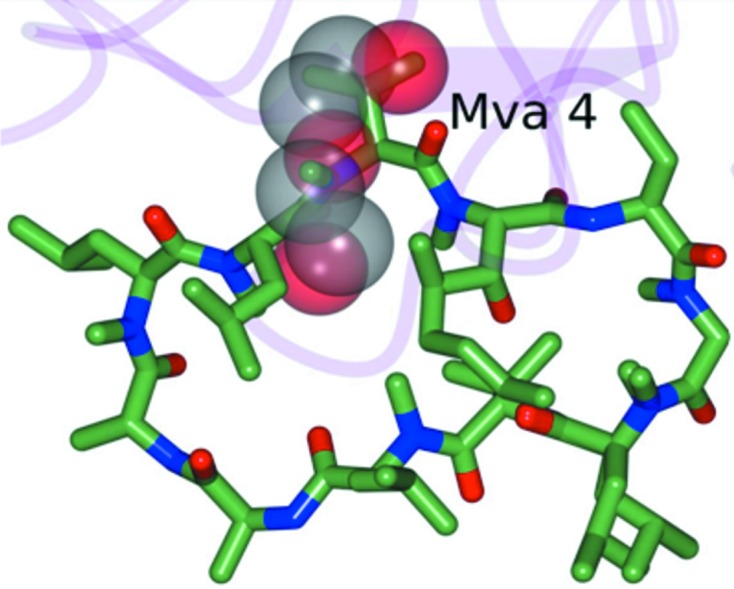
Superposition of the partially disordered PEG 400 molecule in CypD-t (transparent spheres) with cyclosporine A (green) bound to CypD as observed in PDB entry 2z6w (Kajitani et al., 2008 ▶). The partially disordered PEG 400 molecule occupies a position similar to Mva4 of cyclosporine A as indicated. The r.m.s.d. between CypD-t and 2z6w is 0.33 Å between Cα atoms (164 residues).
The crystal structure of CypD-o is very similar to that of CypD-t (Fig. 2 ▶ b), with an r.m.s.d. of 0.25 Å (164 residues, Cα atoms) as determined by alignment with GESAMT. Similar to CypD-t, a large region of positive difference electron density was observed near the hydrophobic residues Val93–Val97, which was assigned as a PEG 400 molecule. However, the entire PEG molecule could not be built as a portion appeared to be disordered. Notably, the electron density present in CypD-t near Arg55 and Gln63, which was modeled as a PEG 400 fragment, was not observed in CypD-o.
Comparing the unit-cell dimensions of CypD-o (a = 40.56, b = 57.01, c = 57.34 Å; P212121) with those of CypD-t (a = 57.02, b = 57.02, c = 87.16 Å; P41212), one can see that the b and c axial lengths of the former are similar to the a and b axes of the latter. Applying the re-indexing operator (−l, −k, −h) to the unit cell of CypD-o would give a = 57.34, b = 57.01, c = 40.56 Å. Given that a and b axes in this transformed unit cell have a similar magnitude, one suspects that the CypD-o crystal form could be indexed in a tetragonal P lattice. If this were indeed correct, the point symmetry would be 4 with space groups P4, P41, P42 or P43, as the tetragonal space groups with 422 point symmetry would yield an unrealistic Matthews coefficient (V M = 0.9 Å3 Da−1, solvent content −32.3%; Matthews, 1968 ▶). However, doubling of the c axis in the transformed unit cell would produce a lattice similar to that obtained for CypD-t, in which case space groups with 422 point symmetry could be a possibility. Analysis of the Laue symmetry using POINTLESS (Evans, 2006 ▶) yielded a multiplicity-weighted R factor (R meas) of 22 and 28% for 4/m and 4/mmm, respectively. By contrast, the R factor for the mmm Laue class was 5% and systematic absences for 21 screw axes along a, b and c were present, indicating that the orthorhombic P lattice was indeed correct.
4. Conclusions
Here, we present high-resolution crystal structures of CypD bound with PEG 400 and refined using diffraction data processed to 1.45 Å resolution and to the ultrahigh resolution of 0.85 Å. The CypD-o form crystallized in a primitive orthorhombic unit cell and represents a new crystal form of CypD. The CypD-t form, at 0.85 Å resolution, is the highest to date for any CypD structure. Our previous work has provided evidence that CypD plays a major role in Aβ-induced synaptic dysfunction in AD, making it an attractive target for drug development. Thus, the ability to acquire high-resolution structural data for various crystal forms of CypD will be useful in the initial phase of AD therapeutic development as detailed information regarding the binding mode of our lead compounds can be readily obtained.
Supplementary Material
PDB reference: CypD, 4o8h
PDB reference: 4o8i
Acknowledgments
This study was supported by grants from the National Institute of Aging (R37AG037319), the National Institute of General Medical Science (R01GM095355) and the National Institute of Neurological Disorder and Stroke (R01NS65482), and use of the University of Kansas Protein Structure Laboratory was supported by grants from the National Center for Research Resources (5P20RR017708-10) and the National Institute of General Medical Sciences (8P20GM103420-10). Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman–Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The authors have no conflict of interest to disclose.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Andreeva, L., Heads, R. & Green, C. J. (1999). Int. J. Exp. Pathol. 80, 305–315. [DOI] [PMC free article] [PubMed]
- Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., Brunskill, E. W., Sayen, M. R., Gottlieb, R. A., Dorn, G. W., Robbins, J. & Molkentin, J. D. (2005). Nature (London), 434, 658–662. [DOI] [PubMed]
- Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J. & Molkentin, J. D. (2007). Nature Cell Biol. 9, 550–555. [DOI] [PMC free article] [PubMed]
- Basso, E., Fante, L., Fowlkes, J., Petronilli, V., Forte, M. A. & Bernardi, P. (2005). J. Biol. Chem. 280, 18558–18561. [DOI] [PubMed]
- Bergsma, D. J., Eder, C., Gross, M., Kersten, H., Sylvester, D., Appelbaum, E., Cusimano, D., Livi, G. P., McLaughlin, M. M. & Kasyan, K. (1991). J. Biol. Chem. 266, 23204–23214. [PubMed]
- Broekemeier, K. M., Dempsey, M. E. & Pfeiffer, D. R. (1989). J. Biol. Chem. 264, 7826–7830. [PubMed]
- Brustovetsky, N., Brustovetsky, T., Purl, K. J., Capano, M., Crompton, M. & Dubinsky, J. M. (2003). J. Neurosci. 23, 4858–4867. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Connern, C. P. & Halestrap, A. P. (1994). Biochem. J. 302, 321–324. [DOI] [PMC free article] [PubMed]
- Crompton, M. (2004). Aging Cell, 3, 3–6. [DOI] [PubMed]
- Crompton, M., Barksby, E., Johnson, N. & Capano, M. (2002). Biochimie, 84, 143–152. [DOI] [PubMed]
- Crompton, M., Virji, S. & Ward, J. M. (1998). Eur. J. Biochem. 258, 729–735. [DOI] [PubMed]
- Davis, T. L., Walker, J. R., Campagna-Slater, V., Finerty, P. J., Paramanathan, R., Bernstein, G., MacKenzie, F., Tempel, W., Ouyang, H., Lee, W. H., Eisenmesser, E. Z. & Dhe-Paganon, S. (2010). PLoS Biol. 8, e1000439. [DOI] [PMC free article] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., Yan, Y., Wang, C., Zhang, H., Molkentin, J. D., Gunn-Moore, F. J., Vonsattel, J. P., Arancio, O., Chen, J. X. & Yan, S. D. (2008). Nature Med. 14, 1097–1105. [DOI] [PMC free article] [PubMed]
- Du, H., Guo, L., Wu, X., Sosunov, A. A., McKhann, G. M., Chen, J. X. & Yan, S. S. (2013). Biochim Biophys Acta, 10.1016/j.bbadis.2013.03.004. [DOI] [PMC free article] [PubMed]
- Du, H., Guo, L., Zhang, W., Rydzewska, M. & Yan, S. (2011). Neurobiol. Aging, 32, 398–406. [DOI] [PMC free article] [PubMed]
- Du, H. & Yan, S. S. (2010). Biochim. Biophys. Acta, 1802, 198–204. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evans, P. (2012). Science, 336, 986–987. [DOI] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Fischer, G., Wittmann-Liebold, B., Lang, K., Kiefhaber, T. & Schmid, F. X. (1989). Nature (London), 337, 476–478. [DOI] [PubMed]
- Galat, A. & Metcalfe, S. M. (1995). Prog. Biophys. Mol. Biol. 63, 67–118. [DOI] [PubMed]
- Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., Downward, J., Latchman, D. S., Tabrizi, S. J., Wood, N. W., Duchen, M. R. & Abramov, A. Y. (2009). Mol. Cell, 33, 627–638. [DOI] [PMC free article] [PubMed]
- Guo, L., Du, H., Yan, S., Wu, X., McKhann, G. M., Chen, J. X. & Yan, S. S. (2013). PLoS One, 8, e54914. [DOI] [PMC free article] [PubMed]
- Halestrap, A. (2005). Nature (London), 434, 578–579. [DOI] [PubMed]
- Halestrap, A. P. (2006). Biochem. Soc. Trans. 34, 232–237. [DOI] [PubMed]
- Halestrap, A. P. & Davidson, A. M. (1990). Biochem. J. 268, 153–160. [DOI] [PMC free article] [PubMed]
- Halestrap, A. P., McStay, G. P. & Clarke, S. J. (2002). Biochimie, 84, 153–166. [DOI] [PubMed]
- Halestrap, A. P., Woodfield, K. Y. & Connern, C. P. (1997). J. Biol. Chem. 272, 3346–3354. [DOI] [PubMed]
- Kabsch, W. (2010a). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kajitani, K., Fujihashi, M., Kobayashi, Y., Shimizu, S., Tsujimoto, Y. & Miki, K. (2008). Proteins, 70, 1635–1639. [DOI] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., MacGregor, G. R. & Wallace, D. C. (2004). Nature (London), 427, 461–465. [DOI] [PMC free article] [PubMed]
- Krissinel, E. (2012). J. Mol. Biochem. 1, 76–85. [PMC free article] [PubMed]
- Maire, A. le, Gelin, M., Pochet, S., Hoh, F., Pirocchi, M., Guichou, J.-F., Ferrer, J.-L. & Labesse, G. (2011). Acta Cryst. D67, 747–755. [DOI] [PubMed]
- Martin, L. J., Gertz, B., Pan, Y., Price, A. C., Molkentin, J. D. & Chang, Q. (2009). Exp. Neurol. 218, 333–346. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCommis, K. S. & Baines, C. P. (2012). Biochim. Biophys. Acta, 1818, 1444–1450. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mikol, V., Kallen, J., Pflügl, G. & Walkinshaw, M. D. (1993). J. Mol. Biol. 234, 1119–1130. [DOI] [PubMed]
- Mikol, V., Kallen, J. & Walkinshaw, M. D. (1994a). Proc. Natl Acad. Sci. USA, 91, 5183–5186. [DOI] [PMC free article] [PubMed]
- Mikol, V., Kallen, J. & Walkinshaw, M. D. (1994b). Protein Eng. 7, 597–603. [DOI] [PubMed]
- Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., Inohara, H., Kubo, T. & Tsujimoto, Y. (2005). Nature (London), 434, 652–658. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Pastorino, J. G., Chen, S. T., Tafani, M., Snyder, J. W. & Farber, J. L. (1998). J. Biol. Chem. 273, 7770–7775. [DOI] [PubMed]
- Potterton, L., McNicholas, S., Krissinel, E., Gruber, J., Cowtan, K., Emsley, P., Murshudov, G. N., Cohen, S., Perrakis, A. & Noble, M. (2004). Acta Cryst. D60, 2288–2294. [DOI] [PubMed]
- Price, E. R., Zydowsky, L. D., Jin, M. J., Baker, C. H., McKeon, F. D. & Walsh, C. T. (1991). Proc. Natl Acad. Sci. USA, 88, 1903–1907. [DOI] [PMC free article] [PubMed]
- Rao, V. K., Carlson, E. A. & Yan, S. S. (2013). Biochim. Biophys. Acta, 10.1016/j.bbadis.2013.09.003.
- Schinzel, A. C., Takeuchi, O., Huang, Z., Fisher, J. K., Zhou, Z., Rubens, J., Hetz, C., Danial, N. N., Moskowitz, M. A. & Korsmeyer, S. J. (2005). Proc. Natl Acad. Sci. USA, 102, 12005–12010. [DOI] [PMC free article] [PubMed]
- Schlatter, D., Thoma, R., Küng, E., Stihle, M., Müller, F., Borroni, E., Cesura, A. & Hennig, M. (2005). Acta Cryst. D61, 513–519. [DOI] [PubMed]
- Sileikyte, J., Petronilli, V., Zulian, A., Dabbeni-Sala, F., Tognon, G., Nikolov, P., Bernardi, P. & Ricchelli, F. (2011). J. Biol. Chem. 286, 1046–1053. [DOI] [PMC free article] [PubMed]
- Takahashi, N., Hayano, T. & Suzuki, M. (1989). Nature (London), 337, 473–475. [DOI] [PubMed]
- Tanveer, A., Virji, S., Andreeva, L., Totty, N. F., Hsuan, J. J., Ward, J. M. & Crompton, M. (1996). Eur. J. Biochem. 238, 166–172. [DOI] [PubMed]
- Trandinh, C. C., Pao, G. M. & Saier, M. H. Jr (1992). FASEB J. 6, 3410–3420. [DOI] [PubMed]
- Valaasani, K. R., Sun, Q., Hu, G., Li, J., Du, F., Guo, Y., Carlson, E. A., Gan, X. & Yan, S. S. (2014). Curr. Alzheimer Res. 11, 128–136. [DOI] [PMC free article] [PubMed]
- Valasani, K. R., Chaney, M. O., Day, V. W. & Shidu Yan, S. (2013). J. Chem. Inf. Model. 53, 2033–2046. [DOI] [PubMed]
- Valasani, K. R., Hu, G., Chaney, M. O. & Yan, S. S. (2013). Chem. Biol. Drug Des. 81, 238–249. [DOI] [PMC free article] [PubMed]
- Valasani, K. R., Vangavaragu, J. R., Day, V. W. & Yan, S. S. (2014). J. Chem. Inf. Model. 54, 902–912. [DOI] [PMC free article] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Zamzami, N., Larochette, N. & Kroemer, G. (2005). Cell Death Differ. 12, 1478–1480. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: CypD, 4o8h
PDB reference: 4o8i