

Abstract
Current multiple sclerosis (MS) therapies only partially prevent chronically worsening neurological deficits, which are largely attributable to progressive loss of CNS axons. Prior studies of experimental autoimmune encephalomyelitis (EAE) induced in C57BL/6 mice by immunization with myelin oligodendrocyte glycoprotein peptide 35–55 (MOG peptide), a model of MS, documented continued axon loss for months after acute CNS inflammatory infiltrates had subsided, and massive astroglial induction of CCL2 (MCP-1), a chemokine for CCR2+ monocytes. We now report that conditional deletion of astroglial CCL2 significantly decreases CNS accumulation of classically activated (M1) monocyte-derived macrophages and microglial expression of M1 markers during the initial CNS inflammatory phase of MOG peptide EAE, reduces the acute and long-term severity of clinical deficits and slows the progression of spinal cord axon loss. In addition, lack of astroglial-derived CCL2 results in increased accumulation of Th17 cells within the CNS in these mice, but also in greater confinement of CD4+ lymphocytes to CNS perivascular spaces. These findings suggest that therapies designed to inhibit astroglial CCL2-driven trafficking of monocyte-derived macrophages to the CNS during acute MS exacerbations have the potential to significantly reduce CNS axon loss and slow progression of neurological deficits.
Keywords: astroglia, axonopathy, chemokine, EAE, macrophage, myelin
Introduction
Experimental autoimmune encephalomyelitis (EAE) is a widely used multiple sclerosis (MS) model. CCL2, a chemokine that, in peripheral tissues, regulates the release of CCR2+Ly6C+ monocytes from the bone marrow and their trafficking to regions of inflammation (Geissmann et al., 2003; King et al., 2009), is also synthesized in the CNS, mainly by astrocytes, and to a lesser extent by microglia, endothelial cells, and neurons (Ransohoff et al., 1993; Berman et al., 1996; Banisadr et al., 2005a; de Haas et al., 2007; Mélik-Parsadaniantz and Rostène, 2008). Constitutive genetic ablation of either CCL2 (Huang et al., 2001) or of its receptor, CCR2 (Fife et al., 2000), reduces monocyte-derived macrophage recruitment into the CNS in mice with EAE and diminishes the severity of their neurological deficits. Studies in radiation bone marrow chimeric mice showed that CCL2 produced by radioresistant CNS cells is critical for the development of severe EAE in mice immunized with myelin oligodendrocyte glycoprotein peptide 35–55 (MOG peptide) (Dogan et al., 2008). More recently, the extent of acute neurological deficits in mice with MOG peptide EAE was reported to be diminished by conditional deletion of astroglial CCL2 (Paul et al., 2014). Furthermore, estrogen receptor α (ERα) on astrocytes reduces astrocyte expression of CCL2 and mediates neuroprotection in EAE (Spence et al., 2011, 2013). Together, these findings underscore the importance of CCL2 in EAE, and possibly also in MS.
In the present study, we examined the effects of conditional ablation of astroglial CCL2 on CNS inflammation and loss of myelin and axons in mice with MOG peptide EAE. Our results indicate that MOG peptide-immunized mice in which wild-type CCL2 alleles (CCL2WT/WT) had been replaced by loxP-flanked CCL2 alleles (CCL2flx/flx), which were disrupted by a mouse glial fibrillary acidic protein promoter-driven cre transgene (mGFAPcre) exhibited milder clinical neurological deficits throughout the course of their illness, and substantially less long-term CNS demyelination and axon loss, than did their littermate mGFAPcre/CCL2WT/WT controls. Collectively, these data support key roles for astroglial CCL2 in modulating CNS inflammation and triggering long-term axon loss in MOG peptide EAE.
Materials and Methods
Mice.
Mouse glial fibrillary acidic protein promoter-driven cre (mGfap-cre, B6.Cg-Tg 73.12Mvs/J; stock number 012886) and Rosa26-STOP-enhanced yellow fluorescent protein (EYFP) (RosaEYFP, B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J; stock number 006148) mice were purchased (The Jackson Laboratory, RRID:nlx_63162) and maintained on a C57BL/6 background. We constructed CCL2flx/flx mice, containing loxP sites flanking exons 2–3 of the chemokine ccl2 gene. These CCL2flx/flx mice were crossed with mice carrying the mGFAPcre transgene and the recombination marker RosaEYFP to create mGFAPCre/CCL2flx/flx/RosaEYFP mice, which were deficient in astroglial CCL2 synthesis. mGFAPCre/CCL2WT/WT/RosaEYFP (mGFAPCre/RosaEYFP) littermates were used as controls. Mice homozygous for CCL2flx/flx appeared healthy. Confocal and stereological analyses were used to quantify cre-mediated recombination in astrocytes, calculated by dividing the number of EYFP+GFAP+ cells by the total number of GFAP+ cells. All mice were maintained in a pathogen-free, Association for Assessment and Accreditation of Laboratory Animal Care approved, veterinarian-staffed vivarium, and all study protocols and procedures were approved by the University of California Davis Institutional Animal Care and Use Committee.
EAE induction.
MOG peptide-EAE was induced in 3 month postnatal male and female mGFAPcre/CCL2flx/flx/RosaEYFP and control mGFAPcre/RosaEYFP mice by subcutaneous flank administration of 300 μg of rodent MOG peptide (amino acids 35–55, New England Peptides) in complete Freund's adjuvant (CFA) containing 5 mg/ml killed Mycobacterium tuberculosis (DIFCO catalog #DF3114-33-8) on day 0, with intraperitoneal administration of 200 ng of pertussis toxin on days 0 and 2. Wild-type CFA control mice received CFA and pertussis toxin, but no MOG peptide. Mice were scored daily as follows: 0, no detectable signs of EAE; 0.5, distal limp tail; 1.0, limp tail or waddling gait; 1.5, limp tail and waddling gait; 2.0, unilateral partial hindlimb paresis; 2.5, bilateral partial hindlimb paresis; 3.0, complete bilateral hindlimb paresis; 3.5, partial hindlimb paralysis; 4.0, complete hindlimb paralysis; and 5, moribund.
Tissue and cell preparation and immunohistochemistry.
Mice were anesthetized by intraperitoneal administration of ketamine (150 mg/kg) and xylazine (16 mg/kg). For immunohistology, animals were perfused with 4% PFA (v/v) in PBS. Tissues were cryoprotected in PBS with 30% sucrose and cryostat cut into 10- to 14-μm-thick sections. Sections were blocked with 5% normal serum (depending upon the species of origin for the secondary antibodies) in PBS for 1 h at room temperature and incubated overnight at 4°C with antibodies against CCL2 (Millipore, catalog #AB1834P RRID:AB_90984, 1:200), CD4 (BD Biosciences catalog #550280 RRID:AB_393575, 1:100), Iba-1 (Wako Chemicals catalog #019-19741 RRID:AB_2313566, 1:1500), GFAP (Dr. Virginia Lee catalog #GFAP RRID:AB_2314536, 1:300), GFP (Abcam catalog #ab13970 RRID:AB_300798, 1:400), or opalin (Novus Biologicals catalog #NBP1-81656 RRID:AB_11028865, 1:2000), or a monoclonal pan-phosphorylated NF-H epitope antibody (SMI-312, Covance Research Products, catalog #SMI-312R-100 RRID:AB_10119994, 1:500), followed by secondary species-specific fluorescent antibodies conjugated to AlexaFluor-488 or AlexaFluor-594 (Jackson ImmunoResearch Laboratories, 1:500) or biotinylated secondary antibodies for 2 h at room temperature.
RNA isolation and real-time PCR analysis.
Mice were perfused with ice-cold PBS (Invitrogen) and lumbar (L4-L5) spinal cords were extracted, stored overnight in RNAlater (Ambion, RRID:nif-0000-30092) at 4°C, and then stored at −20°C. RNA was isolated using the RNeasy Lipid Tissue Mini Kit (QIAGEN, RRID:nif-0000-31384) following the manufacturer's instructions. The samples were treated with DNaseI (QIAGEN), and 500 ng of RNA was transcribed into cDNA using the Omniscript RT Kit (QIAGEN). cDNA (1 μl) was transferred into a 96-well PCR-plate (Thermo Fisher Scientific, RRID:nlx_152478), and 12.5 μl of SYBER Green Master Mix (QuantiTect SYBER Green PCR Kit (QIAGEN) plus 9.6 μl of H2O was added. The PCR was performed as previously described (Soulika et al., 2009). mRNA levels of assayed genes were normalized to mRNA levels of the housekeeping gene GAPDH.
Axon counting and quantification of demyelination.
Semiautomated axon counting and integrated density (IntDen) measurements were obtained using ImageJ software version 1.47 (Schneider et al., 2012) according to a published protocol (Marina et al., 2010). Images of single confocal optical slices were used for SMI-312+ axon counting and immunoreactive opalin intensity measurements. Briefly, to quantify the number of axons, images of white matter tracts of individual spinal cords were manually outlined and converted to grayscale images, and evaluated with the automated “cell counter” plugin. To quantify immunoreactivity, white matter tracts of individual spinal cords were manually outlined and the IntDen was obtained. IntDen provided a measure of intensity proportional to total volume and was calculated using area (μm) × immunoreactivity. Sections were imaged using identical gain and offset settings on a laser scanning confocal microscope. All intensity measurements were performed in triplicate, and values were averaged.
Protein extraction and Western blotting.
Lumbar spinal cords were extracted and homogenized in RIPA buffer containing protease inhibitors (Santa Cruz Biotechnology, RRID:nlx_152453). Tissues were homogenized at 4°C for 1 h. After centrifugation (15,000 × g) for 30 min, supernatants were collected and stored at −80°C. Protein concentrations were determined using the Pierce Micro BCA Protein Assay (Thermo Fisher Scientific). Equal amounts of total protein (50 μg) were denatured in 2× Laemmli buffer at 100°C for 5 min, separated on denaturing SDS-PAGE (Any kD gels, Bio-Rad, RRID:nif-0000-30176), and transferred to nitrocellulose membranes (Bio-Rad) at 30 V overnight at 4°C. Membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences, RRID:nlx_152397) for 1 h at room temperature and probed with an antibody recognizing the post-translationally modified form of CCL2 (Lubkowski et al., 1997) (Santa Cruz Biotechnology catalog #sc-28879 RRID:AB_2070877, 1:200), followed by a HRP-conjugated secondary antibody. Blots were also probed for β-actin as a loading control.
Isolation of CNS mononuclear cells.
Mice killed by ketamine/xylazine intraperitoneal injections were perfused with ice-cold PBS. Brains and spinal cords were minced in PBS, digested at 37°C for 30 min in PBS containing 0.04 units of Liberase R1 (Roche) and 10 μg of DNase I (Roche, RRID:nlx_152451), and passed through a 40 μm mesh. CNS infiltrating cells were isolated via a discontinuous 40%/70% (v/v) Percoll gradient (GE Healthcare, RRID:nlx_152368).
Flow cytometry.
CNS mononuclear cells were immunostained after incubation at 37°C for 3 h in RPMI 1640 containing 10% FBS, 2 mm l-glutamine, 0.1 mm nonessential amino acids, 100 U/ml penicillin-streptomycin, 50 μm 2-mercaptoethanol, and 1 mm sodium pyruvate (Invitrogen) in the presence of brefeldin A (GolgiPlug, BD Biosciences, RRID:nlx_152292). Immediately before immunostaining, Fc receptors were blocked for 10 min with anti-CD16/32 (BD Biosciences, catalog #553142 RRID:AB_394657). Macrophages were identified by brilliant violet (BV)-650 labeled anti-mouse CD11b (BioLegend, catalog #101239 RRID:AB_11125575) and phycoerythrin-cyanine7 (PE-Cy7; BioLegend, catalog #103114 RRID:AB_312979) or allophycocyan (APC)-labeled anti-mouse CD45 (BD Biosciences, catalog #559864 RRID:AB_398672). Classically activated M1 macrophages were identified by BV-510 labeled anti-mouse CD86 (BioLegend, catalog #105039), PE-labeled anti-mouse Ly6C (BioLegend, catalog #128008 RRID:AB_1186132), APC-Cy7-labeled anti-mouse MHCII (BioLegend, catalog #107627 RRID:AB_1659252), and PE-labeled anti-mouse iNOS (Santa Cruz Biotechnology, catalog #sc-651 RRID:AB_2298577). Alternatively activated M2 macrophages were identified by PE-labeled anti-mouse CD206 (BioLegend, catalog # 141705 RRID:AB_10896421), APC-labeled anti-mouse Arginase-1 (R&D Systems, RRID:nlx_152445 catalog #IC5868A), and biotinylated-labeled anti-mouse Ym1 (R&D Systems, catalog #BAF2446 RRID:AB_2260451). For T helper cell subset analysis, cells were stained with Pacific Blue (PB)-labeled anti-mouse CD4 (BD Biosciences, catalog #558107 RRID:AB_397030), fixed, permeabilized using the Cytofix/Cytoperm Plus Kit (BD Bioscience) according to the manufacturer's protocol, and intracellularly stained with APC-labeled anti-mouse IFNγ (BD Biosciences, catalog #554413 RRID:AB_398551), and PE-labeled anti-mouse IL17 (BD Biosciences, catalog #559502 RRID:AB_397256).
Transmission electron microscopy.
Mice anesthetized with a xylazine/ketamine mixture were briefly perfused with 500 units of heparin in 0.1 m phosphate buffer followed sequentially by freshly prepared 4% PFA and then by 3% glutaraldehyde, all in 0.1 m phosphate buffer, pH 7.4. Transverse spinal segments (1–2 mm in thickness) from the L4-L5 spinal cord enlargement were harvested. The tissues were washed with sodium cacodylate buffer (0.2 m), then postfixed with 2% aqueous osmium tetroxide for 2 h. Samples were again washed with cacodylate buffer, then dehydrated through ascending alcohols, washed with propylene oxide, and embedded in EMBed-812 resin (Electron Microscopy Sciences). Semithin sections were stained with toluidine blue. Ultrathin sections (70–80 nm) were cut on a Leica EM UC7 microtome and collected on 1 × 2 mm Formvar-coated copper slot grids. Sections were double stained with uranyl acetate and lead citrate and examined on a Philips CM120 electron microscope.
Statistics.
Results are expressed as mean ± SEM. Significance was measured by unpaired t test or one-way ANOVA followed by Bonferroni's Multiple Comparison Test when data were normally distributed, or by the Mann–Whitney U test in the case of non-normal distribution. For statistical evaluation of EAE clinical scores, the Mann–Whitney ranking U test was used. All p values were two-tailed and subjected to a significance level of 0.05.
Results
CCL2 is efficiently and specifically deleted in astrocytes
To assess the efficiency of mGFAPcre-mediated astroglial recombination, mGFAPCre/CCL2flx/flx mice were crossed to the ROSA26 reporter mouse line, in which cre activity induces excision of a STOP sequence, thus allowing expression of EYFP (Soriano, 1999). The recombination frequency, detected by combined confocal and stereological analysis and calculated as the number of EYFP+GFAP+ cells divided by total number of GFAP+ cells, was 91.5 ± 9.2% in the spinal cords of healthy adult mice (8 weeks of age, n = 5) (Fig. 1A,B).
Figure 1.

Conditional deletion of CCL2 from astrocytes. A, Immunohistochemisty for EYFP (green) and GFAP (red) in naive mGFAPCre/RosaEYFP mouse spinal cord. Scale bar, 10 μm. B, Quantification of recombined astrocytes (GFP+GFAP+ over total GFAP+ cells) in spinal cord was determined to be 91.5 ± 9.18%. C, D, Representative immunohistochemistry image and quantification of integrated density (IntDen) at 14 dpi showing ablation of astroglial CCL2 from a mGFAPCre/CCL2flx/flx/RosaEYFP mouse lumbar spinal cord. n = 6 for control; n = 8 for mGFAPCre/CCL2flx/flx/RosaEYFP. *p = 0.0335 (unpaired t test). Scale bar: C, 100 μm. mRNA (E) and protein quantification (F) in lumbar spinal cords of mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with controls at 14 dpi. Data are mean ± SEM of at least two independent experiments (n = 8). E, *p = 0.0335 (unpaired t test). F, *p = 0.0385 (unpaired t test).
We did not immunohistologically detect CCL2 in astroglia in normal adult mouse spinal cords, whereas astroglia were intensely immunoreactive in mGFAPcre/RosaEYFP mice by the time that clinical neurological deficits had first appeared. Immunoreactive astroglial CCL2 was almost completely ablated in mGFAPCre/CCL2flx/flx/RosaEYFP mice 14 d after MOG peptide immunization (14 dpi) (Fig. 1C,D). Comparisons between mGFAPCre/CCL2flx/flx/RosaEYFP and mGFAPCre/RosaEYFP littermate control mice by qPCR and Western blot analyses showed an approximate 50% reduction in spinal cord CCL2 mRNA and protein expression in the mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with the control mice (Fig. 1E,F). The spinal cord immunoreactive CCL2 that remained in the mGFAPCre/CCL2flx/flx/RosaEYFP mice was presumably derived from other CNS cellular sources (endothelial cells, microglia, and neurons) (Banisadr et al., 2005a; Beutner et al., 2013; Paul et al., 2014).
Depletion of astrocyte CCL2 diminishes severity of clinical deficits in MOG peptide EAE
Mice immunized with MOG peptide were observed daily for neurological deficits. In agreement with a previous study (Paul et al., 2014), there was a delay in onset and a diminution in long-term severity of clinical neurological deficits in the mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with littermate mGFAPCre/RosaEYFP control mice (Fig. 2).
Figure 2.

Deletion of astroglial CCL2 diminishes EAE clinical severity. A, Clinical scores were recorded daily post-MOG peptide immunization through day 85. n = 12 for control; n = 16 for mGFAPCre/CCL2flx/flx/RosaEYFP mice. *p = 0.137 (day 10), 0.0480 (day 12), 0.0116 (day 14), and 0.0427 (day 15) (Mann–Whitney test). B, Mean cumulative clinical scores from A. *p = 0.0351 (unpaired t test). C, Table summarizing EAE clinical parameters in mGFAPCre/RosaEYFP and mGFAPCre/CCL2flx/flx/RosaEYFP mice. Data are mean ± SEM for at least five independent experiments.
Accumulation of inflammatory monocytes in the CNS and expression of inflammatory markers in microglia are diminished by deletion of astroglial CCL2
Macrophages, the major effector cells mediating neurotoxicity in EAE (Bhasin et al., 2008; Sinha et al., 2008), are a heterogeneous population that exerts multiple inflammatory and anti-inflammatory functions (Mosser, 2003; Li et al., 2009). The two extreme macrophage phenotypes are denoted M1 (classically activated, “inflammatory”) and M2 (alternatively activated). M1 macrophages produce potent proinflammatory mediators (e.g., TNFα, IL-1, and NO) (Plüddemann et al., 2011) and are characterized by heightened expression of MHC Class II and costimulatory molecules (e.g., CD86), which are important for the activation and stimulation of CD4 T cells (Mills, 2012; Wynn et al., 2013). Conversely, M2 macrophages produce factors that facilitate tissue repair, such as Ym1 (chitinase), Arg1 (arginase 1), and mannose receptor (CD206) as well as anti-inflammatory and regulatory factors (Gordon and Martinez, 2010; Mills, 2012; Tugal et al., 2013). Transcriptional profiling has added a new element to the characterization of alternate macrophage activation. Various alternatively activated M2 macrophages, referred to as M2a, M2b, and M2c, express different repertoires reflecting their distinct biological roles (Mosser, 2003; Mantovani et al., 2004).
To examine the effects of astroglial CCL2 deletion on CNS macrophage recruitment and expression of M1 and M2 markers during MOG peptide EAE, mGFAPCre/CCL2flx/flxRosaEYFP and mGFAPCre/RosaEYFP mice were immunized with MOG peptide (Soulika et al., 2009). At 16 and 35 dpi, brains and spinal cords were pooled and mechanically/enzymatically disaggregated to prepare single-cell suspensions. Monocyte-derived macrophages (CD11b+CD45hiLy6G−), microglia (CD11b+CD45loLy6G−), and neutrophils (CD11b+CD45hiLy6G+) were quantified by flow cytometry (Fig. 3A–D). Monocyte-derived macrophages were further classified by their expression of the inflammatory marker, Ly6C. In mice lacking astroglial CCL2, there was a 50% reduction in the number of total CNS macrophages (Fig. 3E) at 16 dpi. At that same time point, percentages of CD11b+Ly6G+ neutrophils were increased from 8% to 20% of total CNS infiltrating cells, but percentages of CD11b+/Ly6Chi macrophages were decreased from 60% in the mGFAP/RosaEYFP mice to 30% in the mGFAPCre/CCL2flx/flx/RosaEYFP mice (Fig. 3F). Our observations of increased neutrophil populations in mGFAPCre/CCL2flx/flxRosaEYFP mice were associated with increased mRNA expression of the neutrophil chemoattractants CXCL1 and CXCL2 (Fig. 3G). Also, at 16 dpi, the mGFAPCre/CCL2flx/flx/RosaEYFP mice demonstrated lower percentages of Ly6ChiMHCII+ and Ly6ChiMHCII+CD86+iNOS+ M1 macrophages, but not of MHCII− macrophages, than their littermate controls (Fig. 3H,I). By 35 dpi, numbers of macrophages had declined sharply in both sets of mice and were no longer significantly different between the two groups of mice. Percentages of M2 (alternatively activated) macrophages did not differ significantly between the two groups of mice at either 16 or 35 dpi (Fig. 3J). We concluded that the reduction in classically activated M1 macrophage marker expression in the absence of astroglial CCL2 was not accompanied by a detectable shift to alternatively activated M2 macrophage marker expression; rather, lack of astroglial CCL2 selectively inhibited accumulation of classically activated M1 macrophages in the CNS.
Figure 3.
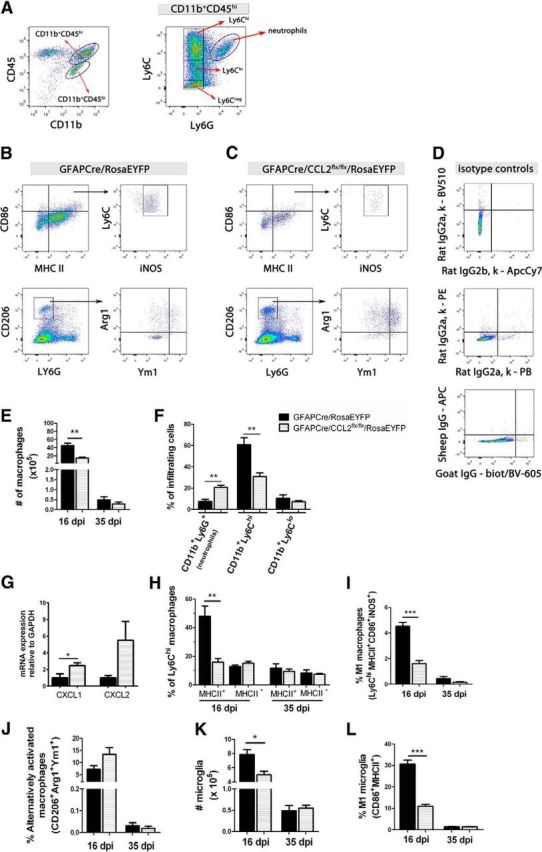
Flow cytometric analysis of macrophage populations in the CNS of mice with EAE. A, Analysis of CD11b+CD45hi (monocyte-derived macrophages) and CD11b+CD45lo (microglia) from pooled brain/spinal cords. CD11b+CD45hi cells were further classified by Ly6C and Ly6G expression. Cells from mGFAPCre/RosaEYFP (B) and mGFAPCre/CCL2flx/flx/RosaEYFP (C) mice were analyzed at 16 dpi. D, Isotype controls are shown for CD86-BV510 and MHC II-ApcCy7, CD206-PE and Ly6G-PB, and Arg1-APC and Ym1-biotinylated-BV605 (biot/BV-605). E, Numbers of CNS macrophages are decreased in mGFAPCre/CCL2flx/flxRosaEYFP mice compared with mGFAPCre/RosaEYFP controls at day 16 dpi, but not at 35 dpi. F, Increased percentages of CD11b+Ly6G+ neutrophils, but fewer CD11b+/Ly6Chi macrophages, were observed in mGFAPCre/CCL2flx/flx/RosaEYFP compared with control mice at 16 dpi. G, mRNA of CXCL1 and CXCL2 were quantified using 16 dpi lumbar spinal cord RNA extracts and real-time RT-PCR. CXCL1 mRNA was increased in mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with controls. *p = 0.0439 (unpaired t test). H, I, Lower percentages of Ly6ChiMHCII+ and Ly6ChiMHCII+CD86+iNOS+ macrophages, but not of MHCII− macrophages, were observed in mGFAPCre/CCL2flx/flx/RosaEYFP mice versus littermate controls, but by 35 dpi, percentages of both sets of macrophages were not significantly different in the two groups of mice. J, No significant difference was observed in the expression of the alternatively activated macrophage M2 markers CD206, Arg1, or Ym1 between the two groups of mice. K, L, There were reduced total numbers of CD86+MHCII+ microglia and percentages of microglia expressing the M1 markers CD86 and MHCII in mGFAPCre/CCL2flx/flx/RosaEYFP mice than in littermate controls at 16 dpi, but not at 35 dpi. Results shown are mean ± SEM for at least two independent experiments (n = 6–11 mice at each time point). *p < 0.05 (unpaired t test). **p < 0.01 (unpaired t test). ***p < 0.001 (unpaired t test).
The frequency of microglia expressing the classically activated M1 markers CD86 and MHC II was decreased in mGFAPCre/CCL2flx/flxRosaEYFP mice compared with their littermate controls at 16 dpi (Fig. 3L). The reduced number of microglial cells (CD11b+CD45lo) (Fig. 3K) in CNS of the mice in which astroglial CCL2 had been deleted may have been the result of a decrease in microglial proliferation because CCL2 has been reported to promote microglial proliferation and growth in vitro (Rezaie et al., 2002).
qRT-PCR analyses of spinal cord macrophage M1 and M2 marker mRNAs
To supplement these flow cytometric analyses of macrophage subsets in whole CNS, spinal cord extracts from MOG peptide-immunized mGFAPCre/CCL2flx/flx/RosaEYFP and mGFAPCre/RosaEYFP littermate control mice were assayed by qRT-PCR for mRNAs encoding classically activated M1 and alternatively activated M2 macrophage subtype-specific constituents (Fig. 4A). Arg1 mRNA and immunoreactive Arg1 protein (Fig. 4A–C) were significantly increased in spinal cords from mice lacking astroglial CCL2. Arg1 is responsible for metabolism of arginine to ornithine and polyamines in alternatively activated M2 macrophages, resulting in diminished expression of the M1 macrophage marker iNOS (Mahbub et al., 2012). We did not observe a difference between mGFAPCre/CCL2flx/flx/RosaEYFP and control mGFAPCre/RosaEYFP mice with MOG peptide EAE in levels of mRNAs encoding classically activated M1 macrophage markers, including CD86, and iNOS, or the alternatively activated M2 macrophage markers, including Ym1 and IL-4R (CD124) (Fig. 4A). The apparent discrepancy between these qRT-PCR results and our flow cytometric findings may be attributable to differences in the composition of inflammatory infiltrates between brain and spinal cord (Pierson et al., 2012) and/or to expression of these markers by CNS cells other than macrophages and microglia.
Figure 4.

Expression of M1 and M2 markers in the CNS of mice with EAE. A, mRNA of classically activated M1 and alternatively activated M2 macrophage markers were quantified using 16 dpi lumbar spinal cord RNA extracts and real-time RT-PCR. Arginase1 (Arg1) mRNA was increased in mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with controls. *p = 0.0439 (unpaired t test). B, Immunofluorescent labeling for Arg1 and CD11b+ macrophages/microglia shows an increase of Arg1 expression in macrophage/microglia populations from mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with controls. Scale bar, 100 μm. C, IntDen of Arg1 was quantified in 14-μm-thick frozen sections of lumbar spinal cord using ImageJ analysis (triplicate measurements were performed for each mouse). n = 6 for control; n = 8 for mGFAPCre/CCL2flx/flx/RosaEYFP. **p = 0.0095 (unpaired t test). Vertical bars represent SEM.
Astroglial CCL2 deletion increases CNS accumulation of Th17 cells and restricts compartmentalization of CD4+ T cells within CNS perivascular spaces
CD4+ T helper 1 (Th1) and T helper 17 (Th17) lymphocytes play a central role in initiating and orchestrating CNS inflammation during EAE (Baxter, 2007; McFarland and Martin, 2007). To examine the effect of astroglial CCL2 inhibition on CD4+ lymphocyte populations within the CNS during EAE, mGFAPCre/CCL2flx/flxRosaEYFP and mGFAPCre/RosaEYFP mice were immunized with MOG peptide as previously described. Mononuclear cells prepared from these two sets of mice were characterized by flow cytometry as total CD4 (CD4+), Th1 (IFNγ+), and Th17 (IL-17+) lymphocytes (Fig. 5). CNS Th17 cell numbers were significantly increased in mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with controls (Fig. 5A). CNS IL-6 mRNA levels were more than threefold higher in mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with controls. The elevated levels of IL-6 mRNA in mice lacking astroglial CCL2 were not associated with increased levels of TGF-β (Fig. 5B). Our observations of increased levels of IL-6 in the mice that lacked astroglial production of CCL2 may have contributed to the increased numbers of Th17 lymphocytes in those mice because IL-6 induces the retinoid-related orphan receptor-γt, triggering the developmental program of Th17 cells (Bettelli et al., 2006).
Figure 5.

Flow cytometric analysis and spatial distribution of CNS CD4 T cells. A, Mononuclear cells were isolated from pooled brain/spinal cord and analyzed by flow cytometry at 16 dpi. Total CD4 (CD4+), and Th1 (IFNγ+IL-17−), did not show statistically significant changes between the two groups of mice. The number of Th17 (IFNγ−IL-17+) cells were upregulated in the CNS of mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with controls. B, IL-6 and TGF-β RNAs were quantified using real-time RT-PCR of lumbar spinal cord RNA extracts at 16 dpi. IL-6 mRNA was increased, but TGF-β mRNA was unchanged, in mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with littermate controls. n = 6 for mGFAPCre/RosaEYFP; n = 8 for mGFAPCre/CCL2flx/flx/RosaEYFP. *p = 0.0119 (unpaired t test). C, Retention of CD4+ T cells (green) within the perivascular space of lumbar spinal cord shown by laminin (red) in mGFAPCre/CCL2flx/flx/RosaEYFP mice compared with controls. D, Quantification of CD4+ T cells observed within spinal cord ventral blood vessels was determined using ImageJ analysis (triplicate measurements were performed for each mouse). *p = 0.0229.
CCL2 induces uropod formation by T lymphocytes, thus facilitating T-cell passage from perivascular spaces into the CNS parenchyma (del Pozo et al., 1997; Carrillo-de Sauvage et al., 2012). To determine the effects of astroglial CCL2 on CD4+ T cell infiltration during EAE, we immunohistologically compared the distributions of CD4+ T cells between perivascular spaces and the parenchyma of the spinal cord at 14 dpi in astroglial CCL2 disrupted versus littermate control mice (Fig. 5C,D). In the absence of astroglia-derived CCL2, CD4+ T cells were confined to the perivascular spaces to a greater extent than in the controls.
MOG peptide EAE-induced demyelination and axon loss are diminished by deletion of astroglial CCL2
By 85 dpi, the lumbar spinal cords of GFAPCre/RosaEYFP mice had developed scattered lesions that were depleted of axons and myelin; these lesions were chiefly located in superficial white matter and were more frequently seen in ventral than dorsal spinal cord. Such lesions were less frequently detected in lumbar spinal cords of the GFAPCre/CCL2flx/flx/RosaEYFP mice (Fig. 6A,B). At 14 dpi, lumbar spinal cord content of the myelin protein opalin (Golan et al., 2008) was not significantly diminished in either GFAPCre/RosaEYFP or GFAPCre/CCL2flx/flx/RosaEYFP mice. By 85 dpi, however, there was a substantial loss of lumbar spinal cord opalin immunoreactivity in the GFAPCre/RosaEYFP mice, but not in the GFAPCre/CCL2flx/flx/RosaEYFP mice (Fig. 6C). Numbers of axons in lumbar spinal cord ventral fasciculi, visualized by immunostaining with SMI-312 (Szaro et al., 1990; Frischer et al., 2009), were not diminished in either GFAPCre/RosaEYFP or GFAPCre/CCL2flx/flx/RosaEYFP mice at 14 dpi. By 85 dpi, there was a substantial loss of axons from both groups of mice, but this loss was significantly greater in GFAPCre/RosaEYFP than GFAPCre/CCL2flx/flx/RosaEYFP mice (Fig. 6D,E).
Figure 6.

Light and transmission electron microscopic analysis of spinal cord pathology. A, Semithin sections (0.5–1 μm) of lumbar spinal cord ventral white matter from mGFAPCre/RosaEYFP and mGFAPCre/CCL2flx/flx/RosaEYFP mice were stained with toluidine blue to delineate preservation of myelin and axons. A large demyelinating lesion observed in mGFAPCre/RosaEYFP spinal cord is shown. Red arrows indicate demyelinated area. Normal myelin was observed in mGFAPCre/CCL2flx/flx/RosaEYFP mice. Scale bars, 50 μm. B, Transmission electron micrographs from ultrathin cross sections (70–80 nm) of spinal cord lumbar enlargement ventral fasciculi comparing mGFAPCre/RosaEYFP and mGFAPCre/CCL2flx/flx/RosaEYFP mice during late EAE (85 dpi). Macrophages (M) were observed in the specimens prepared from both groups of mice. Red arrows indicate the presence of small axons (B, right), which were less prevalent in the control mice (B, left). Original magnification ×20,217. Scale bars, 10 μm. C, IntDen of opalin fluorescence within ventral fasciculi of 14 μm lumbar spinal cord sections from WT mice injected with CFA without MOG peptide (gray bars), and mGFAPCre/RosaEYFP and mGFAPCre/CCL2flx/flx/RosaEYFP mice injected with MOG peptide (black and white dotted bars, respectively), at 14 and 85 dpi. Triplicate measurements were performed for each mouse and averaged. n = 3 WT CFA; n = 5 for mGFAPCre/RosaEYFP and mGFAPCre/CCL2flx/flx/RosaEYFP mice. At 85 dpi. **p = 0.0016, WT CFA versus mGFAPCre/RosaEYFP. ##p = 0.0071, mGFAPCre/RosaEYFP versus mGFAPCre/CCL2flx/flx/RosaEYFP (one-way ANOVA followed by Bonferroni's Multiple Comparison Test). D, Immunofluorescent labeling for SM1-312 showed equivalent numbers of axons in the 3 groups of mice at 14 dpi, but at 85 dpi there was a substantial loss of axons in the lumbar spinal cord ventral white matter of the mGFAPCre/RosaEYFP mice; this loss was less evident in mGFAPCre/CCL2flx/flx/RosaEYFP mice. SMI312+ axon counts are quantified in E and F. n = 3 for WT CFA; n = 5 for mGFAPCre/RosaEYFP and mGFAPCre/CCL2flx/flx/RosaEYFP mice. **p = 0.0013, WT CFA versus mGFAPCre/RosaEYFP. *p = 0.0358, mGFAPCre/RosaEYFP versus mGFAPCre/CCL2flx/flx/RosaEYFP. ##p = 0.0042, WT CFA versus mGFAPCre/CCL2flx/flx/RosaEYFP (one-way ANOVA followed by Bonferroni's Multiple Comparison Test).
Discussion
CNS axon loss, a key pathological feature of MS, is refractory to currently available MS therapies (DeLuca et al., 2004, 2006; Weiner, 2009; Franklin et al., 2012) and is a major contributor to nonremitting neurological deficits (Tallantyre et al., 2010). CNS axon loss is also prominent in MOG peptide EAE. A noteworthy feature of this autoimmune MS model is that relatively little spinal cord axon loss occurs during the first week after onset of clinical deficits, a period during which spinal cord inflammatory infiltrates are most prominent. Instead, spinal cord axon loss progresses after most inflammatory infiltrates have been cleared from the spinal cord (Herrero-Herranz et al., 2008; Soulika et al., 2009). Comparable data on the time course of inflammatory infiltrates versus that of axon loss in patients with MS are not available. The mechanisms responsible for axon loss in MS and MOG peptide EAE have not been fully elucidated but likely involve axonal mitochondrial dysfunction, free radical/reactive oxygen toxicity, and axonal calcium overload (Stys, 2005; Trapp and Nave, 2008; Witte et al., 2014).
Immunohistological and in situ hybridization studies demonstrate robust expression of CCL2 by hypertrophic astroglia in and around active MS plaques and in EAE (McManus et al., 1998; Mahad and Ransohoff, 2003; Tanuma et al., 2006; Soulika et al., 2009). CCL2 signaling was first established as important for the development and progression of neuroinflammation in mice constitutively lacking the CCL2 receptor, CCR2; these mice were observed to be resistant to EAE (Izikson et al., 2000). Subsequently, constitutive deletion of CCL2 was found to diminish the severity of clinical deficits in EAE (Huang et al., 2001), and a study using bone marrow chimeric mice demonstrated the greater impact of CCL2 derived from radiation-resistant CNS cells than from the periphery in eliciting severe MOG peptide EAE (Dogan et al., 2008). In a recent MOG peptide EAE study, conditional deletion of endothelial CCL2 was found to markedly delay the onset and slightly diminish the severity of clinical deficits, whereas conditional deletion of astroglial CCL2 slightly delayed the onset and markedly diminished the severity of clinical deficits (Paul et al., 2014).
To determine the effects of astroglial CCL2 deletion on axon survival and immune cell trafficking in MOG peptide EAE, we constructed a loxP-flanked CCL2 allele and bred mice to be homozygous for this allele and to carry an astrocyte-specific mGFAPCre transgene (Garcia et al., 2004). Molecular and immunohistological characterization of the spinal cords of these mice showed that astroglial CCL2 was efficiently deleted and that astroglia were responsible for approximately half of all spinal cord CCL2 mRNA and CCL2 protein expression at the clinical peak of MOG peptide EAE. In these mice, the onset of clinical neurological deficits after MOG peptide immunization was delayed, and the severity of clinical neurological deficits was diminished. Notably, histological analysis showed that chronic spinal cord demyelination and axon loss were substantially diminished by astroglial CCL2 deletion. As previously reported in MOG peptide EAE (Jones et al., 2008), remyelinated axons, identifiable by transmission electron microscopic visualization of axons surrounded by thin myelin sheaths, were rarely present in mice either lacking or retaining the capacity for astroglial CCL2 synthesis.
Treatment with CCL2 is known to increase excitability of CCR2+ neurons, particularly those in pain pathways (Banisadr et al., 2005b; Belkouch et al., 2011). However, we are not aware of published evidence indicating that direct neuronal CCL2/CCR2 signaling can elicit neuronal or axonal loss and thus have focused our attention on the effects of astroglial CCL2 deletion on recruitment of potentially neurotoxic immune cells to the CNS.
Monocytes express CCR2 and are the best characterized cellular targets for CCL2-mediated chemotaxis (Geissmann et al., 2003). Transected axons in active MS plaques are often in close proximity to macrophages (Trapp et al., 1998; Kuhlmann et al., 2002; Huizinga et al., 2012), although the extent to which these macrophages are derived from invading monocytes versus from microglia has not been established. Arguing for an important role for monocyte-derived macrophages in EAE, M1 polarized monocyte-derived macrophages can produce potentially neurotoxic inflammatory mediators (Brenner et al., 1997; Brown and Bal-Price, 2003; Pacher et al., 2007; Dogan et al., 2008; Colombo et al., 2012), and depletion of circulating monocytes by systemic infusion of silicon particles or clodronate-containing liposomes, or anti-monocyte antibodies diminishes EAE severity (Brosnan et al., 1981; Tran et al., 1998; Mildner et al., 2009). Our data indicated that CNS accumulation of CD45hiLy6ChiMHCII+CD86+iNOS+ M1 monocyte-derived macrophages during the acute phase of MOG peptide EAE was substantially decreased by astroglial CCL2 deletion. In contrast, accumulation of M2 monocyte-derived macrophages during the acute phase of MOG peptide EAE, cells capable of exerting immunosuppressive and neuroprotective effects (Nahrendorf et al., 2007; Liu et al., 2013; Shechter et al., 2013), was not altered by astroglial CCL2 ablation.
Microglia, self-renewing CNS CD11b+CD45lo macrophages derived from embryonic yolk sac progenitors (Ginhoux et al., 2013), can also undergo polarization to an M1 phenotype and may also be key players in MS and EAE neurodegeneration (Bitsch et al., 2000; Rasmussen et al., 2007; Starossom et al., 2012). Microglia expresses CCR2 mRNA, although at lower levels than monocyte-derived macrophages, and thus may be responsive to CCL2 (Rezaie et al., 2002; Chiu et al., 2013; Hickman et al., 2013). Our flow cytometric data indicated that ablation of astroglial CCL2 diminished numbers of M1 polarized CD11b+CD45lo microglia during the acute phase of MOG peptide EAE.
Some CD4+ lymphocytes, including Th17 cells, express CCR2 (Inoue et al., 2012). Of potential relevance to the etiology of axonopathy in MOG peptide EAE, Th17 lymphocytes can establish direct, immune synapse-like contacts with axons and elicit potentially toxic alterations in neuronal intracellular Ca2+ concentrations (Siffrin et al., 2010). However, our flow cytometry data indicated that there were greater CNS accumulations of Th17 lymphocytes in the absence than in the presence of astrocyte-derived CCL2. Possibly explaining this increase, IL-6 mRNA abundance was considerably higher during the acute phase of MOG peptide EAE in mice in which astroglial CCL2 had been ablated than in littermate controls. IL-6 and TGF-β are critical factors that shift the T-cell polarization status from a T regulatory toward a Th17 response (Korn et al., 2007). Our observation of increased IL-6 expression was not accompanied by an increase in TGF-β in mice lacking astroglial CCL2. It is likely that astroglia themselves contributed to this augmented IL-6 induction because lipopolysaccharide-induced IL-6 expression has been shown to be greater in astrocytes cultured from CCL2−/− than from WT mice (Semple et al., 2010). We also found that astroglial CCL2 ablation diminished the capacity of CD4+ lymphocytes that entered the spinal cord during the acute phase of MOG peptide EAE to transit from perivascular spaces to the spinal cord parenchyma.
CNS infiltrates of neutrophils are prominent in EAE, less so in MS, and may exert immunomodulatory effects (Zehntner et al., 2005; Wu et al., 2010; Steinbach et al., 2013). As previously observed in constitutive CCR2 knock-out mice (Saederup et al., 2010), CNS neutrophil accumulations during the acute phase of MOG peptide EAE were considerably greater in mice in which astroglial CCL2 had been deleted than in littermate controls.
To summarize the effects of astroglial CCL2 ablation on immune cell trafficking that we observed, CNS accumulations of potentially neurotoxic M1 polarized monocyte-derived macrophages, and expression of M1 markers by microglia, were substantially diminished during the acute phase of MOG peptide EAE. In contrast, acute CNS Th17 lymphocyte and neutrophil infiltrates were increased in the absence of astroglial CCL2. The neutrophil chemoattractants CXCL1 and CXCL2 were both elevated in the spinal cords of mice lacking astroglial CCL2. Overall numbers of CD4+ lymphocytes in the acutely inflamed CNS were not altered in the absence of astroglial CCL2, but their capacity to exit spinal cord perivascular spaces and enter the spinal cord parenchyma was reduced.
Our results, in conjunction with prior studies showing that depletion of circulating monocytes diminishes EAE severity, support the hypothesis that M1-polarized macrophages and/or microglia play a key role in the axonal loss that characterizes MOG peptide EAE and, by extension, in the axonal loss in MS. These findings suggest that therapeutic interventions to limit monocyte-derived macrophage trafficking to the CNS, or to diminish M1 polarization of monocyte-derived macrophages and microglia in the CNS, would reduce axon loss and slow the progression of nonremitting clinical neurological deficits in patients with MS.
Footnotes
This work was supported by Shriners Hospitals for Children and National Institutes of Health Grant R01 NS025044.
The authors declare no competing financial interests.
References
- Banisadr G, Gosselin RD, Mechighel P, Kitabgi P, Rostène W, Parsadaniantz SM. Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: evidence for its colocalization with neurotransmitters and neuropeptides. J Comp Neurol. 2005a;489:275–292. doi: 10.1002/cne.20598. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Gosselin RD, Mechighel P, Rostène W, Kitabgi P, Mélik Parsadaniantz S. Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J Comp Neurol. 2005b;492:178–192. doi: 10.1002/cne.20729. [DOI] [PubMed] [Google Scholar]
- Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol. 2007;7:904–912. doi: 10.1038/nri2190. [DOI] [PubMed] [Google Scholar]
- Belkouch M, Dansereau MA, Réaux-Le Goazigo A, Van Steenwinckel J, Beaudet N, Chraibi A, Mélik-Parsadaniantz S, Sarret P. The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gβγ-dependent mechanism. J Neurosci. 2011;31:18381–18390. doi: 10.1523/JNEUROSCI.3386-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman JW, Guida MP, Warren J, Amat J, Brosnan CF. Localization of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J Immunol. 1996;156:3017–3023. [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Beutner C, Linnartz-Gerlach B, Schmidt SV, Beyer M, Mallmann MR, Staratschek-Jox A, Schultze JL, Neumann H. Unique transcriptome signature of mouse microglia. Glia. 2013;61:1429–1442. doi: 10.1002/glia.22524. [DOI] [PubMed] [Google Scholar]
- Bhasin JM, Chakrabarti E, Peng DQ, Kulkarni A, Chen X, Smith JD. Sex specific gene regulation and expression QTLs in mouse macrophages from a strain intercross. PLoS One. 2008;3:e1435. doi: 10.1371/journal.pone.0001435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Brück W. Acute axonal injury in multiple sclerosis: correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Brenner T, Brocke S, Szafer F, Sobel RA, Parkinson JF, Perez DH, Steinman L. Inhibition of nitric oxide synthase for treatment of experimental autoimmune encephalomyelitis. J Immunol. 1997;158:2940–2946. [PubMed] [Google Scholar]
- Brosnan CF, Bornstein MB, Bloom BR. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. J Immunol. 1981;126:614–620. [PubMed] [Google Scholar]
- Brown GC, Bal-Price A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol. 2003;27:325–355. doi: 10.1385/MN:27:3:325. [DOI] [PubMed] [Google Scholar]
- Carrillo-de Sauvage MA, Gómez A, Ros CM, Ros-Bernal F, Martín ED, Perez-Vallés A, Gallego-Sanchez JM, Fernández-Villalba E, Barcia C, Sr, Barcia C, Jr, Herrero MT. CCL2-expressing astrocytes mediate the extravasation of T lymphocytes in the brain: evidence from patients with glioma and experimental models in vivo. PloS One. 2012;7:e30762. doi: 10.1371/journal.pone.0030762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, Myers RM, Maniatis T. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4:385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo E, Cordiglieri C, Melli G, Newcombe J, Krumbholz M, Parada LF, Medico E, Hohlfeld R, Meinl E, Farina C. Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J Exp Med. 2012;209:521–535. doi: 10.1084/jem.20110698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haas AH, van Weering HR, de Jong EK, Boddeke HW, Biber KP. Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol. 2007;36:137–151. doi: 10.1007/s12035-007-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo MA, Cabañas C, Montoya MC, Ager A, Sánchez-Mateos P, Sánchez-Madrid F. ICAMs redistributed by chemokines to cellular uropods as a mechanism for recruitment of T lymphocytes. J Cell Biol. 1997;137:493–508. doi: 10.1083/jcb.137.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca GC, Ebers GC, Esiri MM. Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain. 2004;127:1009–1018. doi: 10.1093/brain/awh118. [DOI] [PubMed] [Google Scholar]
- DeLuca GC, Williams K, Evangelou N, Ebers GC, Esiri MM. The contribution of demyelination to axonal loss in multiple sclerosis. Brain. 2006;129:1507–1516. doi: 10.1093/brain/awl074. [DOI] [PubMed] [Google Scholar]
- Dogan RN, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF- and iNOS-expressing macrophages and myeloid dendritic cells. J Immunol. 2008;180:7376–7384. doi: 10.4049/jimmunol.180.11.7376. [DOI] [PubMed] [Google Scholar]
- Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:899–905. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ, ffrench-Constant C, Edgar JM, Smith KJ. Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol. 2012;8:624–634. doi: 10.1038/nrneurol.2012.200. [DOI] [PubMed] [Google Scholar]
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan N, Adamsky K, Kartvelishvily E, Brockschnieder D, Möbius W, Spiegel I, Roth AD, Thomson CE, Rechavi G, Peles E. Identification of Tmem10/Opalin as an oligodendrocyte enriched gene using expression profiling combined with genetic cell ablation. Glia. 2008;56:1176–1186. doi: 10.1002/glia.20688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Herrero-Herranz E, Pardo LA, Gold R, Linker RA. Pattern of axonal injury in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neurobiol Dis. 2008;30:162–173. doi: 10.1016/j.nbd.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizinga R, van der Star BJ, Kipp M, Jong R, Gerritsen W, Clarner T, Puentes F, Dijkstra CD, van der Valk P, Amor S. Phagocytosis of neuronal debris by microglia is associated with neuronal damage in multiple sclerosis. Glia. 2012;60:422–431. doi: 10.1002/glia.22276. [DOI] [PubMed] [Google Scholar]
- Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109:10480–10485. doi: 10.1073/pnas.1201836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Nguyen TT, Deboy CA, Griffin JW, Whartenby KA, Kerr DA, Calabresi PA. Behavioral and pathological outcomes in MOG 35–55 experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;199:83–93. doi: 10.1016/j.jneuroim.2008.05.013. [DOI] [PubMed] [Google Scholar]
- King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113:3190–3197. doi: 10.1182/blood-2008-07-168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Brück W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202–2212. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- Li H, Ciric B, Yang J, Xu H, Fitzgerald DC, Elbehi M, Fonseca-Kelly Z, Yu S, Zhang GX, Rostami A. Intravenous tolerance modulates macrophage classical activation and antigen presentation in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2009;208:54–60. doi: 10.1016/j.jneuroim.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Yu J, Feng L, Hou S, Liu Y, Guo M, Xie Y, Meng J, Zhang H, Xiao B, Ma C. Targeting the shift from M1 to M2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PloS One. 2013;8:e54841. doi: 10.1371/journal.pone.0054841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubkowski J, Bujacz G, Boqué L, Domaille PJ, Handel TM, Wlodawer A. The structure of MCP-1 in two crystal forms provides a rare example of variable quaternary interactions. Nat Struct Biol. 1997;4:64–69. doi: 10.1038/nsb0197-64. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/S1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- Mahbub S, Deburghgraeve CR, Kovacs EJ. Advanced age impairs macrophage polarization. J Interferon Cytokine Res. 2012;32:18–26. doi: 10.1089/jir.2011.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Marina N, Bull ND, Martin KR. A semiautomated targeted sampling method to assess optic nerve axonal loss in a rat model of glaucoma. Nat Protoc. 2010;5:1642–1651. doi: 10.1038/nprot.2010.128. [DOI] [PubMed] [Google Scholar]
- McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. 1998;86:20–29. doi: 10.1016/S0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- Mélik-Parsadaniantz S, Rostène W. Chemokines and neuromodulation. J Neuroimmunol. 2008;198:62–68. doi: 10.1016/j.jneuroim.2008.04.022. [DOI] [PubMed] [Google Scholar]
- Mildner A, Mack M, Schmidt H, Brück W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132:2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32:463–488. doi: 10.1615/CritRevImmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D, Ge S, Lemire Y, Jellison ER, Serwanski DR, Ruddle NH, Pachter JS. Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte- and endothelial-derived CCL2 in neuroinflammation. J Neuroinflammation. 2014;11:10. doi: 10.1186/1742-2094-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248:205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plüddemann A, Mukhopadhyay S, Gordon S. Innate immunity to intracellular pathogens: macrophage receptors and responses to microbial entry. Immunol Rev. 2011;240:11–24. doi: 10.1111/j.1600-065X.2010.00989.x. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- Rasmussen S, Wang Y, Kivisäkk P, Bronson RT, Meyer M, Imitola J, Khoury SJ. Persistent activation of microglia is associated with neuronal dysfunction of callosal projecting pathways and multiple sclerosis-like lesions in relapsing–remitting experimental autoimmune encephalomyelitis. Brain. 2007;130:2816–2829. doi: 10.1093/brain/awm219. [DOI] [PubMed] [Google Scholar]
- Rezaie P, Trillo-Pazos G, Everall IP, Male DK. Expression of β-chemokines and chemokine receptors in human fetal astrocyte and microglial co-cultures: potential role of chemokines in the developing CNS. Glia. 2002;37:64–75. doi: 10.1002/glia.1128. [DOI] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PloS One. 2010;5:e13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Frugier T, Morganti-Kossmann MC. CCL2 modulates cytokine production in cultured mouse astrocytes. J Neuroinflammation. 2010;7:67. doi: 10.1186/1742-2094-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim KW, Klein E, Kalchenko V, Bendel P, Lira SA, Jung S, Schwartz M. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38:555–569. doi: 10.1016/j.immuni.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siffrin V, Radbruch H, Glumm R, Niesner R, Paterka M, Herz J, Leuenberger T, Lehmann SM, Luenstedt S, Rinnenthal JL, Laube G, Luche H, Lehnardt S, Fehling HJ, Griesbeck O, Zipp F. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity. 2010;33:424–436. doi: 10.1016/j.immuni.2010.08.018. [DOI] [PubMed] [Google Scholar]
- Sinha S, Kaler LJ, Proctor TM, Teuscher C, Vandenbark AA, Offner H. IL-13-mediated gender difference in susceptibility to autoimmune encephalomyelitis. J Immunol. 2008;180:2679–2685. doi: 10.4049/jimmunol.180.4.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D. Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:14965–14979. doi: 10.1523/JNEUROSCI.3794-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ, Cao Y, Bondar G, Lam J, Ao Y, Sandoval F, Suriany S, Sofroniew MV, Voskuhl RR. Neuroprotection mediated through estrogen receptor-alpha in astrocytes. Proc Natl Acad Sci U S A. 2011;108:8867–8872. doi: 10.1073/pnas.1103833108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence RD, Wisdom AJ, Cao Y, Hill HM, Mongerson CR, Stapornkul B, Itoh N, Sofroniew MV, Voskuhl RR. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERα signaling on astrocytes but not through ERβ signaling on astrocytes or neurons. J Neurosci. 2013;33:10924–10933. doi: 10.1523/JNEUROSCI.0886-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starossom SC, Mascanfroni ID, Imitola J, Cao L, Raddassi K, Hernandez SF, Bassil R, Croci DO, Cerliani JP, Delacour D, Wang Y, Elyaman W, Khoury SJ, Rabinovich GA. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37:249–263. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach K, Piedavent M, Bauer S, Neumann JT, Friese MA. Neutrophils amplify autoimmune central nervous system infiltrates by maturing local APCs. J Immunol. 2013;191:4531–4539. doi: 10.4049/jimmunol.1202613. [DOI] [PubMed] [Google Scholar]
- Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci. 2005;233:3–13. doi: 10.1016/j.jns.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Szaro BG, Whitnall MH, Gainer H. Phosphorylation-dependent epitopes on neurofilament proteins and neurofilament densities differ in axons in the corticospinal and primary sensory dorsal column tracts in the rat spinal cord. J Comp Neurol. 1990;302:220–235. doi: 10.1002/cne.903020204. [DOI] [PubMed] [Google Scholar]
- Tallantyre EC, Bø L, Al-Rawashdeh O, Owens T, Polman CH, Lowe JS, Evangelou N. Clinico-pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult Scler. 2010;16:406–411. doi: 10.1177/1352458510364992. [DOI] [PubMed] [Google Scholar]
- Tanuma N, Sakuma H, Sasaki A, Matsumoto Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006;112:195–204. doi: 10.1007/s00401-006-0083-7. [DOI] [PubMed] [Google Scholar]
- Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol. 2013;33:1135–1144. doi: 10.1161/ATVBAHA.113.301453. [DOI] [PubMed] [Google Scholar]
- Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65:239–248. doi: 10.1002/ana.21640. [DOI] [PubMed] [Google Scholar]
- Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med. 2014;20:179–187. doi: 10.1016/j.molmed.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Wu Y, Ye XH, Guo PP, Xu SP, Wang J, Yuan SY, Yao SL, Shang Y. Neuroprotective effect of lipoxin A4 methyl ester in a rat model of permanent focal cerebral ischemia. J Mol Neurosci. 2010;42:226–234. doi: 10.1007/s12031-010-9355-8. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehntner SP, Brickman C, Bourbonnière L, Remington L, Caruso M, Owens T. Neutrophils that infiltrate the central nervous system regulate T cell responses. J Immunol. 2005;174:5124–5131. doi: 10.4049/jimmunol.174.8.5124. [DOI] [PubMed] [Google Scholar]