

A 2.0 Å resolution neutron data set and a 1.6 Å resolution X-ray data set were collected for joint X-ray/neutron refinement of the ecDHFR–folate–NADP+ complex in order to study the reaction mechanism of dihydrofolate reductase.
Keywords: Escherichia coli, dihydrofolate reductase, protonation state
Abstract
A crystal of Escherichia coli dihydrofolate reductase (ecDHFR) complexed with folate and NADP+ of 4 × 1.3 × 0.7 mm (3.6 mm3) in size was obtained by sequential application of microseeding and macroseeding. A neutron diffraction data set was collected to 2.0 Å resolution using the IMAGINE diffractometer at the High Flux Isotope Reactor within Oak Ridge National Laboratory. A 1.6 Å resolution X-ray data set was also collected from a smaller crystal at room temperature. The neutron and X-ray data were used together for joint refinement of the ecDHFR–folate–NADP+ ternary-complex structure in order to examine the protonation state, protein dynamics and solvent structure of the complex, furthering understanding of the catalytic mechanism.
1. Introduction
Dihydrofolate reductase (DHFR) is an NADPH-dependent enzyme that catalyzes the reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF), which is required in the biosynthesis of purine, thymine and several amino acids (Sawaya & Kraut, 1997 ▶; Reyes et al., 1995 ▶; Chen et al., 1994 ▶). In rapidly proliferating cells, DNA synthesis greatly increases the demand for dNTPs. THF is the one-carbon carrier during dNTP synthesis. Hence, DHFR is a drug target for cancer, malaria and rheumatoid arthritis to inhibit cell proliferation (Yuthavong et al., 2012 ▶; Sharma & Chauhan, 2012 ▶; Schweitzer et al., 1990 ▶).
During reduction of DHF, a proton is donated to the N5 atom of the DHF pterin ring with a hydride transfer occurring from the nicotinamide ring of NADPH to the C6 atom of DHF. Previously, several catalytic mechanisms have been proposed based on experimental and theoretical studies of DHFR (Shrimpton & Allemann, 2002 ▶; Cummins & Gready, 2001 ▶; Falzone et al., 1994 ▶; Chen et al., 1994 ▶; Bajorath et al., 1991 ▶). Most of these mechanisms involve a proton relay that includes the catalytic Asp27, a structurally conserved water molecule and the O4 atom of the pterin ring, which may tautomerize from the keto to the enol form during catalysis. Some mechanisms invoke a second water molecule to be directly involved in the protonation of the N5 atom (Chen et al., 1994 ▶; Shrimpton & Allemann, 2002 ▶). Cummins & Gready (2001 ▶) have proposed that hydride transfer from the nicotinamide C4 atom of NADPH to the C6 atom of DHF occurs synchronously with the protonation of N5 to convert DHF to THF (Cummins & Gready, 2001 ▶). It is generally proposed that the deprotonated Asp27 is particularly important for the increase in the value of pK a of the N5 atom of the substrate from 2.4 to 6.5 (Khavrutskii et al., 2007 ▶; Chen et al., 1994 ▶; Bajorath et al., 1991 ▶; Maharaj et al., 1990 ▶; Fig. 1 ▶). The ability to differentiate between the mechanisms proposed above is limited because the protonation states of the ligand and catalytic residues are difficult to determine using X-ray crystallography even at atomic resolution (Fisher et al., 2012 ▶).
Figure 1.

A proposed mechanism of dihydrofolate (DHF) reduction. The N5 atom of folate might be directly protonated by a neighboring water molecule. Another proposed mechanism involves a proton relay from the catalytic Asp27 to a water molecule and the tautomerized O4 atom of DHF. Hydride transfer occurs from NADPH to the C6 of DHF to convert DHF to tetrahydrofolate (THF).
Neutron crystallography (NC) can be used to locate H atoms and can readily provide information on the protonation states of amino-acid residues and ligands, the identity of solvent molecules and the nature of bonds involving hydrogen (Niimura & Bau, 2008 ▶; Blakeley et al., 2008 ▶; Niimura, 1999 ▶). In addition, NC can also be used to study protein dynamics by analyzing the backbone amide hydrogen/deuterium-exchange (HDX) pattern: higher exchange rates have been correlated to higher dynamics (Sukumar et al., 2010 ▶; Bennett et al., 2006 ▶, 2008 ▶). To understand the catalytic mechanism of DHFR, it is imperative to visualize both H atoms and protein dynamics. Therefore, H atoms, which are normally invisible in X-ray crystal structures of DHFR, should become visible in neutron structures.
Here, we report the crystallization of Escherichia coli DHFR (ecDHFR) in complex with folate and NADP+. As folate is a slow-turnover substrate and NADP+ is inactive, the ecDHFR–folate–NADP+ complex has been considered to be a pseudo-Michaelis complex useful for the understanding of the catalytic mechanism of DHFR (Falzone et al., 1994 ▶; Reyes et al., 1995 ▶; Sawaya & Kraut, 1997 ▶; Bhabha et al., 2011 ▶). We obtained a ∼3.6 mm3 (4 × 1.3 × 0.7 mm) crystal by optimizing the de novo crystallization conditions, followed by microseeding and macroseeding techniques. A 2.0 Å resolution neutron data set was collected with 79.3% completeness on the IMAGINE beamline (Meilleur et al., 2013 ▶) located at the High Flux Isotope Reactor (HFIR) within Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA. A 1.6 Å resolution room-temperature X-ray data set was also collected from a smaller crystal harvested from the same crystallization condition. Both data sets were used for joint X-ray/neutron (XN) refinement (Afonine et al., 2010 ▶) to study the reaction mechanism of DHFR.
2. Materials and methods
2.1. Protein expression and purification
E. coli DHFR (ecDHFR) was cloned, expressed and purified using the previously described protocol for the recombinant production of Bacillus anthracis DHFR (Bennett et al., 2007 ▶). Briefly, the cDNA of ecDHFR was cloned into a pET-SUMO vector (Invitrogen) with SUMO and a 6×His tag at the exact N-terminus of the construct. The advantage of this fusion protein system is that, after cleavage, no exogenous residues are left on the target protein. After expression in competent E. coli BL21(DE3) cells, the protein was extracted using sonication and centrifugation. The His-tagged protein was purified via immobilized metal-affinity chromatography (IMAC) using a nickel column. Next, the SUMO protease from yeast, Ulp1, was added to the purified fusion protein to remove SUMO and the His tag. The protease-treated protein mixture was loaded onto a new nickel-affinity column and the flowthrough contained the pure wild-type ecDHFR (Fig. 2 ▶). Because of its poor solubility, folate was added to ecDHFR at a molar ratio of 3:1 when the protein was at only 1 mg ml−1 (Sawaya & Kraut, 1997 ▶). After concentration to 30 mg ml−1, NADP+ was added to the protein solution at a molar ratio of 3:1 and the protein was further concentrated to 40 mg ml−1. The solution of the concentrated protein complex (ecDHFR–folate–NADP+) was clarified at 15 000g for 45 min, aliquoted and stored at −80°C.
Figure 2.

Purification of ecDHFR. After two-step purification using IMAC, ecDHFR can be obtained with more than 90% purity. Lane 1, protein markers (labeled in kDa). Lane 2, fusion protein from the first step of purification. Lane 3, Ulp1 protease-treated protein mixture. Lane 4, wild-type ecDHFR after the second purification.
2.2. Crystallization
The ecDHFR–folate–NADP+ complex was crystallized de novo using the hanging-drop method at 4°C: 1 µl protein solution was added to 1 µl reservoir solution (18% PEG 400, 100 mM MnCl2, 20 mM imidazole pH 7.0). Crystals appeared in 1 d and continued to grow for 5 d until they reached their largest dimensions. Although the crystallization condition was optimized, most of the crystals were still not single and formed dense clusters (Fig. 3 ▶ a). Hence, the microseeding technique (Bergfors, 2003 ▶) was applied to obtain single crystals using a cat whisker in the following condition: 15%(v/v) PEG 400, 100 mM MnCl2, 20 mM imidazole pH 7.0. The size of these resultant crystals was usually 1.5 × 0.1 × 0.2 mm (∼0.03 mm3; Fig. 3 ▶ b), which is too small for neutron diffraction (Blakeley et al., 2008 ▶; Bennett et al., 2005 ▶). To obtain a crystal of >1 mm3 in size and to reduce the data-collection time, the macroseeding technique (Bergfors, 2003 ▶) was applied. A small single crystal was picked from the original crystallization drop and transferred to 2 µl washing solution [20%(v/v) PEG 400, 100 mM MnCl2, 20 mM imidazole pH 7.0]. After four consecutive rounds of washing using the washing solution, the crystal was transferred into a large-volume crystallization drop [30 µl protein solution (40 mg ml−1) was added to 30 µl reservoir solution as a sitting drop], which was pre-equilibrated against 1000 µl reservoir solution (15% PEG 400, 100 mM MnCl2, 20 mM imidazole pH 7.0) overnight at 4°C. This crystal continued to grow and reached a final size of 4 × 1.3 × 0.7 mm (∼3.6 mm3) in one month (Fig. 3 ▶ c).
Figure 3.

Crystallization of the ecDHFR–folate–NADP+ complex. (a) De novo crystallization using the hanging-drop method. (b) Single crystals obtained using the microseeding technique. (c) The 4 × 1.3 × 0.7 mm (3.6 mm3) size crystal obtained from the macroseeding technique.
2.3. Data collection and processing
The large crystal was mounted in a quartz capillary containing the same reservoir solution listed above except it was formulated in 100% D2O (Fig. 4 ▶ a). The labile H atoms were allowed to exchange with D by vapor diffusion for several weeks before starting data collection. Quasi-Laue neutron diffraction data were collected to 2.0 Å resolution at room temperature on the IMAGINE beamline (Meilleur et al., 2013 ▶) located at HFIR (Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA; Fig. 4 ▶ b). The crystal was held stationary at a different ϕ setting for each exposure. A total of 33 images were collected with an average exposure time of 12 h per image from four different crystal orientations. The neutron data were processed using LAUEGEN (Campbell, 1995 ▶), which was modified to account for the cylindrical geometry of the detector (Campbell et al., 1998 ▶). LSCALE (Arzt et al., 1999 ▶) was used to determine the wavelength-normalization curve using the intensities of symmetry-equivalent reflections measured at different wavelengths. No explicit absorption corrections were applied. These data were then merged in SCALA, which is incorporated in the CCP4 program suite (Winn et al., 2011 ▶). The statistics of the neutron data collection are shown in Table 1 ▶.
Figure 4.

Neutron data collection of the large ecDHFR crystal. (a) The crystal mounted in a quartz capillary. (b) A Laue neutron diffraction pattern of the crystal.
Table 1. Data collection and processing.
Values in parentheses are for the highest resolution shell.
| Data collection | Neutron | X-ray |
|---|---|---|
| Space group | P212121 | |
| Unit-cell parameters (Å, °) | a = 34.3, b = 45.7, c = 98.9, α = β = γ = 90 | a = 34.3, b = 45.6, c = 99.0, α = β = γ = 90 |
| Resolution (Å) | 2.0 | 1.6 |
| Unique reflections | 8745 | 20795 |
| Multiplicity | 7.5 (5.8) | 4.1 (4.0) |
| Completeness (%) | 79.3 (61.3) | 97.7 (94.0) |
| R merge † | 0.188 (0.305) | 0.097 (0.446) |
| 〈I/σ(I)〉 | 5.3 (3.2) | 13.7 (2.97) |
R
merge = 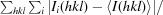
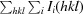 , where Ii(hkl) is the measured intensity and 〈I(hkl)〉 is the mean intensity of all measured observations of reflection hkl.
, where Ii(hkl) is the measured intensity and 〈I(hkl)〉 is the mean intensity of all measured observations of reflection hkl.
A small crystal obtained using the microseeding technique and appropriate for room-temperature X-ray diffraction was mounted and equilibrated against the D2O-containing reservoir solution in the same fashion as the crystal used for neutron diffraction. The 1.6 Å resolution X-ray diffraction data were collected using a Rigaku HomeFlux X-ray diffractometer equipped with a MicroMax-007 HF X-ray generator, Osmic VariMax optics and an R-AXIS IV++ image-plate detector. The diffraction data were indexed, integrated and scaled using the HKL-3000 software suite (Minor et al., 2006 ▶). The statistics of X-ray data collection are shown in Table 1 ▶.
3. Results and discussion
Joint XN refinement (Adams et al., 2009 ▶) of the ternary-complex structure is in progress utilizing PHENIX (Adams et al., 2010 ▶), which allows the use of both the neutron and the X-ray data for complete structural refinement (Afonine et al., 2010 ▶). The preliminary results show that D atoms exchanged on the amide backbone and many side chains were visible in the 2F o − F c and F o − F c nuclear density maps. Owing to their incoherence and contribution to negative neutron scattering, chemically non-exchangeable H atoms at aliphatic and aromatic CH groups were detected as troughs in the 2F o − F c nuclear density map. Some dynamic side chains of residues, such as Lys and Arg, were not clear in the nuclear density map owing to the low data completeness, but are clearly visible in the electron-density map because of its higher resolution and data completeness. Detailed structural analysis of the ternary complex will be reported elsewhere.
The reaction catalyzed by DHFR has been studied in depth over the past 50 years using different spectroscopic, structural and theoretical methods. However, without direct observation of H atoms, the mechanistic details still remain controversial. Here, we used NC to study the structure of ecDHFR–folate–NADP+ ternary complex, a pseudo model of the Michaelis complex. Elucidating the protonation states of the ligands, key catalytic residues and solvent molecules should be helpful to solve the longstanding puzzle.
To collect neutron diffraction data with reasonable resolution (higher than 2.2 Å) for H-atom identification and within a reasonable length of time, it is important to obtain a large crystal (>0.1 mm3), one of the bottlenecks for NC (Blakeley et al., 2008 ▶). We first used the microseeding technique to obtain small single crystals and then applied the macroseeding technique to enhance the three-dimensional growth of one crystal in a large volume of a pre-equilibrated crystallization drop. No other crystals appeared in the drop used for macroseeding; thus, the protein in solution in the equilibrated drop did not form new nucleations and was incorporated into the lattice of the single transferred crystal. We found that during the crystal transfer it is important to remove potential microcrystals by washing the seed thoroughly. Otherwise, these invisible crystals attached to the seed surface would become nuclei and consume free protein in the crystallization drop that would grow into larger crystals, thus not allowing the macroseed to grow to a large volume. As has been detailed previously, we also found that it is important to slightly decrease the precipitant concentration during seeding in order to prevent the appearance of new crystals (Bergfors, 2003 ▶).
The flux on currently available neutron beamlines is considerably lower than that of synchrotron X-ray beamlines or even in-house X-ray diffractometers. Thus, the time required for data collection can range from days to weeks (Niimura & Bau, 2008 ▶; Blakeley et al., 2008 ▶; Bennett et al., 2008 ▶). In this work, we limited the data collection to 33 frames (12 h per frame) to keep the total data-collection time to around two weeks (16.5 d). The overall data completeness is 79.3% and the data completeness of the highest resolution shell is 61.3% (Table 1 ▶). In this respect, the inclusion of the X-ray diffraction data (data completeness of 97.7 and 94.0% overall and for the highest resolution shell, respectively) in structure refinement was helpful for increasing the data-to-parameter ratio and also for improving the accuracy of the final model (Adams et al., 2009 ▶; Table 1 ▶). Combined with the previous computational, structural and biophysical studies, we expect that the XN refined structure will provide new understanding of the reaction mechanism of DHFR.
Acknowledgments
This work was partly supported by the Center for Structural Molecular Biology supported by the US Office of Biological and Environmental Research, US Department of Energy, under FWP ERKP752. PL was partly supported by an NIH–NIGMS-funded consortium (R01GM071939) between ORNL and LBNL to develop computational tools for neutron protein crystallography. MAW is supported by NIH grant R01GM092999. This research used facilities sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy. QW was partly supported by grant 2013CXJ083 from Yangzhou University, China.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Adams, P. D., Mustyakimov, M., Afonine, P. V. & Langan, P. (2009). Acta Cryst. D65, 567–573. [DOI] [PMC free article] [PubMed]
- Afonine, P. V., Mustyakimov, M., Grosse-Kunstleve, R. W., Moriarty, N. W., Langan, P. & Adams, P. D. (2010). Acta Cryst. D66, 1153–1163. [DOI] [PMC free article] [PubMed]
- Arzt, S., Campbell, J. W., Harding, M. M., Hao, Q. & Helliwell, J. R. (1999). J. Appl. Cryst. 32, 554–562.
- Bajorath, J., Kraut, J., Li, Z. Q., Kitson, D. H. & Hagler, A. T. (1991). Proc. Natl Acad. Sci. USA, 88, 6423–6426. [DOI] [PMC free article] [PubMed]
- Bennett, B. C., Gardberg, A. S., Blair, M. D. & Dealwis, C. G. (2008). Acta Cryst. D64, 764–783. [DOI] [PubMed]
- Bennett, B., Langan, P., Coates, L., Mustyakimov, M., Schoenborn, B., Howell, E. E. & Dealwis, C. (2006). Proc. Natl Acad. Sci. USA, 103, 18493–18498. [DOI] [PMC free article] [PubMed]
- Bennett, B. C., Meilleur, F., Myles, D. A. A., Howell, E. E. & Dealwis, C. G. (2005). Acta Cryst. D61, 574–579. [DOI] [PubMed]
- Bennett, B. C., Xu, H., Simmerman, R. F., Lee, R. E. & Dealwis, C. G. (2007). J. Med. Chem. 50, 4374–4381. [DOI] [PubMed]
- Bergfors, T. (2003). J. Struct. Biol. 142, 66–76. [DOI] [PubMed]
- Bhabha, G., Tuttle, L., Martinez-Yamout, M. A. & Wright, P. E. (2011). FEBS Lett. 585, 3528–3532. [DOI] [PMC free article] [PubMed]
- Blakeley, M. P., Langan, P., Niimura, N. & Podjarny, A. (2008). Curr. Opin. Struct. Biol. 18, 593–600. [DOI] [PMC free article] [PubMed]
- Campbell, J. W. (1995). J. Appl. Cryst. 28, 228–236.
- Campbell, J. W., Hao, Q., Harding, M. M., Nguti, N. D. & Wilkinson, C. (1998). J. Appl. Cryst. 31, 496–502.
- Chen, Y. Q., Kraut, J., Blakley, R. L. & Callender, R. (1994). Biochemistry, 33, 7021–7026. [DOI] [PubMed]
- Cummins, P. L. & Gready, J. E. (2001). J. Am. Chem. Soc. 123, 3418–3428. [DOI] [PubMed]
- Falzone, C. J., Wright, P. E. & Benkovic, S. J. (1994). Biochemistry, 33, 439–442. [DOI] [PubMed]
- Fisher, S. J., Blakeley, M. P., Cianci, M., McSweeney, S. & Helliwell, J. R. (2012). Acta Cryst. D68, 800–809. [DOI] [PubMed]
- Khavrutskii, I. V., Price, D. J., Lee, J. & Brooks, C. L. (2007). Protein Sci. 16, 1087–1100. [DOI] [PMC free article] [PubMed]
- Maharaj, G., Selinsky, B. S., Appleman, J. R., Perlman, M., London, R. E. & Blakley, R. L. (1990). Biochemistry, 29, 4554–4560. [DOI] [PubMed]
- Meilleur, F., Munshi, P., Robertson, L., Stoica, A. D., Crow, L., Kovalevsky, A., Koritsanszky, T., Chakoumakos, B. C., Blessing, R. & Myles, D. A. A. (2013). Acta Cryst. D69, 2157–2160. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Niimura, N. (1999). Curr. Opin. Struct. Biol. 9, 602–608. [DOI] [PubMed]
- Niimura, N. & Bau, R. (2008). Acta Cryst. A64, 12–22. [DOI] [PubMed]
- Reyes, V. M., Sawaya, M. R., Brown, K. A. & Kraut, J. (1995). Biochemistry, 34, 2710–2723. [DOI] [PubMed]
- Sawaya, M. R. & Kraut, J. (1997). Biochemistry, 36, 586–603. [DOI] [PubMed]
- Schweitzer, B. I., Dicker, A. P. & Bertino, J. R. (1990). FASEB J. 4, 2441–2452. [DOI] [PubMed]
- Sharma, M. & Chauhan, P. M. (2012). Future Med. Chem. 4, 1335–1365. [DOI] [PubMed]
- Shrimpton, P. & Allemann, R. K. (2002). Protein Sci. 11, 1442–1451. [DOI] [PMC free article] [PubMed]
- Sukumar, N., Mathews, F. S., Langan, P. & Davidson, V. L. (2010). Proc. Natl Acad. Sci. USA, 107, 6817–6822. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yuthavong, Y. et al. (2012). Proc. Natl Acad. Sci. USA, 109, 16823–16828.