

Abstract
The classical estrogen receptors, estrogen receptor-α and estrogen receptor-β are well established in the regulation of body weight and energy homeostasis in both male and female mice, whereas, the role for G protein-coupled estrogen receptor 1 (GPER) as a modulator of energy homeostasis remains controversial. This study sought to determine whether gene deletion of GPER (GPER KO) alters body weight, body adiposity, food intake, and energy homeostasis in both males and females. Male mice lacking GPER developed moderate obesity and larger adipocyte size beginning at 8 weeks of age, with significant reductions in energy expenditure, but not food intake or adipocyte number. Female GPER KO mice developed increased body weight relative to WT females a full 6 weeks later than the male GPER KO mice. Female GPER KO mice also had reductions in energy expenditure, but not significant increases in body fat content. Consistent with their decrease in energy expenditure, GPER KO males and females showed significant reductions in two brown fat thermogenic proteins. GPER KO females, prior to their divergence in body weight, were less sensitive than WT females to the feeding-inhibitory effects of leptin and CCK. Additionally, body weight was not as modulated by ovariectomy or estradiol replacement in GPER KO mice. Estradiol treatment activated phosphorylated extracellular signal-regulated kinase (pERK) in WT but not GPER KO females. For the first time, GPER expression was found in the adipocyte but not the stromal fraction of adipose tissue. Together, these results provide new information elucidating a sexual dimorphism in GPER function in the development of postpubertal energy balance.
Keywords: G protein coupled receptor 30, GPER; estrogen receptor-α, ERα/ERS1; adiposity; Cholecystokinin, CCK; leptin
Introduction
The role of sex hormones in regulating metabolic function has become a topic of interest and is of great importance, largely due to the function of sex hormones in regulating the sexual dimorphisms seen in body weight, food intake, obesity, reproduction, and eating disorders. Estrogens (17β-estradiol) have been demonstrated to play a role in regulating adiposity, reproduction [1] and insulin sensitivity [2,3]. Additionally, recent papers have demonstrated a critical role for three putative estrogen receptors in modulating adiposity, insulin sensitivity, and energy homeostasis. Specifically, estrogen receptor alpha (ERα/ERS1), estrogen receptor beta (ERβ/ERS2), as well as G protein-coupled estrogen receptor (GPER) have been demonstrated to regulate energy homeostasis [3-6]. Importantly, the findings with respect to GPER and its effects on metabolic homeostasis differ between publications. In the late 1990s, four independent laboratories cloned a putative G protein-coupled receptor (GPCR) using different approaches [7-10]. Expression studies indicate GPER mRNA is expressed in numerous tissues throughout the body (e.g. placenta, lung liver, prostate, ovary, and brain), although there are contradictions in patterns of tissue expression [7,9,10].
Estrogens bind to GPER and the selective estrogen antagonists, ICI 182,780 and tamoxifen, block GPER’s estrogenic effects [11]. Despite demonstrations of the estrogenic activity of GPER, controversy about the cellular and organismal function of GPER remains. GPER knockout (GPER KO) mice have been used to explore the in vivo estrogenic activation of GPER to mediate thymus size (mice were 8-10 weeks old [12]), vascular disease (mice were 10-11 months old [13]), glucose tolerance (mice were 6 months of age [14]), pancreatic islet survival (mice were 8 weeks old [15], and reproductive function (mice were 8-10 weeks old [16]). Importantly, the role for GPER in regulating energy homeostasis remains controversial. Haas et al. [13] reported increased body weight and visceral adiposity in male and female GPER KO mice using the Wang et al. mouse model (mice were 10-11 months of age [12]); however, these findings contrasted those of Liu et al. using the same mutant mouse model from a different laboratory (mice were 7-9 weeks in the Liu et al. experiments [15]). Other laboratories have reported that neither male nor female mice show significant differences in body weight or visceral adiposity when compared to their WT littermates (mice were 3-4 months of age [16,17]). Martensson et al., [14] reported female, but not male mutant mice present with reduced body weight and skeletal growth (mice were 3-4 months of age). In contrast, Isensee et al., reported no significant differences in body weight or fat mass between WT and GPER KO animals when exposed to either a normal or high fat diet (mice were 6 months old [18]). And lastly, Sharma et al., [6] recently demonstrated male GPER mice have increased body adiposity (mice were 12 months old), insulin resistance (mice were 12-18 months old), increased proinflammatory cytokines (mice were 12-24 months old), and reductions in circulating adiponectin levels (mice were 12-24 months old). These disparate findings with respect to the role of GPER in modulating energy homeostasis suggest that environmental factors, method of gene deletion, age of mice, and the genetic background of GPER KO mouse models may contribute to controversies associated with the role of GPER in body weight homeostasis.
Recently, Sharma et al., demonstrated that male GPER KO mice have increased body weight, adiposity, inflammation, and energy expenditure [6]. Importantly, the temporal development of body weight gain and sexual dimorphisms in the onset of weight gain have not been determined. Here we present data demonstrating a metabolic phenotype of GPER KO mice developed by Wang et al., [12]. Our mouse model was generated using a heterozygous breeding strategy. We compared the phenotype of GPER KO mice directly to WT littermates obtained from these heterozygous intercrosses. Additionally, our GPER KO mice have been backcrossed for more than 10 generations onto the C57BL6 background indicating a 99% inbred background strain. We determined that GPER KO males and females differ from WT males and females with respect to circulating concentrations of leptin, insulin, adiponectin, thermogenic brown adipose tissue proteins, and sensitivity to leptin, CCK and estradiol. Lastly, we demonstrate the intracellular location of GPER within adipose tissue.
Methods
Animals
All work was conducted in accordance with the animal care committee at UT Southwestern Medical Center. The GPER KO mice and wild-type (WT) littermates were a generous gift of Dr. Janice S. Rosenbaum (Proctor & Gamble, Cincinnati, OH) [6,12,13]. GPER KO mice were generated by transfecting embryonic stem cells with a targeting construct containing the GPER (gene encoding GPER) locus followed by verification of successful gene targeting [12]. The GPER mutant mice used in these studies were bred in-house using a heterozygous breeding strategy; homozygosity was confirmed via quantitative real-time PCR. We directly compared WT and mutant littermates obtained from heterozygous intercrosses. All mice were housed in individual cages at 23-24°C on a 12-h light, 12-h dark cycle (lights on at 0600 h) and fed a phytoestrogen free, low fat (4% kcal from fat) pellet diet (#2016, Teklad Global, Harlan Laboratories) ad libitum. All studies were carried out in GPER KO mice and their WT littermates at the ages indicated.
Food intake and body weight measurements
Daily food intake and body weight measurements were made in individually housed male and female GPER KO mice and their respective WT littermates (n = 18-19/genotype for males representing different litters; n = 12/genotype for females) for 16 weeks. Food intake was tracked daily, and week 13 is represented in all figures. All mice were maintained on the above-described phytoestrogen free diet for the entire duration of the study.
Body adiposity
Body adiposity was assessed by nuclear magnetic resonance (NMR) (EchoMRI; EchoMedical Systems, Houston TX) as described previously [19]. Briefly, 13 week old mice (n = 8/genotype for males; n = 8/genotype for females from different litters) were placed in a restraint tube and inserted into the NMR to provide estimates of total lean tissue, fat tissue, and water. This time point was chosen because male GPER KO mice had slightly diverged from WT mice and GPER KO female mice had not yet significantly diverged from WT mice with respect to body weight.
Measurements of energy expenditure
Energy expenditure was measured with a Physioscan System (Accuscan Instruments, Columbus, OH) in WT and GPER KO male (n = 8) and female (n = 8) age and body weight-matched (10 week old) mice. Whole animal heat production was determined as calories per hour. Volume of oxygen consumed (VO2) was determined as milliliters per kilogram body weight per minute (ml*kg−1.min−1) and was subsequently adjusted to kilograms of lean mass. Flow rates were set at 0.5 l/min for each mouse. All animals were acclimatized to the chambers for at least 48h to mitigate the stress of the new environment. VO2 and VCO2 samples were measured following acclimatization at 10 min intervals for each animal and subsequently binned into 6h, 12h, or 24h periods. Animals had ad libitum access to chow throughout the experiment and no differences in food intake between the GPER KO and WT mice were noted.
Plasma leptin, SAA3, adiponectin and insulin measurements
Food was removed from the home cages of 13 week old male and female GPER KO and WT mice in the middle of the light phase (1200h) (n = 8/genotype for males; n = 8/genotype for females from different litters in proestrus). The age of mice for this study was chosen because it was prior to their divergence in body weight. Mice were sacrificed 2-hrs prior to lights off and trunk blood was collected. Plasma leptin, serum amyloid A-3 (SAA3), and insulin concentrations were measured using the respective mouse ELISA kits (Crystal Chem Inc., Downers Grove, IL) according to the manufacturer’s instruction. Serum adiponectin levels were measured using western blot analyses. Serum samples were mixed with 2× Laemmli sample buffer. Protein samples were loaded on 10% Bis-Tris NuPAGE gels (1.5 mm; Invitrogen) for analysis with 1:500 polyclonal anti-adiponectin antibody followed by incubation with IRDye 800–coupled goat anti-rabbit secondary antibody (Rockland Immunochemicals). Each band was detected and quantitated by the Odyssey Infrared Imaging System (LI-COR Biosciences).
Adipocyte Cell Size and Number
Gonadal and brown fat pads of male and female, age-matched (13 week old) GPER KO and WT mice (n = 5/genotype from different litters) were isolated. The gonadal fat pad was chosen because of the reported high correlation between gonadal/visceral fat deposition and the medical complications associated with obesity. To determine adipose tissue morphology and adipocyte size, 5μm sections of gonadal and brown fat tissues were stained with hematoxylin and eosin (H&E). The analysis was carried out within a designated field across all the samples. The gonadal fat pad images were viewed under rhodamine fluorescence and imaged using a Leica DM2000 compound epifluorescence microscope equipped with an Optronics Microfire Color CCD Camera and analyzed for adipocyte area using NIH ImageJ software. Eight hundred to one thousand cells from each sample were included in the analysis.
Adipocyte/SV fractionation
To determine where GPER is expressed within adipose tissues, we isolated adipocytes from the stromal vascular component of adipose tissue (SVF) using the collagenase fractionation method as previously described [20]. Briefly, gonadal adipose tissues were excised and minced into fine pieces. The adipose pieces were then digested in adipocyte isolation buffer (100mM HEPES pH7.4, 120mM NaCl, 50mM KCl, 5mM glucose, 1mM CaCl2, 1.5% BSA) containing 1mg/ml collagenase at 37°C with constant slow shaking (∼120rpm) for 2 hours. During the digestion period, the suspension was triturated several times through a pipette to dissociate the clumps. To isolate floated adipocytes, the mixture was passed through a 210μm mesh, centrifuged at 500×g for 10 minutes and floated adipocytes were collected from the top of the centrifuged effluent. The remaining suspension was then passed through an 80μm mesh to remove undigested clumps and debris. The effluent was centrifuged at 500xg for 10 minutes and the pellet washed once in 5ml PBS. After re-centrifugation, the pellet was collected (SVF).
Basal hypothalamic neuropeptide expression
Following a 12-hr fast, 13 week old WT and GPER KO mice (n = 5/genotype for males; n = 5/genotype for females in proestrus) were sacrificed 30-mins prior to the onset of the dark phase at a time point at which body weight did not significantly differ. The basal medial hypothalamus (an area critical for body weight homeostasis known to express GPER) was dissected and RNA was isolated via the Trizol method (Tri-Reagent: MRC, Cincinnati, OH) followed by a reverse transcription reaction (High capacity cDNA reverse transcription kits: Perkin Elmer, Applied Biosystems, Foster City, CA). Hypothalamic mRNA expression was analyzed for L32 (house keeping gene), Leptin receptor (long signaling form, Ob-Rb), ERα, ERβ, POMC and AgRP (Perkin Elmer, Applied Biosystems, Foster City, CA, sequences for the primers are available upon request). The cycle number at which the fluorescence exceeds the threshold of detection of L32 is subtracted from the gene of interest (ΔCT). The average DCT for each experimental group is derived from the average ΔCT of each mouse in that group. The percent change in gene expression, relative to the reference group, is defined as 100*2ΔΔCT, where ΔΔCT equals the group ΔCT minus the ΔCT of the reference group.
Effect of leptin on food intake and body weight
On the test day, 13 week old male (n = 8/genotype from different litters) and female (n = 8/genotype from different litters in proestrus) age and weight-matched GPER KO and WT mice had their food removed 2- hrs prior to lights off. The mice received a single intraperitoneal (ip) injection of 5mg/kg leptin (Human Leptin, Amylin) or vehicle (saline) 2-hrs prior to dark onset. The leptin dose was selected based on previous studies demonstrating the efficacy for the 5mg/kg dose in reducing 24h food intake [21]. Food was returned at the onset of dark and food intake was measured over the subsequent 4 and 24-hrs following the injections. All mice received leptin or saline in a counterbalanced design, with subsequent injections occurring after complete recovery of food intake and body weight to baseline levels (generally 5 days).
Effect of CCK on food intake
Male and female 14 week old GPER KO and WT mice (n = 8/genotype from different litters; females were in proestrus) were food deprived for 24-hrs. Mice received an ip injection of 4 μg/kg CCK-8 or vehicle (saline) 4-hrs into the light phase in a counterbalanced design. The selection of dose and the experimental paradigm were based on previously published studies [22,23]. Food was returned following the ip injection and intake was measured over the subsequent 15, 30, 60 and 120 minutes. All mice received CCK or saline in a counterbalanced design, with subsequent injections occurring after complete recovery of food intake and body weight to baseline levels (generally 3 days).
Effect of 17β-Estradiol replacement following Ovariectomy
Ovariectomy (OVX) or sham surgeries were performed in 10 week old weight-matched WT and GPER KO mice (n = 8/group which consisted of OVX +17β-estradiol or OVX + vehicle). The females were anesthetized, and bilateral dorsal incisions were made through the skin, such that the ovary and oviduct could be rapidly removed. In the sham operation, the ovary and oviduct were visualized but left intact before the incisions were sutured. 17β-estradiol or placebo pellets were placed subcutaneously (Innovative Research of America, Sarasota FL; 17β-estradiol 0.03 mg/pellet 60 day release (a dose that provides physiologically relevant levels of 17β-estradiol into circulation)). Food intake, body weight, and body adiposity were monitored following the surgery. The success of the OVX procedure was confirmed at sacrifice by measuring uterine weights.
Effect of 17β-estradiol on adipocyte size following ovariectomy
14 week old weight-matched WT and GPER KO mice (n = 8/group which consisted of OVX + 17β-estradiol or OVX + vehicle) were sacrificed as detailed above. Gonadal adipose tissue was isolated, weighed, and fixed with 10% formalin overnight and then stored in 50% ethanol. The fixed fat pads were sent to Richardson Molecular Pathology Core at UT Southwestern Medical Center, where the tissues were embedded with paraffin, sectioned and stained with haematoxylin and eosin (H&E). Images were viewed under rhodamine fluorescence and imaged using the Leica DM2000 compound epifluorescence microscope equipped with an Optronics Microfire Color CCD Camera and analyzed for adipocyte area using NIH ImageJ software. Eight hundred to one thousand cells from each sample were included in the analysis.
Effect of 17β-estradiol on glucose homeostasis following ovariectomy
13 week old weight-matched WT and GPER KO mice (n = 8/group which consisted of OVX +17β-estradiol or OVX + vehicle) were fasted for 3 h (starting at 8 a.m.) prior to administration of glucose (2.5 g/kg body weight) by gavage. At the indicated time points, venous blood samples were collected in heparin-coated capillary tubes from the tail vein. Food was restricted throughout the experiment.
I3vt surgery
In a separate cohort of 10 week old GPER KO and WT OVX (n = 4/ genotype) mice, a stereotaxic i3vt cannula (Plastics One, Roanoke, VA) was placed as described previously [19]. Briefly, the cannula was placed 0.825mm posterior to bregma and on the midline and the cannula tip was 4.8mm below bregma. Cannula placement was verified after the mice had regained their presurgical weight (3 or 4 days). To confirm cannula placement, mice had their food removed for 4-hrs following lights-on and were administered i3vt NPY (5 μg/1 μl). The mice that consumed more than 0.5 g of chow in the subsequent 2 h were considered to have the cannula appropriately placed [19,24].
Effect of 17β-estradiol on phosphorylated extracellular signal-related kinase (pERK)
To determine central sensitivity to 17β-estradiol, we measured 17β-estradiol-induced pERK levels in hypothalamic tissue of 13 week old OVX female GPER KO mice and their WT littermates (n = 4/group). Mice were given a single i3vt injection of 0.05 μg/1 μl of 17β-estradiol or vehicle 2-hrs prior to lights off in the fed state, and were sacrificed 15 min after the i3vt injection. The basal medial hypothalamus was processed for protein extraction. Proteins were extracted by homogenizing samples in lysis buffer [150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, protease inhibitors (Sigma), and phosphatase inhibitors (Sigma)], and centrifuged at 20,000 g for 15 min. The extracts were then subjected to SDS-PAGE followed by immunoblotting with primary antibodies against pERK1/2 (1:1000, Cell Signaling, Beverly, MA) and β-actin (1:10,000 dilution, Abcam Inc., Cambridge, MA) followed by an incubation in HRP-linked secondary antibodies (Bio-Rad, Hercules, CA). Data were expressed and analyzed relative to β-actin.
Statistical analysis
All data are presented as mean ± SEM. Experiments were analyzed by mixed ANOVAs with food intake/body weight/drug/plasma hormone levels as between-subject variables and time as a within-subjects variable. Post-hoc tests of individual groups were made using Tukey’s HSD. Significance was set at p < 0.05.
Results
Food intake and body weight
There were no differences between the GPER KO and WT mice with respect to breeding capabilities, litter size, or pup weights at time of birth. Male and female GPER KO mice were weaned at 3-4 wks of age and their food intake and body weights were determined weekly. Data presented are from a minimum of 20 different litters of mice, and there were no differences between mice within a litter or from different litters. Body weights of GPER KO mice are indistinguishable at weaning; however, as the males approach 8-10wks of age they were significantly heavier than the WT littermates (Fig. 1A). The female GPER KO mice do not differ in body weight when compared to their WT littermates (Fig. 1B) until 13 weeks when body weights began to diverge, and female GPER KO mice weighed more than WT mice. The average daily food intake in GPER KO mice (both males and females) did not differ significantly from their WT littermates (Fig. 1C).
Figure 1.

Body weight, food intake, and energy expenditure of GPER KO mice. A. Weekly body weights of male WT and GPER KO mice. B. Weekly body weights of female WT and GPER KO mice. C. Average daily food intake in male and female WT and GPER KO mice tracked at 13 weeks. D. Fat mass measured by NMR in 13 week old male and female WT and GPER KO mice. E. Energy expenditure was measured and represented by average VO2 for 10 week old male WT and GPER KO mice. F. Energy expenditure was measured and represented by average VO2 for 10 week old female WT and GPER KO mice. All data are expressed as mean ± SEM and n=12-19 per group. Values significantly different from sex- and age-matched WT controls are designated by asterisks above the column. * P<0.05.
Body adiposity
Male GPER KO mice had increased fat mass at 13 weeks of age relative to WT males ((P<0.05) Fig. 1D,). Female GPER KO mice had a trend for increased fat mass compared to WT littermates; however, this trend did not reach statistical significance prior to the divergence of body weight (Fig. 1D).
Energy expenditure
Male and female 10 week old mice were placed in a metabolic chamber to determine if differences in body weight were due to differences in energy expenditure or ambulatory activity. Energy expenditure was measured as an average VO2 during the light (12 hours), dark (12 hours) and total 24 hour cycles. Total VO2 was significantly reduced in male GPER KO mice ((P<0.05) Fig 1E). There was a trend in female GPER KO mice for reductions in VO2, and this reached significance during the dark phase ((P<0.05) Fig. 1F). There were no differences in homecage locomotor activity between GPER KO and WT mice regardless of sex (data not shown), similar to what was reported by Sharma et al., [6]. These data suggest that GPER KO mice have reduced energy expenditure which is not a result of reductions in physical activity.
Plasma Leptin, SAA3, Adiponectin and Insulin Measurements
There were no differences in fasting insulin for males (0.63 +/−0.11 ng/ml WT; 0.63 +/−0.1 ng/ml GPER KO) or females (0.35 +/−0.03 ng/ml WT; 0.38 +/−0.06 ng/ml GPER KO) at 13 weeks of age. Plasma leptin levels also did not differ for males (7.45 +/−2.22 ng/ml WT; 6.93 +/−1.66 ng/ml GPER KO) or females (4.16 +/−1.17 ng/ml WT; 4.72 +/−1.81 ng/ml GPER KO). Consistent with recent reports by Sharma et al., [6], male and female GPER KO mice have increased circulating SAA3, a marker of inflammation (Fig 2A), and reduced plasma adiponectin levels relative to WT males and females (Fig. 2B) (P<0.05).
Figure 2.
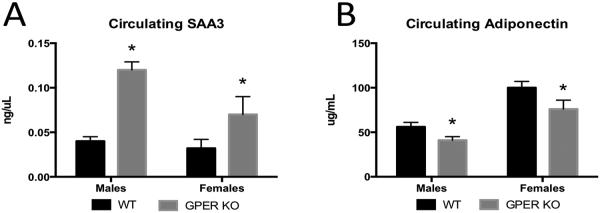
Circulating inflammatory factors and adipokines. A. Circulating SAA3 levels measured from serum in male and female 13 week old WT and GPER KO mice. B. Circulating adiponectin levels measured from serum in male and female 13 week old WT and GPER KO mice. All data are expressed as mean ± SEM and n=8-10 per group. Values significantly different from sex- and age-matched WT controls are designated by asterisks above the column. * P<0.05.
Adipocyte Cell Size and Number
To further characterize the increased adiposity in GPER KO, visceral (gonadal) adipose histology was analyzed. Male GPER KO mice had fewer adipocytes (GPER KO (272±44.5) vs. WT (358±38.2)) per μm2 tissue. The area of gonadal adipocytes was 35% greater in GPER KO male mice when compared to WT mice (Fig 3A). For the females, there was a trend for increased adipocyte size; however, this did not reach statistical significance (Fig. 3A). Representative images are shown in figure 3B.
Figure 3.

White adipose morphology and GPER expression in adipose tissues. A. Average adipocyte area as measures from H&E sections from gonadal/visceral adipose tissues in 13 week old male and female WT and GPER KO mice. B. Representative H&E sections of adipose tissues used to calculate adipocyte area from male, female WT and GPER KO mice. C. Expression of GPER in whole adipose tissue, isolated adipocytes, and the stromal vascular (SV) fraction in 13 week old male and female WT mice. All data are expressed as mean ± SEM and n=8-10 per group. Values significantly different from sex- and age-matched WT controls are designated by asterisks above the column. * P<0.05.
GPER Expression Within Adipose Tissues
To understand how GPER influences adipocyte size and adiposity, we examined the expression of GPER within different adipose tissue cell populations. We report for the first time that GPER is found predominately in the adipocyte fraction of adipose tissue and there was virtually no expression in the stromal vascular cell fraction (Fig 3C).
Brown Adipose Tissue
It is known that brown adipose tissue contributes to energy expenditure; therefore following our observation that GPER KO mice have reductions in energy expenditure, we sought to determine the morphology of the brown adipose tissue. We found both male and female GPER KO mice had significantly more lipid accumulation within brown adipose tissues than WT mice (Fig. 4A) and this is consistent with reductions energy expenditure. To further explore how GPER might influence energy expenditure, we performed qPCR and assayed genes associated with thermogenic activity in brown adipose tissue. We found a significant reduction in uncoupling protein-1 (UCP1) and β-3 adrenergic receptor (β-3AR) gene expression in male GPER KO compared to WT mice (Fig. 4B), again consistent with reductions in energy expenditure. For GPER KO females (Fig. 4C) we found significant reductions in β-3AR. Together, these findings indicate GPER has a functional role in the thermogenic properties of brown adipose tissue.
Figure 4.

Brown adipose tissue. A. Representative H&E sections of brown adipose tissues from 13 week old WT and GPER KO male and female mice. B. Gene expression of brown adipose tissue genes involved in lipid accumulation and energy expenditure in 13 week old male WT and GPER KO mice. C. Gene expression of brown adipose tissue genes involved in lipid accumulation and energy expenditure in 13 week old female WT and GPER KO mice. Uncoupling protein-1 (UCP1), Peroxisome proliferator activated receptor gamma (PPARg), PPARg coactivator-1 alpha (PGC-1a), beta-3 adrenergic receptor (b3-AdR), PRD1-BF1-RIZ1 homologous domain containing 16 (PRDM16). All data are expressed as mean ± SEM and n=8-10 per group. Values significantly different from sex- and age-matched WT controls are designated by asterisks above the column. * P<0.05.
Hypothalamic Gene Expression
To determine differences in hypothalamic gene expression for genes known to regulate energy homeostasis [25], we compared medial basal hypothalamic gene expression in 14 week old male and proestrus female GPER KO mice with WT littermates prior to their divergence in body weight. Female GPER KO mice have decreased hypothalamic ERα mRNA and increased ERβ mRNA (Fig. 5A, B) relative to WT mice. No significant differences in Ob-Rb, AgRP or POMC levels were observed in GPER KO and WT female mice (data not shown). There were no differences in hypothalamic gene expression in the males (Fig 5A, B). These data suggest that deletion of GPER does not significantly alter basal hypothalamic gene expression in males, but plays some role in the regulation of expression of ERα and ERβ in females, which may influence body weight as they age.
Figure 5.

Expression of other ERs and effects of GPER on leptin and CCK sensitivity. A. Basal medial hypothalamic expression of ERα in 13 week old male and female WT and GPER KO mice. B. Basal medial hypothalamic expression of ERβ in 13 week old male and female WT and GPER KO mice. C. Average food intake of 13 week old male WT and GPER KO mice 4 and 24 hours after leptin administration. D. Average food intake of 13 week old female male WT and GPER KO mice 4 and 24 hours after leptin administration. E. Average food intake of 14 week old male WT and GPER KO mice 30 min after CCK administration. F. Average food intake of 14 week old female WT and GPER KO mice 30 min after CCK administration. All data are expressed as mean ± SEM and n=8-10 per group. Values significantly different from sex- and age-matched WT controls are designated by asterisks above the column. * P<0.05.
Anorexic Effect of Peripherally Injected Leptin
Previously, we and others have determined that 17β-estradiol enhances the anorexigenic effects of leptin [19,26]. Therefore, we determined whether GPER plays a role in the anorexic effects of leptin. Age and weight- matched male and female GPER KO and WT littermates were injected with leptin intraperitoneally. As previously mentioned, there were no baseline differences in serum leptin levels between GPER KO and WT mice of either sex. Leptin-treated male GPER KO mice showed a 22% and 15% reduction in 2 and 24-hr food intake, respectively, which was comparable to WT mice (Fig 5C). Leptin significantly reduced food intake in the WT females; however, leptin treated female GPER KO mice failed to show a significant reduction in food intake at 2-hr or 24-hr compared to vehicle-injected female WT mice (Fig 5D). These data suggest loss of GPER impacts leptin mediated reductions in food intake in females but not males.
Anorexic Effect of IP CCK in GPER KO vs. WT mice
Estrogens have been demonstrated to influence meal size and short-term food intake by increasing sensitivity to the satiation signal cholecystokinin (CCK) [27,28]. Due to the rapid effects on feeding induced by CCK, we hypothesized activation of GPER may mediate CCK-induced satiation. Additionally, prandial secretion of CCK is sexually dimorphic, with females having greater secretion than males [29]. We therefore determined whether male and female GPER KO mice would have impairments in sensitivity to CCK-induced satiety. To do so, we compared food intake at 15, 30, 60, 90, and 120-mins following ip CCK injections in male and female GPER KO and WT mice (data are shown for the 30 min time point). Male WT and GPER KO mice had a significant reduction (P<0.05) in food intake at 15, 30, 60, 90, and 120-mins (30 min time point is shown (Fig 5E)) following CCK. WT females responded to CCK, while female GPER KO mice failed to show significant reductions in food intake at any of the time points (30 min time depicted (Fig. 5F)). These data suggest loss of GPER impacts CCK-induced reductions in food intake in females but not males.
17β-Estradiol-Induced Reductions in Body Weight Following OVX
To determine a potential role for GPER in mediating 17β-estradiol regulation of body weight, we compared food intake and body weights in OVX +/− 17β-estradiol-replaced WT and GPER KO OVX mice. We found 17β-estradiol significantly reduced body weights of OVX WT mice (Fig 6A); however, there was an attenuated reduction in body weight in OVX GPER KO mice treated with 17β-estradiol ((p<0.05) Fig 6B). Further, the decrease in body weight in WT mice was due to a decrease in body fat (Fig 6C), which was attenuated in 17β-estradiol treated GPER KO mice, further suggesting that GPER participates in estrogen-induced weight regulation in females.
Figure 6.

The effects of 17β-estradiol on body weight, composition, adipocyte morphology and glucose homeostasis in OVX WT and GPER KO females. A. Daily body weight change after OVX in WT mice treated with vehicle or 17β-estradiol. B. Body weight change after OVX in GPER KO mice treated with vehicle or 17β-estradiol. C. Fat mass in 14 week old OVX WT and GPER KO treated with vehicle or 17β-estradiol. D. Oral glucose tolerance test in 13 week old OVX WT mice treated with vehicle or 17β-estradiol. E. Oral glucose tolerance test in 13 week old OVX GPER KO mice treated with vehicle or 17β-estradiol. F. Average adipocyte area as measures from H&E sections from visceral adipose tissues in 14 week old OVX WT and GPER KO mice treated with vehicle or 17β-estradiol. G. Representative H&E sections of adipose tissues measured in F. All data are expressed as mean ± SEM and n=8-12 per group. *P<0.05 compared with vehicle treatment.
17β-Estradiol-Induced Improvements in Glucose Homeostasis Following OVX
To determine a potential role for GPER in mediating 17β-estradiol regulation of glucose homeostasis following OVX, we compared glucose homeostasis in OVX +/− 17β-estradiol-replaced WT and GPER KO OVX mice. We found 17β-estradiol significantly improved glucose homeostasis in OVX WT mice (Fig 6D); however, there were no improvements in glucose homeostasis in OVX GPER KO mice treated with 17β-estradiol (Fig 6E), suggesting that GPER participates in estrogen-induced improvements in glucose homeostasis in females.
17β-Estradiol-Induced Reductions in Adipocyte Size Following OVX
To assess GPER’s role in mediating 17β-estradiol reductions in adipocyte size following OVX, we compared adipocyte size/morphology in OVX +/− 17β-estradiol-replaced WT and GPER KO OVX mice. We found 17β-estradiol significantly reduced adipocyte size in OVX WT mice; however, there was an attenuated reduction in adipocyte size in OVX GPER KO mice treated with 17β-estradiol ((p<0.05) Fig 6F, G), suggesting that GPER participates in 17β-estradiol-induced alterations in adipose tissue function/morphology in females.
17β-Estradiol-Induced pERK Following OVX
Previous studies have demonstrated that estrogenic activation of the MAP kinase Erk 1/2 pathway is mediated by ERα [30]. Moreover, there are data indicating estrogenic activation of GPER induces pERK [31]. 17β-estradiol induced pERK in the medial basal hypothalamus of OVX WT but not OVX GPER KO mice (Fig 7). These data suggest GPER KO mice are less sensitive to 17β-estradiol-induced pERK in the CNS and these data are consistent with the attenuated response to 17β-estradiol-induced improvements in body weight, glucose homeostasis, and reductions in fat cell size following OVX in the GPER KO mice.
Figure 7.

17β-estradiol induced phosphorylation of ERK in the basal medial hypothalamus in WT and GPER KO OVX females. A single i3vt injection of 0.05 μg/μl 17β-estradiol increased pErk 1/2 in OVX WT but not OVX GPER KO females (Fig. 7). All data are expressed as mean ± SEM and n=5 per group. *P<0.05 compared with vehicle treatment.
Discussion
The main findings of this study are that there is a strong sexual dimorphism in the temporal onset of body weight gain in GPER KO mice. We also found that: 1) male GPER KO mice develop moderate obesity as they age and this is associated with reductions in energy expenditure, increased fat cell size, and increased lipid in brown adipose tissue; 2) female GPER KO do not differ with respect to adiposity when compared to WT mice initially; however, as the mice age there is a divergence in body weight, with GPER KO females having increased body weight relative to WT females; 3) prior to the divergence of body weight, female GPER KO mice are less sensitive to modulators of food intake such as CCK and leptin; 4) ovariectomy induces weight gain in WT but not GPER KO mice and 17β-estradiol replacement was less affective in modulating body weight and glucose homeostasis in the GPER KO relative to WT mice. Lastly, central administrations of 17β-estradiol activate pERK in WT but not OVX GPER KO mice. These data indicate that GPER KO females are less sensitive to the effects of estrogens on food intake and energy homeostasis.
Body adiposity/body weight measurements
We found male GPER KO mice are significantly heavier than WT littermates as they attained puberty. With respect to the male phenotype, our results replicate those reported by Haas et al., [13] and demonstrate 10-11 week old GPER KO males have increased body weight and fat mass when compared to WT littermates. Additionally, our results are consistent with recent findings from Sharma et al., [6]; however, we extend these findings and demonstrate GPER KO females also diverge in body weight as they age.
Decreased metabolic rate
Deficits in energy expenditure or ambulatory activity can cause increased body weight and are influenced by estrogens [32-34]. We found male GPER KO mice have increased body weight due to reductions in energy expenditure. Interestingly, female GPER KO mice, prior to the divergence in body weight, had reductions in energy expenditure that achieved significance only during the dark phase. It is possible that as GPER KO females age, reductions in energy expenditure facilitate body weight gain.
Plasma leptin, SAA3, insulin and adiponectin
Sharma et al., [6] found that weight gain in male GPER KO mice was associated with increased fasting plasma concentrations of cholesterol, triglycerides, and insulin, as well as reductions in adiponectin. Here we confirm their findings and demonstrate both GPER KO males and females have significantly lower adiponectin levels when compared to weight-matched WT mice. Additionally, Sharma et al., [6] determined male GPER KO mice had increased immunomodulatory cytokines; our data extend their findings and demonstrate higher circulating levels of SAA3, a marker of inflammation, in both GPER KO males and females when compared to WT mice.
Adipocyte size, number
Recently, GPER was reported to be expressed in adipose tissue of both male and female WT mice [13], and in human adipose tissue [35]. Here we demonstrate GPER is expressed predominately in the adipocyte, and not in the stromal vascular fraction, within adipose tissues. Additionally, we found that increased body weights in male GPER KO mice were accompanied by a significant increase in adiposity and number of large adipocytes. We also confirmed our previous findings that both lean and fat mass is increased in GPER KO males [36], which is consistent with the recently published findings by Sharma et al., [6]. GPER KO females showed a trend towards increased fat mass prior to the divergence of body weight. In the future, it will be important to determine adipose tissue mass and histology once body weights have diverged in the GPER KO females. We have recently characterized ERα in the adipocyte fraction of adipose tissue [33] and uncovered a potential cross talk between estrogen receptors in regulating adipose tissue function. It is possible the absence of GPER in adipose tissue may result in changes in the levels of the other estrogen receptors. To address this, we have bred the GPER KO to ERαKO and ERβKO mice; data will be forthcoming.
Hypothalamic gene expression
NPY/AgRP and POMC neurons are major hypothalamic targets for the anorectic actions of estrogens [36-38]. We did not observe differences in gene expression for AgRP or POMC in GPER KO mice. Interestingly, we saw reduced expression of ERα and increased ERβ expression in the GPER KO females when compared to the WT mice. These data suggest crosstalk between GPER and the expression of ERα and ERβ. Shi et al., [38] have suggested an important interaction between estrogen receptors in modulating energy homeostasis; our data support this hypothesis. Specifically, it is possible that reductions in hypothalamic ERα facilitates weight gain in the GPER KO females, an idea consistent with our previous publication [39] demonstrating that reductions in hypothalamic ERα are associated with changes in energy expenditure and weight gain.
Anorexic effect of ip leptin in GPER KO vs. WT mice
17β-estradiol enhances the anorexigenic effects of leptin [19,26]; therefore, we compared the sensitivity to leptin in GPER KO vs. WT mice. In males, both WT and GPER KO had an anorectic response to leptin at 2-hrs and 24-hrs. In females, however, GPER KO mice were less responsive than WT mice at 2-hrs and 24-hrs following leptin injections. Plasma insulin and leptin levels (along with estrogen and testosterone, data not shown) did not differ between GPER KO and WT littermates in males or females. Additionally there were no significant differences in hypothalamic leptin receptor expression between GPER KO and WT mice (data not shown). These data suggest that GPER mediates leptin-induced satiety in females, but not in males. The potential crosstalk between estrogens and GPER, and their influence on leptin sensitivity, remains unknown; however, it is possible reductions in hypothalamic ERα in GPER KO females may be associated with the attenuation in sensitivity to leptin we observed in the GPER KO females.
Anorexic effect of ip CCK in GPER KO vs. WT mice
Apart from leptin, there are many other hormonal signals that control feeding behavior. CCK is released from the small intestine, particularly in response to lipid rich emulsions, and leads to rapid satiation in humans and mice. Additionally, prandial secretion of CCK is sexually dimorphic and is greater in females than males [29]. Our findings suggest female GPER KO mice are less sensitive to anorexia induced by peripheral CCK administration than WT mice; however there are no differences in CCK sensitivity between the male GPER KO and the WT mice. Additionally, we found changes in food intake across the female WT estrus cycle, which were not observed in the GPER KO mice (data not shown). These data suggest the rapid estrogenic enhancement of CCK-induced satiety may be mediated through GPER.
17β-estradiol effect on body weight following OVX
Following OVX there is a significant increase in food intake and body weight, which is attenuated by 17β-estradiol [33,39,40]. Previously, Otto et al. demonstrated 17β-estradiol responses in OVX GPER KO do not differ from WT mice with respect to 17β-estradiol’s impact on the uterus, thymus and the mammary gland [16]. Furthermore, Windahl et al. found no impairments in the GPER’s response to 17β-estradiol [17]. Importantly, none of these studies focused on the estrogenic effect on body weight following OVX in GPER KO mice. Here we present data suggesting an attenuated body weight and adiposity response to 17β-estradiol in OVX GPER KO mice when compared to WT mice. These findings support the idea that estrogens interact with GPER and are important in body weight regulation. Further, we demonstrate that the estrogenic effects of improved glucose homeostasis and decreased adipocyte size are also, in part, regulated by GPER. An alternative explanation is that reductions in hypothalamic ERα seen in the GPER KO mice may reduce 17β-estradiol’s ability to modulate glucose homeostasis and body weight.
17β-estradiol effect on pERK levels following OVX
Studies in cell lines demonstrate estrogens promote rapid activation of Erk 1/2 [41-43]. Moreover, studies in breast cancer cells transfected with GPER show estrogens can activate Erk 1/2 in the absence of ERα and ERβ [44,45]. Several studies have implicated hypothalamic Erk 1/2 in the regulation of energy homeostasis [46,47] and activation of the Erk 1/2 signaling pathway in the CNS mediates the anorectic effects of leptin [48] and CCK [23]. Canonaco et al., reported overlapping distribution of Erk 1/2 with GPER in most brain areas [49] of the hamster. Here, we demonstrate for the first time that female OVX GPER KO mice have reduced 17β-estradiol-induced pErk 1/2 when compared to WT mice. These findings may also provide a further explanation for the reduced sensitivity to CCK and leptin in the GPER KO mice.
In summary, we report a potential role for GPER in regulating body weight, body adiposity, and energy expenditure. The importance of estrogenic activity in regulating body weight in males has previously been demonstrated in the ERαKO mice [31], the aromatase knockout mice (ArKO) [50], as well as our recent findings with tissue specific knockdown of ERα [32,39]. Here, we extend these findings and demonstrate that GPER may be critical in males through its influence on brown and white adipose tissues. This is consistent with a recent publication by Finkelstein et al. [51], and demonstrates an important role for estrogens in adipose tissue in men, which may also be GPER mediated. Changes in the ratio of ERα to ERβ may influence sensitivity to modulators of food intake such as CCK and leptin and therefore influence weight gain. Additionally, the attenuated pERK response in the GPER KO females following 17β-estradiol may also contribute to reductions in sensitivity to pERK dependent modulators of food. In conclusion, our findings indicate a sexual dimorphism with respect to the role of GPER in mediating body weight regulation, energy expenditure, and adiposity; however, the timing and tissues which GPER directly influences have yet to be determined.
Highlights.
We investigate the sexually dimorphic role of GPER in body weight and energy homeostasis
We examine the role of GPER in modulating estrogen-induced metabolic changes in females
GPER KO males have increased body weight and adiposity and decreased energy expenditure while female body weights do not diverge until later age.
Female GPER KO mice have decreased sensitivity to leptin and CCK while male GPER KO mice are indistinguishable from their WT littermates.
GPER KO females have reduced sensitivity to estrogenic effects on body weight and glucose homeostasis.
Acknowledgements
We also thank Dr. Roger Unger for providing us the human leptin. We thank Dr. Nedungadi for performing the leptin and insulin assays. This work was supported by National Institute of Health Grant DK073689 and DK088761.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors have nothing to declare
References
- 1.Bellefontaine N, Elias CF. Minireview: Metabolic Control of the Reproductive Physiology: insights from genetic mouse models. Hormones and Behavior. 2014 doi: 10.1016/j.yhbeh.2014.04.007. DOI: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ropero AB, Alonso-Magdalena P, Quesada I, et al. The role of estrogen receptors in the control of energy and glucose homeostasis. Steroids. 2008;73:874–879. doi: 10.1016/j.steroids.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Brown LM, Clegg DJ. Central effects of estradiol in the regulation of food intake, body weight, and adiposity. J Steroid Biochem Mol Biol. 2010;122:65–73. doi: 10.1016/j.jsbmb.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi H, Seeley RJ, Clegg DJ. Sexual differences in the control of energy homeostasis. Front Neuroendocrinol. 2009;30:396–404. doi: 10.1016/j.yfrne.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34:309–338. doi: 10.1210/er.2012-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma G, Hu C, Brigman JL, et al. GPER Deficiency in Male Mice Results in Insulin Resistance, Dyslipidemia, and a Proinflammatory State. Endocrinology. 2013;154:4136–4145. doi: 10.1210/en.2013-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmeci C, Thompson DA, Ring HZ, et al. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- 8.O’Dowd BF, Nguyen T, Marchese A, et al. Discovery of three novel G-protein-coupled receptor genes. Genomics. 1998;47:310–313. doi: 10.1006/geno.1998.5095. [DOI] [PubMed] [Google Scholar]
- 9.Takada Y, Kato C, Kondo S, et al. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem Biophys Res Commun. 1997;240:737–741. doi: 10.1006/bbrc.1997.7734. [DOI] [PubMed] [Google Scholar]
- 10.Owman C, Nilsson C, Lolait SJ. Cloning of cDNA encoding a putative chemoattractant receptor. Genomics. 1996;37:187–194. doi: 10.1006/geno.1996.0541. [DOI] [PubMed] [Google Scholar]
- 11.Thomas P, Pang Y, Filardo EJ, et al. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Dehghani B, Magrisso IJ, et al. GPR30 contributes to estrogen-induced thymic atrophy. Mol Endocrinol. 2008;22:636–648. doi: 10.1210/me.2007-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haas E, Bhattacharya I, Brailoiu E, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. 2009;104:288–291. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martensson UE, Salehi SA, Windahl S, et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. 2009;150:687–698. doi: 10.1210/en.2008-0623. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Le May C, Wong WP, et al. Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes. 2009;58:2292–2302. doi: 10.2337/db09-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otto C, Fuchs I, Kauselmann G, et al. GPR30 does not mediate estrogenic responses in reproductive organs in mice. Biol Reprod. 2009;80:34–41. doi: 10.1095/biolreprod.108.071175. [DOI] [PubMed] [Google Scholar]
- 17.Windahl SH, Andersson N, Chagin AS, et al. The role of the G protein-coupled receptor GPR30 in the effects of estrogen in ovariectomized mice. Am J Physiol Endocrinol Metab. 2009;296:E490–496. doi: 10.1152/ajpendo.90691.2008. [DOI] [PubMed] [Google Scholar]
- 18.Isensee J, Meoli L, Zazzu V, et al. Expression pattern of G protein-coupled receptor 30 in LacZ reporter mice. Endocrinology. 2009;150:1722–1730. doi: 10.1210/en.2008-1488. [DOI] [PubMed] [Google Scholar]
- 19.Clegg DJ, Brown LM, Woods SC, et al. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes. 2006;55:978–987. doi: 10.2337/diabetes.55.04.06.db05-1339. [DOI] [PubMed] [Google Scholar]
- 20.Tang W, Zeve D, Suh JM, et al. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322:583–586. doi: 10.1126/science.1156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bunger L, Hill WG. Effects of leptin administration on long-term selected fat mice. Genet Res. 1997;69:215–225. doi: 10.1017/s0016672397002814. [DOI] [PubMed] [Google Scholar]
- 22.Asarian L. Loss of cholecystokinin and glucagon-like peptide-1-induced satiation in mice lacking serotonin 2C receptors. Am J Physiol Regul Integr Comp Physiol. 2009;296:R51–56. doi: 10.1152/ajpregu.90655.2008. [DOI] [PubMed] [Google Scholar]
- 23.Sutton GM, Patterson LM, Berthoud HR. Extracellular signal-regulated kinase 1/2 signaling pathway in solitary nucleus mediates cholecystokinin-induced suppression of food intake in rats. J Neurosci. 2004;24:10240–10247. doi: 10.1523/JNEUROSCI.2764-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown LM, Clegg DJ, Benoit SC, et al. Intraventricular insulin and leptin reduce food intake and body weight in C57BL/6J mice. Physiol Behav. 2006;89:687–691. doi: 10.1016/j.physbeh.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez-Lasheras C, Konner AC, Bruning JC. Integrative neurobiology of energy homeostasisneurocircuits, signals and mediators. Front Neuroendocrinol. 2010;31:4–15. doi: 10.1016/j.yfrne.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 26.Pelleymounter MA, Baker MB, McCaleb M. Does estradiol mediate leptin’s effects on adiposity and body weight? Am J Physiol. 1999;276:E955–963. doi: 10.1152/ajpendo.1999.276.5.E955. [DOI] [PubMed] [Google Scholar]
- 27.Butera PC, Bradway DM, Cataldo NJ. Modulation of the satiety effect of cholecystokinin by estradiol. Physiol Behav. 1993;53:1235–1238. doi: 10.1016/0031-9384(93)90387-u. [DOI] [PubMed] [Google Scholar]
- 28.Eckel LA, Geary N. Estradiol treatment increases feeding-induced c-Fos expression in the brains of ovariectomized rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R738–746. doi: 10.1152/ajpregu.2001.281.3.R738. [DOI] [PubMed] [Google Scholar]
- 29.Geary N. Endocrine controls of eating: CCK, leptin, and ghrelin. Physiol Behav. 2004;81:719–733. doi: 10.1016/j.physbeh.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Razandi M, Pedram A, Park ST, et al. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. 2003;278:2701–2712. doi: 10.1074/jbc.M205692200. [DOI] [PubMed] [Google Scholar]
- 31.Filardo EJ, Quinn JA, Bland KI, et al. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 32.Heine PA, Taylor JA, Iwamoto GA, et al. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis KE, M DN, Sun K, et al. The sexually dimorphic role of adipose and adipocyte estrogen receptors in modulating adipose tissue expansion, inflammation, and fibrosis. Mol Metab. 2013;2:227–242. doi: 10.1016/j.molmet.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wade GN, Gray JM. Gonadal effects on food intake and adiposity: a metabolic hypothesis. Physiol Behav. 1979;22:583–593. doi: 10.1016/0031-9384(79)90028-3. [DOI] [PubMed] [Google Scholar]
- 35.Hugo ER, Brandebourg TD, Woo JG, et al. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ Health Perspect. 2008;116:1642–1647. doi: 10.1289/ehp.11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ford J, Hajibeigi A, Long M, et al. GPR30 deficiency causes increased bone mass, mineralization, and growth plate proliferative activity in male mice. J Bone Miner Res. 2011;26:298–307. doi: 10.1002/jbmr.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bohler HC, Jr., Tracer H, Merriam GR, et al. Changes in proopiomelanocortin messenger ribonucleic acid levels in the rostral periarcuate region of the female rat during the estrous cycle. Endocrinology. 1991;128:1265–1269. doi: 10.1210/endo-128-3-1265. [DOI] [PubMed] [Google Scholar]
- 38.Slamberova R, Hnatczuk OC, Vathy I. Expression of proopiomelanocortin and proenkephalin mRNA in sexually dimorphic brain regions are altered in adult male and female rats treated prenatally with morphine. J Pept Res. 2004;63:399–408. doi: 10.1111/j.1399-3011.2004.00134.x. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y, Hill JW, Fukuda M, et al. PI3K signaling in the ventromedial hypothalamic nucleus is required for normal energy homeostasis. Cell Metab. 2010;12:88–95. doi: 10.1016/j.cmet.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clegg DJ, Brown LM, Zigman JM, et al. Estradiol-dependent decrease in the orexigenic potency of ghrelin in female rats. Diabetes. 2007;56:1051–1058. doi: 10.2337/db06-0015. [DOI] [PubMed] [Google Scholar]
- 41.Improta-Brears T, Whorton AR, Codazzi F, et al. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci U S A. 1999;96:4686–4691. doi: 10.1073/pnas.96.8.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Migliaccio A, Di Domenico M, Castoria G, et al. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- 43.Watters JJ, Campbell JS, Cunningham MJ, et al. Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology. 1997;138:4030–4033. doi: 10.1210/endo.138.9.5489. [DOI] [PubMed] [Google Scholar]
- 44.Filardo EJ, Quinn JA, Frackelton AR, Jr., et al. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 45.Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16:362–367. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 46.Morikawa Y, Ueyama E, Senba E. Fasting-induced activation of mitogen-activated protein kinases (ERK/p38) in the mouse hypothalamus. J Neuroendocrinol. 2004;16:105–112. doi: 10.1111/j.0953-8194.2004.01135.x. [DOI] [PubMed] [Google Scholar]
- 47.Ueyama E, Morikawa Y, Yasuda T, et al. Attenuation of fasting-induced phosphorylation of mitogen-activated protein kinases (ERK/p38) in the mouse hypothalamus in response to refeeding. Neurosci Lett. 2004;371:40–44. doi: 10.1016/j.neulet.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 48.Rahmouni K, Sigmund CD, Haynes WG, et al. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes. 2009;58:536–542. doi: 10.2337/db08-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Canonaco M, Giusi G, Madeo A, et al. A sexually dimorphic distribution pattern of the novel estrogen receptor G-protein-coupled receptor 30 in some brain areas of the hamster. J Endocrinol. 2008;196:131–138. doi: 10.1677/JOE-07-0392. [DOI] [PubMed] [Google Scholar]